Abstract
Background
The combination of umeclidinium (UMEC), a long-acting muscarinic receptor antagonist, and vilanterol (VI), a selective long-acting β2 agonist, is in development for the treatment of chronic obstructive pulmonary disease (COPD). This study evaluated the pharmacokinetics, safety and tolerability, and pharmacodynamics of once-daily, inhaled UMEC and UMEC/VI when co-administered with oral verapamil, a moderate P-glycoprotein transporter and moderate cytochrome P450 3A4 (CYP3A4) inhibitor frequently used by patients with COPD and cardiovascular comorbidities.
Methods
Subjects were randomized to one of two 13-day treatment regimens: UMEC 500 μg or UMEC 500 μg/VI 25 μg. All subjects received a single tablet containing 240 mg verapamil on each of days 9–13.
Results
Repeat doses of UMEC and UMEC/VI in combination with and without verapamil were safe and well tolerated. There was no increase in systemic exposure of UMEC when administered in combination with VI compared to UMEC alone. UMEC maximum concentration was similar with or without verapamil; a moderate increase in UMEC area under the curve (approximately 1.4-fold) was observed with verapamil. Verapamil did not increase systemic exposure to VI following administration of the UMEC/VI combination.
Conclusion
Administration of UMEC and UMEC/VI combination was well tolerated and did not show clinically relevant increases in systemic exposure for either drug. The UMEC/VI combination is unlikely to have a clinically meaningful drug–drug interaction with moderate P-glycoprotein transporter and CYP3A4 inhibitor drugs.
Introduction
Chronic obstructive pulmonary disease (COPD) is widespread and affects an estimated 210 million people worldwide.Citation1–Citation3 COPD has a high personal, societal, and economic impact, especially in industrialized countries and working-age populations, often necessitating early retirement.Citation1,Citation2,Citation4,Citation5
Current treatment guidelines recommend using bronchodilators, usually a β2-adrenoceptor agonist, or a long-acting muscarinic antagonist (LAMA) for treatment of symptoms associated with COPD. If symptoms are not adequately controlled by these monotherapies, additional benefit may be provided by combination therapy with different drug classes.Citation6–Citation8 Furthermore, delivery of two agents with a single inhaler, once daily (QD) could provide greater ease of use and increased adherence.Citation9,Citation10
Umeclidinium (UMEC) is a new inhaled LAMA that has been evaluated in clinical studies in healthy volunteersCitation11,Citation12 and patients with COPDCitation13–Citation15 following single- and repeat-dose administration. Vilanterol is a potent and selective long-acting β2 agonist that has been evaluated in healthy volunteers and subjects with asthma and COPDCitation16 following single- and repeat-dose administration and in combination with fluticasone furoate and the LAMA darotropium.Citation17 A UMEC/vilanterol (VI) combination is under development as a QD combination therapy for treatment of COPD.Citation18,Citation19
We report the key results of an open-label, QD, repeat-dose study (GlaxoSmithKline protocol: DB2113950; Clinicaltrials.gov identifier: NCT1128634) evaluating the effects of oral administration of verapamil 240 mg, a moderate P-glycoprotein transporter and cytochrome P450 isozyme 3A4 (CYP3A4) inhibitorCitation20 frequently used by patients with COPD and cardiovascular related comorbidities, on the pharmacokinetics (PK), safety and tolerability, and pharmacodynamics (PD) of inhaled UMEC 500 μg monotherapy and UMEC 500 μg/VI 25 μg combination therapy.
Methods
Subjects
Healthy male and female non-smoking volunteers (of non-childbearing potential and 18–65 years of age) with a body weight > 45 kg and a body mass index within the range 18–28 kg/m2 were enrolled. Subjects were required to have no clinically active and relevant abnormality on a 12-lead electrocardiogram (ECG) or 24-hour Holter ECG, and to have spirometry results within the normal range (forced expiratory volume in 1 second [FEV1] ≥ 80% of predicted, FEV1/forced vital capacity ≥ 70% in 1 second) at screening. Subjects with a QT interval corrected with Bazzet’s formula (QTcB) > 450 msec or an ECG not suitable for QT measurements were excluded.
All volunteers provided written, informed consent prior to screening and the study was conducted in accordance with Good Clinical Practice and the guiding principles of the World Medical Association Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects.Citation21
Study design
This was a randomized, open-label study. The data were collected from Hammersmith Medicines Research (London, UK) between March and April 2010. UMEC and UMEC/VI were delivered by dry powder inhaler. Verapamil was administered orally as a single tablet. The planned sample size was primarily based on feasibility and not on statistical considerations. The randomization schedule was generated by Discovery Biometrics (GlaxoSmithKline, Stevenage, UK), prior to the start of the study, using validated internal software. Subjects were randomized in order to have approximately 14 subjects in each cohort completing the dosing and critical assessments. Subjects were randomized to one of two cohorts, each subject taking part in two treatment periods of 13 days. For Cohort 1, the Period 1 treatment was UMEC 500 μg QD for 8 days, immediately followed by Period 2 treatment for 5 days of UMEC 500 μg QD and verapamil 240 mg QD. For Cohort 2, the Period 1 treatment was UMEC 500 μg/VI 25 μg QD for 8 days, immediately followed by Period 2 treatment for 5 days of UMEC 500 μg/VI 25 μg QD and verapamil 240 mg QD.
Volunteers received a physical examination, laboratory tests, lung function assessments, ECG, and Holter monitoring during the screening visit (≤30 days prior to dosing). Subjects were admitted to the unit on Day 1 for completion of baseline safety tests prior to dose administration on Day 1, and remained resident until the 24-hour assessments post-dosing. The follow-up visit was conducted within 5 to 10 days of the final dose of study drug.
Sample collection
Blood samples were taken via an indwelling cannula or by direct venipuncture, collected into an ethylenediaminetetraacetic acid tube and immediately placed on water ice. Total blood volume for clinical laboratory and PK samples over the duration of the study did not exceed 500 mL. Samples were centrifuged at 1500 rpm for 10 minutes under chilled conditions. Supernatant plasma was transferred to a 3.6 mL Nunc tube and stored at −80°C before shipment. Samples were batched and shipped, frozen on dry ice, to GlaxoSmithKline for analyses.
Urine samples were kept refrigerated between voids until the individual collection periods were finished. Urine volumes were recorded for collections at 4, 8, 12, and 24-hour time points. Following mixing, a 10 mL aliquot from each collection period was transferred to appropriately labeled polypropylene specimen containers. Urine aliquots were immediately frozen at approximately −20°C, until shipped to a central lab for analysis.
Pharmacokinetic analyses
Plasma samples for UMEC and VI were analyzed using a validated analytical method based on protein precipitation, followed by high-performance liquid chromatography with mass spectrometry analysis. The lower limit of quantitation (LLQ) for UMEC and VI were 20 pg/mL and 30 pg/mL, respectively, using a 100 μL aliquot of human plasma. The higher limit of quantification for UMEC and VI was 20,000 pg/mL and 30,000 pg/mL, respectively. Quality control (QC) samples, containing UMEC and VI at three different concentrations and stored with study samples, were analyzed with each batch of samples against separately prepared calibration standards. For the analysis to be acceptable, no more than one-third of the QC results were to deviate from the nominal concentration by more than 15%, and at least 50% of the results from each QC concentration were to be within 15% of nominal. The applicable analytical runs met all predefined run acceptance.
Urine samples were analyzed for UMEC using a validated analytical method based on dilution, followed by high-performance liquid chromatography with mass spectrometry analysis. The LLQ for UMEC was 0.1 ng/mL using a 50 mL aliquot of human urine with a higher limit of quantification of 50 ng/mL. For each analytical method, QC samples, containing UMEC at three different concentrations and stored with study samples, were analyzed with each batch of samples against separately prepared calibration standards. For the analysis to be acceptable, no more than one-third of the QC results were to deviate from the nominal concentration by more than 15%, and at least 50% of the results from each QC concentration should be within 15% of nominal. The applicable analytical runs met all predefined run acceptance criteria.
Concentrations of UMEC and VI in plasma were summarized by treatment and planned sampling time point. The derived PK parameters area under the plasma concentration-time curve from time zero to 0.25 hours (AUC(0–0.25h)), AUC(0–2h), AUC(0–t), AUC(0–∞), AUClast, maximum observed plasma concentration (Cmax), terminal phase half-life (t½), time to Cmax (tmax), and tlast were summarized for the analyte UMEC. The parameters AUC(0–0.25h), AUC(0–0.5h), AUC(0–2h), Cmax, t½, tmax, and tlast were summarized for the analyte VI. Vilanterol AUC(0–∞) was planned; however, due to limitations in the plasma concentration profile, only AUC(0–2h) and previous AUCs were possible.
For the analyte UMEC, log-transformed values of AUC(0–∞) and Cmax were analyzed and for the analyte VI, log-transformed values of AUC(0–0.25h), AUC(0–2 h), and Cmax were analyzed using a mixed effects model. Treatment was fitted as a fixed effect and subject was fitted as a random effect. The ratios and 90% confidence intervals (CIs) for comparing the effects of verapamil after UMEC and UMEC/VI combination were presented. The treatment ratios were calculated by back-transforming the difference between the adjusted means obtained from the analyses. The adjusted geometric means and 90% CIs of these geometric means by treatment for each analyte were also presented.
Blood potassium
Potassium, analyzed in the routine clinical laboratory sample, was monitored to assess the PD effects of VI and verapamil. Minimum (0–4 hours) and weighted mean (0–4 hours) for blood potassium were derived and each of these variables was statistically analyzed using a mixed effects model. The model included no baseline (none available), treatment was fitted as a fixed effect, and subject was fitted as a random effect.
Safety analyses
Adverse event (AE) and serious AE data were collected and recorded starting on Day 1 and continuing through the end of the confinement period and at follow-up. All safety and tolerability endpoints (AEs, heart rate [HR], systolic and diastolic blood pressure, 12-lead ECG [QTcB and QT interval corrected using Friedericia’s formula (QTcF)], lung function FEV1, 24-hour Holter monitoring including maximum and mean HR, and laboratory tests) were summarized. Plots of means and 95% CIs for maximum and mean (0–24-hour) Holter HRs were produced. Maximum (0–4 hours) and weighted mean (0–4 hours) of HR (vital signs), QTcB, and QTcF were derived and each of these variables was separately analyzed using a mixed effects model. Subject-level baseline, period-level baseline, period, and treatment group were fitted as fixed effects and subject was fitted as a random effect.
Results
Baseline characteristics and subject disposition
Thirty-two subjects were enrolled and 29 subjects (91%) completed the study. Subject disposition and demographics are shown in . Three subjects had AEs that led to withdrawal. All were considered possibly treatment related and resolved following treatment discontinuation. One subject was withdrawn on Day 4 of UMEC with verapamil dosing due to several AEs (dyspepsia, nausea, vomiting, and headache). One subject receiving UMEC/VI was withdrawn on Day 7 due to elevated liver function tests. One subject withdrew consent on Day 3 of UMEC treatment after experiencing feelings of paranoia.
Table 1 Summary of subject disposition and demographic characteristics
Pharmacokinetics
Following Day 1 dose administration of UMEC 500 μg monotherapy, UMEC plasma concentrations were below the LLQ in two of 16 subjects at or before 2 hours post-dose, the last sampling time point. UMEC was quantifiable in the plasma of all 16 subjects up to 2 hours post-dose following administration of UMEC 500 μg and VI 25 μg in combination.
Following repeat dose administration of UMEC monotherapy for 8 days, UMEC was below the LLQ in plasma in three of 15 subjects at or before the last sampling time point (Day 8, 24 hours post-dose). UMEC was quantifiable in plasma in all 15 subjects up to 24 hours post-dose following administration of UMEC/VI. Following Day 13 dose administration of UMEC and verapamil 240 mg, UMEC plasma concentrations were below the LLQ in one of 14 subjects at or before the last sampling time point (48 hours post-dose). UMEC was quantifiable in plasma up to 48 hours post-dose following Day 13 administration of UMEC/VI and verapamil in all 15 subjects.
Plasma concentrations of UMEC and derived PK parameters are summarized in with the ratio of adjusted means to evaluate the effect of verapamil administration. Following both single- and repeat-dose administration of either UMEC monotherapy or UMEC/VI combination, with or without oral administration of verapamil, UMEC was rapidly absorbed with the Cmax occurring 5 minutes post-dose. Moderate-to-large intrasubject variability was observed for both treatments with values for the intrasubject coefficient of variation ranging from 46% to 86% for Cmax, and 27% to 117% for the AUC.
Table 2 Pharmacokinetics of UMEC
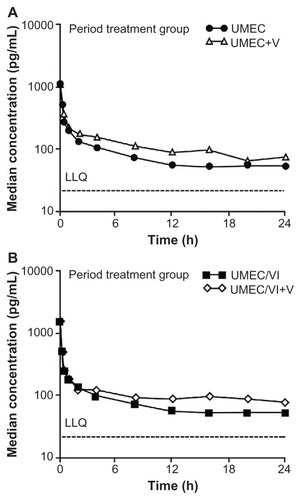
The ratio of adjusted geometric means of Cmax was similar when UMEC or UMEC/VI was administered in the presence or absence of verapamil. The treatment ratio for Cmax for the UMEC treatment group was 1.05 (90% CI: 0.90, 1.22) and for the UMEC/VI treatment group the ratio was 0.89 (90% CI: 0.73, 1.07). The treatment ratio for AUC(0–t), for the UMEC treatment group was 1.39 (90% CI: 1.18, 1.64) and for the UMEC/VI treatment group the ratio was 1.37 (90% CI: 1.29, 1.46). A graphic representation of the effect of verapamil on UMEC systemic exposure as monotherapy (, Panel A) and as UMEC/VI combination (, Panel B) is presented. UMEC urine data were in agreement with plasma data which showed a slight increase in renal clearance of unchanged UMEC with higher plasma concentrations (data not shown).
Figure 1 Median plasma umeclidinium concentration-time semi-log plot at Day 8 (without verapamil) and Day 13 (with verapamil). (A) Umeclidinium treatment group; (B) Umeclidinium/vilanterol treatment group.
Overall, 71% of plasma samples (299/422) showed non-quantifiable (NQ) VI concentrations. Following Day 1 dose administration of VI 25 μg in combination with UMEC, all of the VI plasma concentrations were NQ after 5 minutes post-dose. Following Day 8 dose administration the majority of plasma samples had quantifiable concentrations of VI up to 30 minutes and all were NQ after 2 hours post-dose. Following Day 13 dose administration the majority of plasma samples had quantifiable concentrations of VI up to 30 minutes and all were NQ after 8 hours post-dose.
Plasma concentrations of VI and derived PK parameters are summarized in along with the ratio of adjusted means to evaluate the effect of verapamil. Following both single- and repeat-dose administration of UMEC/VI, and UMEC/VI and verapamil, VI was rapidly absorbed following inhalation, with the majority of the Cmax values occurring at 5 minutes post-dose, followed by a rapid decline. The AUC and apparent t½ could not be determined on Day 1 due to the majority of samples having NQ concentrations of VI. Moderate-to-high intrasubject variability was observed for both treatments, with values for the coefficient of variation ranging from 39% to 79% for Cmax, and 54% to 128% for AUC.
Table 3 Pharmacokinetics of VI
Statistical analysis of VI PK parameters showed no difference in VI Cmax and AUC following UMEC/VI and verapamil co-administration compared with UMEC/VI alone.
Safety and tolerability
Twenty-one (66%) subjects reported at least one AE. Six subjects in each of the UMEC, UMEC with verapamil, and UMEC/VI treatment periods reported AEs. Nine subjects in the UMEC/VI with verapamil treatment period reported AEs. All the AEs were rated as mild or moderate by the investigator. The most frequently reported AE was headache, which occurred in eight subjects (25%) across all regimens.
Three subjects had AEs that led to withdrawal from the study (). All were considered possibly treatment-related and resolved following treatment discontinuation. One subject was withdrawn on Day 4 of UMEC with verapamil dosing due to AEs of dyspepsia, nausea, vomiting, and headache which began on Day 3. One subject receiving UMEC/VI withdrew on Day 7 due to elevated liver function tests. The alanine amino transferase values for this subject (150–172 IU/L [normal range: 8–36 IU/L]) fulfilled the stopping criteria (≥ 3 × upper limit of normal). The subject was asymptomatic and liver chemistry results returned to acceptable limits after approximately 20 days. The full hepatitis profile was normal. A single blood sample was taken for PK analysis following the liver event on Day 7. Plasma UMEC concentration was 89 pg/mL (comparable with 96 pg/mL recorded at 2 hours post-dose on Day 1) and showed that the concentration observed was within the normal range. Plasma VI was below the quantification limit. One subject withdrew consent on Day 3 of UMEC treatment after experiencing feelings of paranoia, not sleeping well, and having nightmares.
There were no clinically significant vital signs, 12-lead ECG, or Holter findings. No subject had a resting HR > 130 bpm (summary of vital signs not shown). No subject had a resting pulse rate increase > 40 bpm from baseline (summary of ECG data not shown). No subject had a QTc > 500 msec or an absolute change from baseline > 60 msec.
A summary of the statistical analysis of vital signs and ECG parameters is shown in . For maximum HR (0–4 hours) and weighted mean HR (0–4 hours), the average changes from baseline were small for both UMEC and UMEC/VI treatment comparisons with and without verapamil. Values of QTcB, QTcB (0–4 hours), and QTcF (0–4 hours) showed increases for comparisons of these two treatments with and without verapamil.
Table 4 Statistical analyses of vital signs and ECG parameters
Pharmacodynamics
Lowered blood potassium is a known PD effect of both β2 agonists and verapamil.Citation20,Citation22 Lowered blood potassium was evaluated by monitoring potassium levels from 0–4 hours post-dose and a summary of the statistical analysis of minimum and weighted mean potassium levels is shown in . The administration of verapamil reduced the mean minimum (0–4 hours) potassium by −0.10 mmoL/L in the UMEC group and −0.13 mmoL/L in the UMEC/VI treatment group. These changes were not considered to be clinically significant. The administration of verapamil decreased the mean weighted mean (0–4 hours) potassium by −0.06 mmoL/L in the UMEC group and −0.10 mmoL/L in the UMEC/VI treatment group. These changes were not considered to be clinically significant. The mean minimum potassium in the UMEC/VI group (4.05 mmoL/L) is essentially the same as the mean minimum potassium for the UMEC group (4.03 mmoL/L). Likewise, the weighted mean potassium for the UMEC/VI group and the UMEC group were equivalent (each 4.14 mmoL/L), indicating no effect of VI on blood potassium.
Table 5 Summary of analysis of minimum and weighted mean potassium
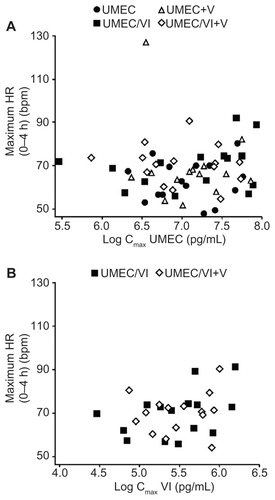
Pooled scatter plots of the PD variable individual change from baseline maximum for supine HR versus the UMEC Cmax (, Panel A) and VI Cmax (, Panel B) indicated that there was no obvious PK/PD relationship.
Figure 2 Plot of individual maximum (0–4 hours) heart rate versus log Cmax (by treatment). (A) Umeclidinium log Cmax; (B) Vilanterol log Cmax.
Discussion
We report the results of a repeat-dose assessment of administration of oral verapamil 240 mg QD on the steady-state PK following repeated doses of UMEC and UMEC/VI, both delivered via a dry powder inhaler device. At steady state there was no increase in UMEC systemic exposure in the presence of VI and systemic exposure was somewhat lower with combination treatment compared with monotherapy. Furthermore, these results were independent of the administration of verapamil.
UMEC AUCs increased by approximately 40% with co-administration of verapamil for both UMEC monotherapy and UMEC/VI. However, the ratio of the adjusted geometric means of Cmax was not different when co-administered with verapamil for either UMEC monotherapy or UMEC/VI. These data suggest that UMEC Cmax is likely driven by rapid absorption from the lung, as the tmax was 5 minutes and therefore not affected by the presence of verapamil, which would have its effect on absorption from the gastrointestinal tract.
The VI PK data also suggested rapid distribution and elimination of drug from the systemic circulation, with VI being NQ in the majority of the later time point samples. Results also indicated that verapamil had no effect on VI Cmax and AUC when verapamil was co-administered with UMEC/VI. While VI systemic exposure in terms of AUC was 14% higher in the presence of verapamil on Day 13, the ratio between Day 13 to Day 8 contained unity, thereby indicating that the increase was not statistically significant. These data suggest that similar to UMEC, Cmax for VI is likely driven by rapid absorption from the lung (tmax = 5 minutes) and this is not affected by the presence of verapamil. The rapid absorption of both UMEC and VI is consistent with observations from previous studies involving healthy volunteersCitation11,Citation12,Citation16 or COPD patientsCitation16 which reported tmax values between 5–15 minutes for UMEC and 5–10 minutes for VI.
There were no obvious trends observed between individual change from baseline maximum for supine HR and UMEC Cmax or VI Cmax when administered as UMEC, UMEC/VI, or UMEC/VI co-administered with verapamil. The general lack of VI PD effects on HR, blood potassium, and QTcF seen with verapamil co-administration is consistent with these effects being predominantly attributable to the VI Cmax.
Repeated doses of inhaled UMEC and UMEC/VI were generally well tolerated alone and when co-administered with verapamil. These safety results are consistent with previous reports of UMEC and UMEC/VI in healthy volunteersCitation11,Citation12,Citation19 and in patients with COPD.Citation13–Citation15,Citation18 There were no deaths or serious AEs reported in the present study, although three subjects experienced AEs that led to discontinuation of the investigational product. There were no clinically significant laboratory values, other than the alanine amino transferase elevation in one subject, and no clinically significant vital signs, 12-lead ECG, or Holter findings. The observed increases in both weighted mean QTcB and QTcF over the 4-hour period after dosing when verapamil was co-administered with UMEC (vs UMEC alone) were consistent with the use of verapamil and were not considered to be clinically significant.Citation22
Verapamil is one of the drugs frequently used by patients with COPD and comorbidities such as hypertension and other cardiovascular ailments. A concern for the co-administration of verapamil and β agonists is the known potential for each to contribute to a lowering of blood potassium levels.Citation23,Citation24 Slight changes in the minimum (0–4 hours) potassium and weighted mean (0–4 hours) potassium following the co-administration of verapamil were not considered clinically significant. There was no reduction of blood potassium in the UMEC/VI treatment group compared to the UMEC treatment group.
These results showing no significant effect on either UMEC or VI Cmax, given that Cmax might potentially be related to a clinical effect on HR or other side effects, and the lack of a clinically significant effect on PD or safety from the modest increase in UMEC AUC, support clinical progression in this patient population. It should also be noted that the 500 μg dose of UMEC in this study was significantly higher than the 125 μg dose progressed in the Phase III program for development of the UMEC/VI combination.
Limitations of this study include that it did not include a monotherapy arm for VI, therefore, the effect of UMEC on VI systemic exposure at steady state could not be examined. The limit of LLQ of the bioanalytical method was such that full characterization of the VI plasma concentration–time profile and analysis of the PK parameters following administration of VI in combination with UMEC was not possible.
In conclusion, repeat inhaled doses of UMEC 500 μg and UMEC 500 μg/VI 25 μg in healthy volunteers was safe and well tolerated and not associated with clinically significant changes in systemic exposure. Although a moderate increase in UMEC AUC in the presence of verapamil suggested a weak interaction, the UMEC/VI combination is unlikely to have a clinically meaningful drug–drug interaction with moderate P-glycoprotein transporter and CYP3A4 inhibitor drugs.
Acknowledgments
This study was sponsored by GlaxoSmithKline. The authors thank the volunteers and staff of Hammersmith Medicines Research, London, UK who participated. The pharmacokinetic analyses were performed by Phast Clinical Data Inc (Raleigh, NC, USA).
RM contributed to the study design, data analysis, and interpretation. DK contributed to the study design and data interpretation. AP contributed to the study design and data collection. SH contributed to data collection. GC was the medical monitor. All authors provided input and/or reviewed drafts of the manuscript and approved the final version for submission.
Editorial support (in the form of writing assistance, assembling tables and figures, collating author comments, grammatical editing, and referencing) was provided by Gloria Boone and Lawrence Boone, Boone Consulting Services and was funded by GlaxoSmithKline. Additional editorial support (in the form of copyediting and final manuscript preparation) was provided by Tara N Miller, PhD at Gardiner-Caldwell Communications and was funded by GlaxoSmithKline. The Open Access publication fee was paid by GlaxoSmithKline.
Disclosure
All authors are full-time employees of, and own stock in, GlaxoSmithKline. The authors report no other conflicts of interest.
References
- FletcherMJUptonJTaylor-FishwickJCOPD uncovered: an international survey on the impact of chronic obstructive pulmonary disease [COPD] on a working age populationBMC Public Health20111161221806798
- ManninoDMBramanSThe epidemiology and economics of chronic obstructive pulmonary diseaseProc Am Thorac Soc20074750250617878461
- HalbertRJNatoliJLGanoABadamgaravEBuistASManninoDMGlobal burden of COPD: systematic review and meta-analysisEur Respir J200628352353216611654
- NgTPNitiMFonesCYapKBTanWCCo-morbid association of depression and COPD: a population-based studyRespir Med2009103689590119136238
- FosterTSMillerJDMartonJPCaloyerasJPRussellMWMenzinJAssessment of the economic burden of COPD in the US: a review and synthesis of the literatureCOPD20063421121817361502
- GOLD: Global initiative for chronic obstructive lung diseaseGlobal Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease2011 Available from: http://www.goldcopd.org/uploads/users/files/GOLD_Report_2011_Feb21.pdfAccessed November 23, 2012
- Gruffydd-JonesKLoveridgeCThe 2010 NICE COPD Guidelines: how do they compare with the GOLD guidelines?Prim Care Respir J201120219920421437566
- FromerLCooperCBA review of the GOLD guidelines for the diagnosis and treatment of patients with COPDInt J Clin Pract20086281219123618547365
- TashkinDPFabbriLMLong-acting beta-agonists in the management of chronic obstructive pulmonary disease: current and future agentsRespir Res20101114921034447
- CazzolaMCalzettaLMateraMGß(2) -adrenoceptor agonists: current and future directionBr J Pharmacol2011163141721232045
- MehtaRHardesKCahnASafety, tolerability and pharmacokinetics (PK) of repeated doses of GSK573719 Inhalation Powder, a new long-acting muscarinic antagonist, in healthy adults [abstract]Eur Respir J2011a38Suppl 5723s
- CahnALovickRNewlandsASafety, tolerability, pharmacodynamics (PD) and pharmacokinetics (PK) of GSK573719 inhalation powder in healthy subjects [abstract]Eur Resp J201138Suppl 55723s
- MehtaRNewlandsAKelleherDPreeceACahnACraterGSafety, pharmacokinetics (PK) and pharmacodynamics (PD) of single doses of GSK573719 inhalation powder, a new long-acting muscarinic antagonist (LAMA), in patients with COPDEur Respir J201138Suppl 55138s
- DonohueJAnzuetoABrooksJMehtaRKalbergCCraterGA randomized, double-blind, dose-ranging study of the novel LAMA GSK573719 in patients with COPDRespir Med2012106797097922498110
- DecramerMMaltaisFFeldmanGBronchodilation of umeclidinium, a new long-acting muscarinic antagonist, in COPD patientsRespir Physiol Neurobiol2013185239339923026438
- KempsfordRNorrisVSiedererSVilanterol trifenatate, a novel inhaled long-acting beta2 adrenoceptor agonist, is well tolerated in healthy subjects and demonstrates prolonged bronchodilation in subjects with asthma and COPDPulm Pharmacol Ther201326225626423232038
- ProcopiouPABarrettVJBevanNJSynthesis and structure-activity relationships of long-acting beta2 adrenergic receptor agonists incorporating metabolic inactivation: an antedrug approachJ Med Chem201053114522453020462258
- FeldmanGWalkerRRBrooksJMehtaRCraterG28-day safety and tolerability of umeclidinium in combination with vilanterol in COPD: a randomized placebo-controlled trialPulm Pharmacol Ther201225646547122955035
- KelleherDLMehtaRSJean-FrancoisBMSafety, tolerability, pharmacodynamics and pharmacokinetics of umeclidinium and vilanterol alone and in combination: a randomized crossover trialPLoS ONE2012712e5071623284643
- US Food and Drug AdministrationGuidance for Industry Drug Interaction Studies – Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations Draft GuidanceSilver SpringUS Food and Drug Administration2012 Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdfAccessed November 23, 2012
- World Medical AssociationWorld Medical Association Declaration of Helsinki – Ethical Principles for Medical Research Involving Human SubjectsSeoul59th WMA General Assembly2008 Available from: http://www.wma.net/en/30publications/10policies/b3/17c.pdfAccessed November 23, 2012
- GD Searle LLCCalan (verapamil hydrochloride) [Package insert]New YorkGD Searle LLC, Division of Pfizer Inc2010 Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/018817s024lbl.pdfAccessed November 23, 2012
- SalpeterSRCardiovascular safety of beta(2)-andrenoceptor agonist use in patients with obstructive airway disease: a systematic reviewDrugs Aging200421640541415084142
- SovaniMPWhaleCITattersfieldAEA benefit-risk assessment of inhaled long-acting beta2-agonists in the management of obstructive pulmonary diseaseDrug Saf2004271068971515350154