
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Mutations of the CFTR gene cause cystic fibrosis (CF), the most common recessive monogenic disease worldwide. These mutations alter the synthesis, processing, function, or half-life of CFTR, the main chloride channel expressed in the apical membrane of epithelial cells in the airway, intestine, pancreas, and reproductive tract. Lung disease is the most critical manifestation of CF. It is characterized by airway obstruction, infection, and inflammation that lead to fatal tissue destruction. In spite of great advances in early and multidisciplinary medical care, and in our understanding of the pathophysiology, CF is still considerably reducing the life expectancy of patients. This review highlights the current development in pharmacological modulators of CFTR, which aim at rescuing the expression and/or function of mutated CFTR. While only Kalydeco® and Orkambi® are currently available to patients, many other families of CFTR modulators are undergoing preclinical and clinical investigations. Drug repositioning and personalized medicine are particularly detailed in this review as they represent the most promising strategies for restoring CFTR function in CF.
Introduction
Cystic fibrosis and the CFTR gene
Cystic fibrosis (CF) is an inherited (recessive autosomal) chronic disease that affects the respiratory, digestive, and reproductive systems. Although intestinal symptoms are usually the first to occur during the life of the patient, it is the progressive lung damage, due to cycles of infection/inflammation, that finally leads to irreversible lung disease and death. With ~90,000 people diagnosed, a prevalence of 1/2,500 and about one carrier among 25 individuals, CF is the most common life-threatening Mendelian disorder worldwide. Advances in research and medical treatments have raised the life expectancy of CF newborns beyond 50 years; however, the current median age of survival for CF patients is still in the late 20s.
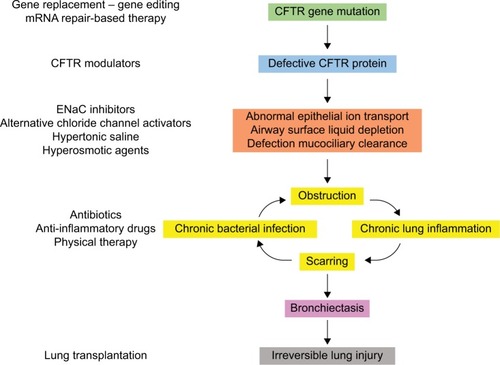
CF is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene, which was cloned and identified as the gene affected in CF in 1989.Citation1 CFTR gene encodes the main anion channel expressed in the epithelium. Additionally, CFTR is also expressed in many other cells types (eg, fibroblasts,Citation2 neurons,Citation3 cardiomyocytes,Citation4 and immune cellsCitation5–Citation7), where its function is not always well known. Among the 2,000+ CFTR mutations identified so far (http://genet.sickkids.on.ca), only a fraction of them causes CF. These CF-causing mutations induce a decrease or a loss of function of CFTR at the plasma membrane. In the lung, the lack of CFTR leads to dehydration of the airway surface liquid and drives the cascade of pathological events characteristic of CF ().
Figure 1 Pathophysiology of CF lung disease and potential therapies targeting the basic defect or the symptoms.
Abbreviations: CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; ENaC, epithelial sodium channel; mRNA, messenger RNA.
Structure of CFTR
The CFTR gene contains 27 exons spanning 250 kb on the long arm of chromosome 7 (7q31.2).Citation8,Citation9 The encoded mRNA is ~6.5 kb long and is translated into a protein of 1,480 amino acids. The CFTR protein belongs to the adenosine triphosphate (ATP)-binding cassette (ABC) transporters and functions as an adenosine 3′,5′-cyclic monophosphate (cAMP)-regulated chloride channel in a variety of polarized epithelial cells.Citation10 The predicted protein structure is shown in .
Figure 2 Predicted topology of CFTR protein.
Abbreviations: CFTR, cystic fibrosis transmembrane conductance regulator; MSD, membrane-spanning domain; NBD, nucleotide-binding domain; TMD: transmembrane domain; R, regulatory domain.
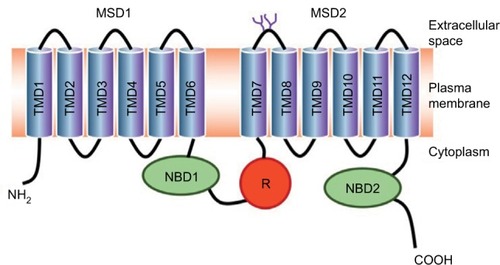
The R domain is a unique structural feature of CFTR as it is not found in other ABC transporters.Citation11 The R domain is highly charged and contains multiple consensus sequences for protein kinase A phosphorylationCitation12,Citation13 as well as target sites for other kinases.Citation14–Citation16 Phosphorylation of the R domain of CFTR is necessary for channel activity: when unphosphorylated, the R domain inhibits CFTR.Citation17,Citation18 Although the phosphorylation of the R region is required, it is not sufficient for opening the CFTR channelCitation13,Citation19–Citation21 nor for the interaction with multiple binding partners.Citation22,Citation23 Moreover, phosphorylation of the R domain also regulates the membrane stability of CFTR by modulating the balance between endocytosis and exocytosis.Citation24
ATP binding and hydrolysis by the nucleotide-binding domains (NBDs) is a prerequisite to anion transport through CFTR channels.Citation25,Citation26 The two NBDs form a head-to-tail dimer with two ATP-binding sites located at the dimer interface.Citation27 Within this dimer, ATP binds to NBD1 but is hydrolyzed at the NBD2 ATP-binding site.Citation27–Citation29 ATP interaction with NBDs facilitates their dimerization and induces conformational changes in the membrane-spanning domains required for the gating of CFTR channel.Citation26
CFTR function(s): not only a chloride channel
CFTR is the only ABC transporter functioning as an ion channel. The characteristic properties of CFTR-associated conductance are a linear current–voltage relationship and a single conductance of 6–11pS.Citation30,Citation31 Although CFTR may also transport negatively charged organic molecules such as gluconateCitation32 and glutathione,Citation33,Citation34 it is mostly selective for monovalent anion. In vivo, it mainly transports Cl− and HCO3−.Citation31,Citation35 Lack of apical Cl− secretion in CF epithelial cells had already been characterized several years before the discovery of the CFTR gene.Citation36 Over the past few years, it has become apparent that CFTR-dependent bicarbonate secretion, required for normal expansion of mucins (the main component of mucus), is also defective in patients with CF.Citation37 Therefore, the role of CFTR in CF pathogenesis is both due to lack of Cl−, resulting in low hydration of the airway surface liquid, and decrease of
transport, which maintain mucins in an aggregated and poorly soluble form.
In addition to the defective apical Cl− and HCO3− secretion (due to the absence or dysfunction of CFTR), the hyperabsorption of Na+ through hyperactive epithelial sodium channel (ENaC) is another hallmark of CF epithelia.Citation36 The failure of mutated CFTR proteins to regulate ENaC activity is proposed to play a major role in the pathophysiology of CF lung disease.Citation38–Citation40 How does CFTR regulate ENaC and how much CFTR is needed to do so is still debated.Citation41,Citation42 It has been reported that CFTR and ENaC physically interact in several cell types.Citation43–Citation45 CFTR could also decrease the open probability of ENaC,Citation46,Citation47 possibly by protecting ENaC against endogenous proteolytic cleavage.Citation48 Finally, CFTR could modulate ENaC stability at the plasma membraneCitation49 or regulate the electric coupling between the two channels.Citation50,Citation51
CFTR controls many other ion channels and transporters.Citation52 Besides its own ability to transport Cl− and HCO3−, CFTR indirectly modulates the transports of these ions by regulating, for instance, several members of the solute carrier 26 (SLC26) family.Citation53 While some of these proteins function as Cl−/HCO3− exchange proteins and participate in pH regulation, SLC26A9 is a chloride channel expressed in the apical membrane of epithelial cells and is constitutively active in human bronchial epithelial cells (HBECs).Citation54–Citation56 It contributes to cAMP-dependent chloride secretion and its activity is maximal when coexpressed with wild-type (WT) CFTR.Citation55
Classes of CFTR mutations
Six classes of CFTR mutations have been described (). Mutations of classes I, II, III, and VI are considered as severe as they are associated with little to no CFTR protein at the plasma membrane, while mutations of classes IV and V generate milder phenotypes as they lead to only partial loss of CFTR activity.Citation57,Citation58
Table 1 Classes of CFTR mutations
Class I mutations are nonsense mutations causing defects in mRNA splicing or premature insertion of a stop codon in the polypeptidic chain synthesis. They account for ~10% of the CFTR mutations worldwide and are particularly prevalent in the Ashkenazi Jewish population where they reach 50% of CFTR alleles, with W1282X being the most frequent mutation of this population.Citation59
Class II mutations cause defective protein processing and trafficking to the plasma membrane. Among these, the most common CF allele F508del-CFTR is found in ~70% of the patients (The Clinical and Functional Translation of CFTR [CFTR2]; http://cftr2.org). The deletion of the phenylalanine at position 508 of the CFTR protein causes CFTR misfolding and prolonged retention of the protein in the endoplasmic reticulum, followed by rapid degradation by the ubiquitin-proteasome pathway.Citation60,Citation61
Class III mutations are relatively rare mutations characterized by altered gating and reduced open probability of the channel. The G551D mutation, also known as the Celtic mutation, is the prototype of class III mutation and represent ~2%–3% of CF alleles in north west and central Europe but is less common in other parts of Europe.Citation62
Class IV mutant proteins are correctly inserted at the plasma membrane but the channel single conductance is altered. The most frequent class IV mutations encountered in patients are R117H (1.3%) and R347P (0.37%).
Class V (eg, A455E and 2789+5G→T) and VI (eg, 4326delTC and 4279insA) mutations lead to reduced amount of CFTR protein at the plasma membrane, by affecting CFTR mRNA (stability, alternative splicing, etc) or increasing the turnover of the CFTR protein, respectively.
Some CFTR mutations display more than one type of dysfunctions. For example, in addition to trafficking defect, F508del-CFTR also presents with characteristic defects of classes III and IV, with a reduced open probabilityCitation63 and decreased membrane stability,Citation64 respectively.
First modulators and natural compounds
The expression and activity of CFTR channels are regulated by many intracellular signaling pathways. The most known modulator of the CFTR chloride channel is intracellular cAMP, and the activity of CFTR is mainly regulated via phosphorylation by various protein kinases and dephosphorylation by protein phosphatases.
Naturally occurring compounds inducing phosphorylation of the channel were among the first CFTR modulators identified.Citation65 Alkylxanthines, such as caffeine, theophylline, and theobromine, are found in plants such as coffee or chocolate beans or tea leaves. Among them, 3-isobutyl-1-methylxanthine inhibits phosphodiesterases (PDEs) to enhance CFTR phosphorylation by preventing its dephosphorylation.Citation66–Citation68 Patch clamp single-channel recordings also suggested that some xanthine derivatives can directly activate CFTR channel to increase open probability of the channel independently of cAMP levels,Citation69 possibly through direct binding to NBD1.Citation70
Soybeans and soy food (eg, tofu, soy flour, and soy milk) contain large amount of isoflavones,Citation71 such as genistein (5,7-dihydroxy-3-(4-hydroxyphenyl)-4H-1-benzopyran-4-one). Genistein is a protein tyrosine kinase inhibitor that was found to activate CFTR, independently from protein kinase A or PDE activity.Citation72 In contrast with 3-isobutyl-1-methylxanthine,Citation73 it does not inhibit PDE activity but it requires CFTR phosphorylation to increase open probability of the channel.Citation72 This is particularly true to mutated protein F508del-Citation74,Citation75 and G551D-CFTRCitation75 channels for which genistein restores phosphorylation-dependent activation of the channel by direct binding to NBD1.Citation76
Curcumin exhibits structural similarities to isoflavones and might bind directly to CFTR proteinCitation77 to rescue F508del-CFTR trafficking in vitro.Citation78,Citation79 In vivo, curcumin increased survival rate of F508del-CFTR mice by preventing gastrointestinal obstruction in treated animals as compared with controls.Citation80 Moreover, curcumin corrects CFTR-dependent Cl− transport across nasal and rectal epithelium of F508del-CFTR mice.Citation80 These effects are still controversial as many other studies failed to reproduce them.Citation81,Citation82
Resveratrol is a natural polyphenol compound with antioxidant and anti-inflammatory properties that has been shown to activate CFTR-mediated chloride transport in epithelial cells in vitroCitation83,Citation84 and in vivoCitation85–Citation87 independently from [cAMP]i or R domain phosphorylation.Citation87 Two independent studies also demonstrated that resveratrol corrects F508del-CFTR trafficking in CF cell linesCitation88 and in CF mouse models.Citation86 However, doses required for such effects might be difficult to achieve in vivo.Citation89
Very low cytotoxicity and high abundance of natural compounds in regular aliment make them an appealing therapeutic option. It seems difficult to achieve sufficiently high concentration of these compounds from food intake only; therefore, administering purified compounds at higher doses could be considered. They may not be selective enough, however, as they often regulate various cellular and biochemical functions.
Drug repositioning
The goal of drug repositioning is to identify new indications of marketed drugs in particular for rare and neglected diseases.Citation90 They have multiple advantages over innovative treatments: they are considered safer, as they have already undergone extensive toxicology and safety assessment, they are often less expensive, and shortage is less likely to occur.
Iminosugars that interfere with N-glycosylation are approved for the treatment of Gaucher disease.Citation91 Although strong in vitroCitation92,Citation93 and preclinical evidenceCitation92,Citation94 demonstrated that N-butyldeoxynojirimycin (miglustat, Zavesca®) corrects both Cl− and Na+ transport by restoring the trafficking defect of F508del-CFTR, a Phase II clinical trial failed to demonstrate significant changes in chloride transport measured by nasal potential difference (NPD), sweat chloride, or force expiratory volume in 1 second (FEV1) in CF patients.Citation95
PDE5 inhibitors (iPDE5) and soluble guanylyl cyclase activators are currently approved for the treatment of erectile dysfunctionCitation96 and pulmonary hypertension.Citation97 They both lead to increased intracellular cGMP content, although the final mechanism of action on CFTR is still unknown. Some data suggested two distinct effects: a cGMP-dependent increase in CFTR activity and a cGMP-independent effect on CFTR trafficking.Citation98 Some in vitro studies required 1,000-fold greater concentration than what is used in the clinic to observe an effect on CFTR trafficking.Citation98,Citation99 In vivo preclinical studies have yet showed that improvement in chloride transport could be achieved with clinical doses of iPDE5, such as sildenafil and vardenafil, in CF mice.Citation100,Citation101 Outcomes of a Phase IIa open-label study aiming at investigating safety and efficacy of sildenafil in CF lung disease were recently published.Citation102 No change in sputum IL-8 was noted, but sputum neutrophil elastase content was significantly reduced after treatment. However, pharmacokinetic profiles of sildenafil suggested that CF patients may eliminate sildenafil at a faster rate than non-CF patients.
Similar to iPDE5, riociguat (BAY 63-2521) increases intracellular cGMP levels in a concentration-dependent manner and in synergy with nitric oxide (NO).Citation103,Citation104 It is a soluble guanylate cyclase activator developed by Bayer, already approved for pulmonary arterial hypertension. A Phase II trial is currently ongoing for adult CF patients homozygous for F508del mutation (NCT02170025).
Ibuprofen has long been known for its anti-inflammatory properties, and has been showed to significantly slow the decline in FEV1 in CF patients.Citation105 This effect was solely attributed to its anti-inflammatory effect. However, a recent in vitro study demonstrated that ibuprofen is also an efficient F508del-CFTR corrector via inhibition of cyclooxygenase-1.Citation106
Approved for the treatment of cystinosis,Citation107 cysteamine is a proteostasis regulator that restores autophagy, which is defective in CF.Citation108,Citation109 This is associated with a rescue and stabilization of F508del-CFTR at the plasma membrane.Citation110,Citation111 Given orally together with epigallocatechin gallate (EGCG, a flavonoid derived from green tea, contained in dietary supplements), cysteamine significantly reduced sweat chloride levels and levels of pro-inflammatory markers TNF-α and IL-8 during a small pilot study in homozygous F508del-CFTR patients.Citation111 An open-label Phase II trial involving 34 patients met the primary end point of efficacy, with a significant reduction in sweat chloride concentration of −18.0 mmol/L, but no significant difference in FEV1 was observed.Citation112
Escin, extracted from horse chestnut tree, possesses anti-inflammatory effects and is already used in patients with chronic venous insufficiency, hemorrhoids, and post-traumatic edema.Citation113 Escin significantly enhanced CFTR function in Fisher rat thyroid cells transfected with different CFTR class I mutants (G542X, W1282X) and in primary HBECs isolated from G542X/F508del and W1282/F508del patients.Citation114 By contrast, escin failed to improve CFTR function in HBECs from a patient homozygous for F508del, demonstrating that Escin acts as a readthrough agent for nonsense mutations.
All these compounds are excellent illustrations that, as for many other rare diseases, CF therapy may benefit from drug repositioning as a strategy to speed up drug development.
Genotype-specific therapies
With the development of high-throughput screening (HTS) assays allowing rapid screening of thousands of small molecules, many families of chemical structures have been identified. Thanks to expanding knowledge of the structure and function of CFTR, and to increased understanding of the different functional consequences of CFTR mutations, structure–activity relationship and optimization of the most promising lead compounds have led to a series of potential pharmacological therapies for CF to correct CFTR defects at different levels.Citation114–Citation117 CFTR modulators can be categorized according to the class of mutation or dysfunction that they aim at targeting ( and ).
Figure 3 Overview of the most advance CFTR modulators in preclinical and clinical studies, with regard to the class of CFTR mutations and the primary defect of the corresponding mutant protein.
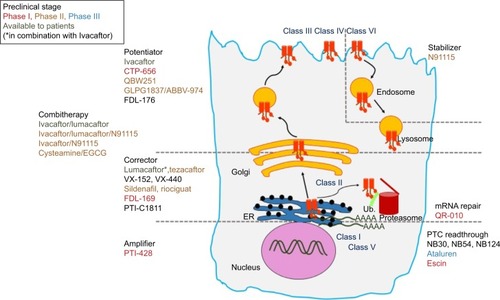
Table 2 Mechanisms of action of pharmacological modulators of CFTR available to CF patients or under preclinical development as mono- and/or combitherapies for CF
Therapies targeting class I
Development of premature termination codon (PTC) “read-through” agents allow ribosomes to continue translation through class I nonsense mutations to produce full-length CFTR protein. Almost 20 years ago, aminoglycosides, such as gentamicin, were first described as a potential pharmacological approach for class I mutations.Citation118–Citation120 In addition to its potent bactericidal activity, gentamicin displayed beneficial effects on electrophysiological parameters assessed by NPD in vivo after topical nasal applicationCitation121,Citation122 or intravenous administrationCitation118,Citation123 in CF patients with at least one class I mutation. However, high inter-individual variability in clinical benefits was observed, in particular between patients carrying only one and those carrying two nonsense mutations.Citation124 In addition, high nephron- and oto-toxicity render per os or systemically administered aminoglycosides not well suited for long-term use. To tackle this, new series of synthetic aminoglycoside derivatives were developed through a systematic structure-based approach.Citation125,Citation126 NB30, NB54, and NB124 had significantly reduced toxicityCitation125,Citation127 and demonstrated superior in vitro readthrough activity in HBE cell lines or primary cells expressing at least one nonsense CFTR mutation.Citation125,Citation128 Moreover, when systemically administrated to cftr−/− mice expressing human CFTR-G542X,Citation129 NB54 and NB124 restored CFTR activity measured ex vivo across intestinal epithelium to at least 5% of the current observed in WT animals.Citation127,Citation128
Through HTS, PTC Therapeutics™ (Dublin, Ireland) identified PTC-124 (3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]-benzoic acid), ataluren. It is an orally bioavailable small molecule inducing complete translation of proteins containing premature nonsense mutations without affecting the normal stop codons.Citation130 In the initial Phase II trial, CF adults with at least one CFTR nonsense mutation received oral treatment with ataluren for 14 days followed by a washout period of 14 days.Citation131 CFTR function measured by NPD was restored and a small decrease in ENaC activity was also recorded. Moreover, patients presented with slight increase in FEV1 and bodyweight, and some of them reported an improvement in pulmonary symptoms such as cough. A pediatric trial was conducted with children of age 6 and older, and demonstrated similar improvements in CFTR function although it did not correlate with FEV1.Citation132 Despite these encouraging data, ataluren did not provide a significant improvement in FEV1 of a Phase III placebo-controlled trial.Citation133 Interaction with chronically inhaled tobramycin could be a cause, as the subgroup of patients not receiving inhaled aminoglycosides showed a more robust improvement in FEV1 (+5.7% predicted) together with fewer pulmonary exacerbations (−40%) in the ataluren group as compared to the placebo. Moreover, variable responses were found among patients with different genotypes suggesting that readthrough agents may not work for all class I mutations.
Therapies targeting class II
The aim of class II targeting compounds is to rescue the trafficking defect of mutant CFTR and therefore increase the quantity of mutated CFTR protein inserted in the plasma membrane. Soon after the identification of the CFTR gene, Denning et alCitation61 demonstrated that low-temperature incubation (eg, 27°C) restores F508del-CFTR expression at the plasma membrane. This was the first evidence that the trafficking defect of F508del-CFTR could be modified to allow partial escape from the endoplasmic reticulum quality control and functional expression on the cell surface. Additional in vitro proofs of mutant CFTR druggability were obtained with chemical chaperonesCitation134,Citation135 or the transcriptional regulator butyrate.Citation136 In vitro, 4-phenylbutyrate (4-PBA), an analog of butyrate, corrects the trafficking defect of F508del-CFTRCitation137 by modulating the interaction with 70 kDa Heat shock protein (Hsp) family Hsc70.Citation138 4-PBA was one of the first corrector to be tested in a pilot clinical trial for CF, where it slightly improved CFTR activity in nasal epithelium but did not reduce sweat chloride concentration.Citation139
With the development of HTS assays and medicinal chemistry, many families of new chemical structures with corrector properties have emerged. Their corrector activities are exerted through direct modulation of protein foldingCitation140,Citation141 or cellular proteostasis,Citation142 or may act as pharmacological chaperons.Citation143 While many of the compounds available so far, such as corr-4aCitation116 or VRT-325,Citation141 will never progress beyond the status of “bench tools”, some hits have been identified and optimized in view of clinical assessments. The most advanced corrector for F508del-CFTR is VX-809 (lumacaftor), developed by Vertex Pharmaceuticals (Boston, MA, USA). VX-809 restores F508del-CFTR trafficking by improving its folding and stabilizing membrane-spanning domain 1.Citation144,Citation145 Four weeks of oral lumacaftor as monotherapy in homozygous F508del-CFTR patients was demonstrated safe and well tolerated.Citation146 Sweat chloride contents were significantly decreased with treatment in a dose-dependent manner. However, lumacaftor failed to demonstrate any therapeutic benefit for lung function as it did not change FEV1 nor modulate NPD parameters. This lack of clinical effect suggested that corrector-based monotherapies are not efficient enough to improve lung function because they do not target the other biological defects of F508del-CFTR, ie, decreased membrane stability and open probability.
Because in vitro studies showed that CFTR potentiator VX-770 (see ivacaftor, classes III and IV) improved the open probability of VX-809-rescued F508del-CFTR,Citation145 a new Phase IICitation147 and a Phase IIICitation148 studies with combination of lumacaftor and ivacaftor were conducted. Overall, the absolute increase in FEV1 was modest (+3%).Citation148 Currently marketed as Orkambi®, the ivacaftor–lumacaftor combination has been heavily challenged because it seems no more efficient that conventional multitherapiesCitation149 for a price outrageously tenfold higher.Citation150 More importantly, two in vitro studies evidenced negative interference between ivacaftor and several correctors including lumacaftor, as prolonged exposure of HBECs with ivacaftor decreases the stability of lumacaftor-corrected F508del-CFTR.Citation151,Citation152 This could explain in part the modest improvement of lung function observed in patients taking lumacaftor/ivacaftor.
Vertex Pharmaceuticals is currently expanding its drug portofolio by developing more correctors such as VX-661 (tezacaftor), for which they claimed slightly better clinical efficacy (+4.8% FEV1) than VX-809 when combined with ivacaftor in patients with two copies of F508del-CFTR.Citation153 Moreover, VX-661 showed additional benefit (+4.6% FEV1) in patients carrying both F508del- and G551D-CFTR mutations and who were already taking ivacaftor.Citation153
More next-generation correctors such as VX-152 and VX-440 will be evaluated in combination with VX-661/ivacaftor as triple combinations (VX-152/VX-661/ivacaftor and VX-440/VX-661/ivacaftor) in homozygous F508del patients and patients with one F508del associated with a second mutation that results in minimal CFTR function. In vitro, these triple combinations resulted in an increase in chloride transport in HBECs approximately threefold higher than with lumacaftor/ivacaftor.
Other drug discovery companies have undertaken development of correctors. Among them, PTI-C1811 (Proteostasis Therapeutics, Cambridge, MA, USA) and FDL-169 (Flatley Discovery Lab, Charlestown, MA, USA) act through different mechanisms than VX-809 and are both claimed to have similar or superior in vitro activity when combined with potentiators.
Unlike CFTR correctors that act at the protein level, ProQR Therapeutics NV (Leiden, the Netherlands) developed QR-010, a single-strand modified RNA specifically designed to repair the F508del mutation at the mRNA level to generate a WT-CFTR transcript. In vivo in a preclinical mouse model, QR-010 demonstrated a robust increase in CFTR activity measured by NPDCitation154 and a restoration of CFTR-dependent salivary secretion rates.Citation155 QR-010 is now being tested in two clinical trials. In a Phase Ib study (NCT02532764), single and multiple ascending doses will assess QR-010 safety and tolerability in F508del homozygous patients. The second study (NCT02564354) is exploratory proof-of-concept study in CF patients with at least one copy of F508del. It will explore whether intranasal administration of QR-010 can restore function of the CFTR protein as measured by NPD.
Therapies targeting class III and IV
Class III and IV mutations are considered mild because they produce full-length CFTR that inserts into the plasma membrane where it can correctly interact with other proteins. However, chloride transport is reduced because the open probability (class III) or the single conductance (class IV) of the channel is altered. Pharmacological compounds that enhance CFTR function at the cell membrane are called potentiators.
VX-770 (ivacaftor) was identified by HTS in epithelial cells expressing G551D-CFTR.Citation156 Early clinical investigations enrolling patients with at least one G551D mutations provided encouraging efficacy data (as measured by NPD and sweat chloride concentration) in patients receiving 150 mg ivacaftor twice a day, together with a safety profile comparable to the placebo group.Citation157 Longer studies (STRIVE and ENVISION), up to 48 weeks, also demonstrated that patients aged 6 years and older treated with ivacaftor gained significantly more weight and their frequency of pulmonary exacerbation was reduced by 55% with ivacaftor as compared to placebo.Citation158,Citation159 During the KONNECTION study, ivacaftor resulted in significant improvement in FEV1 (+10.7% at 8 weeks), sweat chloride, and body mass index in patients carrying one of the following non-G551D alleles: G178R, S549N, S549R, G551S, G970R, G1244E, S1251N, S1255P, or G1349D.Citation160 Results were comparable to those observed during the STRIVE and ENVISION studies (+10.6% FEV1) in patients with G551D mutation.Citation158,Citation159
Because CF lung disease is progressive, treating patients as early as possible was the aim of the KIWI study which enrolled preschoolers (2.5–5 years old) with one G551D mutation. Ivacaftor seemed to be safe in that cohort, although extended results are awaited.Citation161
During the initial screening, ivacaftor was shown to also potentiate activity of rescued F508del-CFTR.Citation156 As expected with a potentiator, a clinical trial with ivacaftor for F508del/F508del patients failed to support its use as a monotherapy for this class of patients.Citation162 As of now, ivacaftor (Kalydeco®; Vertex Pharmaceuticals) is the only potentiator approved for CF patients aged 2 years and older who carry at least one of the following mutations: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R or R117H. Ivacaftor is seen by the CF community as a proof of principle of clinical benefit from a CFTR modulator, and its approval was a very significant milestone in CF treatment.
Concert Pharmaceuticals Inc. (Lexington, MA, USA) is applying deuterium chemistry to enhance the pharmacokinetic properties of ivacaftor. This approach was tested in CF patients with class III mutations in a Phase I crossover comparison of deuterated ivacaftor (CTP-656) vs ivacaftor. CTP-656 demonstrated a longer half-life of the compound in plasma compared to ivacaftor, supporting the possibility to reduce the dosing to once a day.Citation163
QBW251 is a potentiator developed by Novartis Pharmaceuticals. In vitro data showed superior efficacy of QBW251 as compared to ivacaftor when both are combined with lumacaftor. Phase II trial (NCT02190604) has been conducted and some outcomes have been recently presented.Citation164 CF heterozygous patients with at least one class III to VI mutation were enrolled (including patients with one F508del mutation as it can be considered either as class II, III, or VI). A separate arm of the study enrolled only F508del-CFTR homozygous patients. Orally administered QBW251 (150 mg or 450 mg, twice a day) for 2 weeks was safe and well tolerated in the 40 CF patients.Citation164 In patients with a residual function, QBW251 (450 mg) statistically increased FEV1 over placebo by 7.3%, an increase that is considered as clinically relevant for lung function and very similar to that observed with ivacaftor. As for ivacaftor, QBW251 monotherapy did not demonstrate any efficacy in patients with two copies of F508del.
The potentiator GLPG1837/ABBV-974 is codeveloped by Galapagos NV (Mechelen, Belgium) and AbbVie Pharmaceuticals (North Chicago, IL, USA). Phase I has demonstrated that single (up to 2 g) and multiple doses (up to 800 mg twice a day for 14 days) of GLPG1837/ABBV-974 were safe and well tolerated in healthy volunteers.Citation165 Two Phase II open-label studies are ongoing and will explore GLPG1837/ABBV-974 safety, tolerability, and efficacy in CF patients with G551D (SAPHIRA1) and S1251N (SAPHIRA2).
Therapies targeting class V and VI
Currently, there is no clinical data available for class V-specific therapies. For class VI, a new class of compounds increasing the half-life of CFTR protein at the plasma membrane has recently attracted interest. VRT-325 and Corr-4a were prototypes for this type of compounds, so-called “stabilizers”, which are meant to be complementary to existing and future CFTR modulators.
N91115, developed by Nivalis Therapeutics (Boulder, CO, USA), is an inhibitor of S-nitroglutathione (GSNO) reductase and aimed at increasing intracellular levels of GSNO. GSNO induces the S-nitrosylation of the cellular chaperone Hsp70/Hsp90 organizing protein which prevents the association of CFTR with Hsp70/Hsp90 organizing protein.Citation166–Citation168 N91115 was proven safe and well tolerated in CF patients with two F508del alleles.Citation169 N91115 has recently received the status of Orphan Drug designation by the FDA and two Phase II clinical studies are ongoing (see “Combitherapies and personalized medicine” section). In the near future, potentiators may also prove useful to provide maximal activation of class VI mutants.
Combitherapies and personalized medicine
Many pharmacological agents are currently in development to correct mutant CFTR activity in CF. These agents are becoming increasingly specific, and aim at targeting patients with particular genotype. The most advance treatment for CF currently available for patients is ivacaftor. This is a typical example of personalized medicine where only individuals with specific mutations can be treated with this drug. Although ivacaftor provides a significant improvement in lung function, this may not be achievable in every CF patient with a single compound. More specifically, in patients carrying CFTR mutations displaying multiple dysfunctions, such as F508del-CFTR, combination of several molecules will likely lead to better clinical results. Here, the biological defects of F508del-CFTR could be ideally addressed by a triple combination of a corrector to increase the amount of F508del-CFTR protein expressed in the plasma membrane, a potentiator to enhance its open probability and a stabilizer to increase its half-life at the plasma membrane.
To tackle the membrane instability of rescued F508del-CFTR, Nivalis is currently evaluating N91115 safety and efficacy in combination with lumacaftor/ivacaftor in homozygous F508del patients (NCT02589236) and with ivacaftor in patients with one F508del and a gating mutation (NCT02724527) in two Phase II clinical trials.
Another new class of compounds is currently investigated by Proteostasis Therapeutics. They are developing PTI-428, a CFTR amplifier, which aims to selectively increase the amount of immature form of CFTR protein to provide other CFTR modulators with more substrate to act upon.Citation170 PTI-428 received Fast Track designation from the FDA and a Phase I is ongoing to assess its safety, tolerability and pharmacokinetics in CF patients (NCT02718495).
In the near future, one can also envisage combitherapies with activators of alternative chloride channels or with inhibitors of the ENaC.Citation171
One of the biggest challenges to implement personalized medicine for CF will be to develop new in vitro models to better predict the individual response of patient to different combinations of treatments. Development and use of experimental materials based on patient tissues (such as airway and intestinal organoids or induced pluripotent stem cells) will hopefully provide new powerful assays to better anticipate the individual clinical benefit of CFTR modulators.
Conclusion
Many classes of compounds restoring the function of CFTR mutants have been identified; however, most of them, such as natural compound curcumin, were never translated into therapy mainly because of lack of benefit to patients as well as off-target effects or low bioavailability. Drug repositioning, through the exciting examples of the cystamine/EGCG combination or sildenafil, may speed up the development of novel therapies for CF. Currently, ivacaftor alone or in combination with lumacaftor are the only pharmacological modulators of CFTR approved for the treatment of CF. The combination lumacaftor/ivacaftor has been highly challenged as they do not seem to provide significant improvement in lung function as compared to conventional therapies. Ivacaftor targets only a specific CFTR mutant (G551D-CFTR) which is found in <2% of the patients, Finally, these two marketed therapies cost over USD 250,000/year (a tenfold increase as compared to usual multitherapies) for a modest improvement in the quality of life of patients. Thus, there is still a major and urgent need for new molecules and therapeutic approaches to be developed for treating CF.
Acknowledgments
TL and SN received financial support from the European Commission under the H2020-PHC-13-2014 funding program (PRO-CF-MED, ref. 633545) granted to ProQR Therapeutics NV.
Disclosure
The authors report no conflicts of interest in this work.
References
- RiordanJRommensJKeremBIdentification of the cystic fibrosis gene: cloning and characterization of complementary DNAScience19892454922106610732475911
- HuauxFNoelSDhoogheBDysregulated proinflammatory and fibrogenic phenotype of fibroblasts in cystic fibrosisPLoS One201385e6434123734196
- MarcorellesPFriocourtGUguenALedéFFérecCLaquerrièreACystic fibrosis transmembrane conductance regulator protein (CFTR) expression in the developing human brain: comparative immunohistochemical study between patients with normal and mutated CFTRJ Histochem Cytochem2014621179180125062999
- GaoZSunHYLauCPChin-Wan FungPLiGREvidence for cystic fibrosis transmembrane conductance regulator chloride current in swine ventricular myocytesJ Mol Cell Cardiol20074219810517112538
- BonfieldTLHodgesCACottonCUDrummMLAbsence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infectionJ Leukoc Biol20129251111112222859830
- DiABrownMEDeriyLVCFTR regulates phagosome acidification in macrophages and alters bactericidal activityNat Cell Biol20068993394416921366
- PainterRGValentineVGLansonNACFTR expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosisBiochemistry20064534102601026916922501
- ZengerlingSTsuiLCGrzeschikKHOlekKRiordanJRBuchwaldMMapping of DNA markers linked to the cystic fibrosis locus on the long arm of chromosome 7Am J Hum Genet19874032282363472464
- TsuiLBuchwaldMBarkerDCystic fibrosis locus defined by a genetically linked polymorphic DNA markerScience19852304729105410572997931
- GadsbyDCVerganiPCsanadyLThe ABC protein turned chloride channel whose failure causes cystic fibrosisNature2006440708347748316554808
- SebastianARishishwarLWangJOrigin and evolution of the cystic fibrosis transmembrane regulator protein R domainGene2013523213714623578801
- ChengSHRichDPMarshallJGregoryRJWelshMJSmithAEPhosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channelCell1991665102710361716180
- CsanádyLSeto-YoungDChanKWPreferential phosphorylation of R-domain Serine 768 dampens activation of CFTR channels by PKAJ Gen Physiol2005125217118615657296
- BilletAJiaYJensenTRiordanJRHanrahanJWRegulation of the cystic fibrosis transmembrane conductance regulator anion channel by tyrosine phosphorylationFASEB J20152993945395326062600
- HallowsKRKobingerGPWilsonJMWittersLAFoskettJKPhysiological modulation of CFTR activity by AMP-activated protein kinase in polarized T84 cellsAm J Physiol Cell Physiol20032845C1297C130812519745
- SeavillekleinGAmerNEvagelidisAPKC phosphorylation modulates PKA-dependent binding of the R domain to other domains of CFTRAm J Physiol Cell Physiol20082955C1366C137518799655
- CsanádyLChanKWSeto-YoungDKopscoDCNairnACGadsbyDCSevered channels probe regulation of gating of cystic fibrosis transmembrane conductance regulator by its cytoplasmic domainsJ Gen Physiol2000116347750010962022
- ChappeVIrvineTLiaoJEvagelidisAHanrahanJWPhosphorylation of CFTR by PKA promotes binding of the regulatory domainEMBO J200524152730274016001079
- HegedűsTAleksandrovAMengosACuiLJensenTJRiordanJRRole of individual R domain phosphorylation sites in CFTR regulation by protein kinase ABiochim Biophys Acta2009178861341134919328185
- WilkinsonDJStrongTVMansouraMKCFTR activation: additive effects of stimulatory and inhibitory phosphorylation sites in the R domainAm J Physiol Lung Cell Mol Physiol19972731L127L133
- VaisHZhangRReenstraWWDibasic phosphorylation sites in the R domain of CFTR have stimulatory and inhibitory effects on channel activationAm J Physiol Cell Physiol20042873C737C74515140750
- BozokyZKrzeminskiMMuhandiramRRegulatory R region of the CFTR chloride channel is a dynamic integrator of phospho-dependent intra- and intermolecular interactionsProc Natl Acad Sci U S A201311047E4427E443624191035
- BozokyZKrzeminskiMChongPAForman-KayJDStructural changes of CFTR R region upon phosphorylation: a plastic platform for intramolecular and intermolecular interactionsFEBS J2013280184407441623826884
- FarinhaCMSwiatecka-UrbanABrautiganDLJordanPRegulatory crosstalk by protein kinases on CFTR trafficking and activityFront Chem20164126835446
- MoranOOn the structural organization of the intracellular domains of CFTRInt J Biochem Cell Biol20145271424513531
- HwangTCSheppardDNGating of the CFTR Cl(−) channel by ATP-driven nucleotide-binding domain dimerisationJ Physiol2009587pt 102151216119332488
- LewisHABuchananSGBurleySKStructure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulatorEMBO J200423228229314685259
- VerganiPLocklessSWNairnACGadsbyDCCFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domainsNature2005433702887688015729345
- ZhouZWangXLiuHYZouXLiMHwangTCThe two ATP binding sites of cystic fibrosis transmembrane conductance regulator (CFTR) play distinct roles in gating kinetics and energeticsJ Gen Physiol2006128441342216966475
- AndersonMGregoryRThompsonSDemonstration that CFTR is a chloride channel by alteration of its anion selectivityScience199125350162022051712984
- LinsdellPMechanism of chloride permeation in the cystic fibrosis transmembrane conductance regulator chloride channelExp Physiol200691112312916157656
- LinsdellPHanrahanJWAdenosine triphosphate–dependent asymmetry of anion permeation in the cystic fibrosis transmembrane conductance regulator chloride channelJ Gen Physiol199811146016149524141
- GouldNSMinEMartinRJDayBJCFTR is the primary known apical glutathione transporter involved in cigarette smoke induced adaptive responses in the lungFree Radic Biol Med20125271201120622266045
- LinsdellPHanrahanJWGlutathione permeability of CFTRAm J Physiol Cell Physiol19982751C323C326
- TangLFatehiMLinsdellPMechanism of direct bicarbonate transport by the CFTR anion channelJ Cyst Fibros20098211512119019741
- QuintonPMChloride impermeability in cystic fibrosisNature198330158994214226823316
- QuintonPMCystic fibrosis: impaired bicarbonate secretion and mucoviscidosisLancet2008372963641541718675692
- KnowlesMGatzyJBoucherRIncreased bioelectric potential difference across respiratory epithelia in cystic fibrosisN Engl J Med198130525148914957300874
- Bangel-RulandNTomczakKWeberWMTargeting ENaC as a molecular suspect in cystic fibrosisCurr Drug Targets201516995195725544019
- MatsuiHGrubbBRTarranREvidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways diseaseCell1998957100510159875854
- CollawnJFLazrakABebokZMatalonSThe CFTR and ENaC debate: how important is ENaC in CF lung disease?Am J Physiol Lung Cell Mol Physiol201230211L1141L114622492740
- HobbsCADa TanCTarranRDoes epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease?J Physiol2013591pt 184377438723878362
- BerdievBKCormet-BoyakaEToussonAMolecular proximity of cystic fibrosis transmembrane conductance regulator and epithelial sodium channel assessed by fluorescence resonance energy transferJ Biol Chem200728250364813648817913705
- JiHLChalfantMLJovovBThe cytosolic termini of the β- and γ- ENaC subunits are involved in the functional interactions between CFTR and ENaCJ Biol Chem200027536279472795610821834
- KunzelmannKKiserGLSchreiberRRiordanJRInhibition of epithelial Na+ currents by intracellular domains of the cystic fibrosis transmembrane conductance regulatorFEBS Lett199740033413449009227
- KonstasAAKochJPKorbmacherCcAMP-dependent activation of CFTR inhibits the epithelial sodium channel (ENaC) without affecting its surface expressionPflügers Arch2003445451352112548398
- ChinetTCFulltonJMYankaskasJRBoucherRCStuttsMJMechanism of sodium hyperabsorption in cultured cystic fibrosis nasal epithelium: a patch-clamp studyAm J Physiol Cell Physiol19942664C1061C1068
- GentzschMDangHDangYThe cystic fibrosis transmembrane conductance regulator impedes proteolytic stimulation of the epithelial Na(+) channelJ Biol Chem201028542322273223220709758
- LuCJiangCPribanicSRotinDCFTR stabilizes ENaC at the plasma membraneJ Cyst Fibros20076641942217434346
- HorisbergerJ-DENaC–CFTR interactions: the role of electrical coupling of ion fluxes explored in an epithelial cell modelPflügers Arch2003445452252812548399
- KunzelmannKENaC is inhibited by an increase in the intracellular Cl− concentration mediated through activation of Cl− channelsPflügers Arch2003445450451212548397
- KunzelmannKTianYMartinsJRAirway epithelial cells – functional links between CFTR and anoctamin dependent Cl− secretionInt J Biochem Cell Biol201244111897190022710346
- El KhouriETouréAFunctional interaction of the cystic fibrosis transmembrane conductance regulator with members of the SLC26 family of anion transporters (SLC26A8 and SLC26A9): physiological and pathophysiological relevanceInt J Biochem Cell Biol201452586724530837
- LoriolCDulongSAvellaMCharacterization of SLC26A9, facilitation of Cl- transport by bicarbonateCell Physiol Biochem2008221–4015030
- BertrandCAZhangRPilewskiJMFrizzellRASLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epitheliaJ Gen Physiol2009133442143819289574
- OusingsawatJSchreiberRKunzelmannKDifferential contribution of SLC26A9 to Cl− conductance in polarized and non-polarized epithelial cellsJ Gen Physiol2012227623232329
- HaardtMBenharougaMLechardeurDKartnerNLukacsGLC-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis: a novel class of mutationJ Biol Chem199927431218732187710419506
- WelshMJSmithAEMolecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosisCell1993737125112547686820
- QuintALererISagiMAbeliovichDMutation spectrum in Jewish cystic fibrosis patients in Israel: implication to carrier screeningAm J Med Genet A2005136A324624815948195
- ChengSHGregoryRJMarshallJDefective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosisCell19906348278341699669
- DenningGMAndersonMPAmaraJFMarshallJSmithAEWelshMJProcessing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitiveNature199235863897617641380673
- BobadillaJLMacekMFineJPFarrellPMCystic fibrosis: a worldwide analysis of CFTR mutations – correlation with incidence data and application to screeningHum Mutat200219657560612007216
- DalemansWBarbryPChampignyGAltered chloride ion channel kinetics associated with the ΔF508 cystic fibrosis mutationNature199135463545265281722027
- VargaKGoldsteinRFJurkuvenaiteAEnhanced cell surface stability of rescued ΔF508 cystic fibrosis transmembrane conductance regulator by pharmacological chaperonesBiochem J2008410355556418052931
- DeyIShahKBradburyNANatural compounds as therapeutic agents in the treatment cystic fibrosisJ Genet Syndr Gene Ther20167128427081574
- DrummMWilkinsonDSmitLChloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytesScience19912545039179717991722350
- BecqFJensenTJChangXBPhosphatase inhibitors activate normal and defective CFTR chloride channelsProc Natl Acad Sci U S A19949119916091647522329
- BecqFFanjulMMertenMFigarellaCHollandeEGolaMPossible regulation of CFTR-chloride channels by membrane-bound phosphatases in pancreatic duct cellsFEBS Lett199332733373427688697
- ChappeVMetteyYVierfondJMStructural basis for specificity and potency of xanthine derivatives as activators of the CFTR chloride channelBr J Pharmacol199812346836939517388
- CohenBELeeGJacobsonKA8-Cyclopentyl-1,3-dipropylxanthine and other xanthines differentially bind to the wild-type and ΔF508 mutant first nucleotide binding fold (NBF-1) domains of the cystic fibrosis transmembrane conductance regulatorBiochemistry19973621645564619174362
- IllekBFischerHFlavonoids stimulate Cl conductance of human airway epithelium in vitro and in vivoAm J Physiol Lung Cell Mol Physiol19982755L902L910
- FrenchPJBijmanJBotAGBoomaarsWEScholteBJde JongeHRGenistein activates CFTR Cl- channels via a tyrosine kinase- and protein phosphatase-independent mechanismAm J Physiol Cell Physiol19972732C747C753
- IllekBFischerHSantosGFWiddicombeJHMachenTEReenstraWWcAMP-independent activation of CFTR Cl channels by the tyrosine kinase inhibitor genisteinAm J Physiol Cell Physiol19952684C886C893
- HwangTCWangFYangICReenstraWWGenistein potentiates wild-type and delta F508-CFTR channel activityAm J Physiol19972733 pt 1C988C9989316420
- IllekBZhangLLewisNCMossRBDongJYFischerHDefective function of the cystic fibrosis-causing missense mutation G551D is recovered by genisteinAm J Physiol Cell Physiol19992774C833C839
- MoranOGaliettaLJVZegarra-MoranOBinding site of activators of the cystic fibrosis transmembrane conductance regulator in the nucleotide binding domainsCell Mol Life Sci200562444646015719171
- WangWBernardKLiGKirkKLCurcumin opens cystic fibrosis transmembrane conductance regulator channels by a novel mechanism that requires neither ATP binding nor dimerization of the nucleotide-binding domainsJ Biol Chem200728274533454417178710
- EganMEGlockner-PagelJAmbroseCACalcium-pump inhibitors induce functional surface expression of delF508-CFTR protein in cystic fibrosis epithelial cellsNat Med20028548549211984593
- NorezCAntignyFBecqFVandebrouckCMaintaining low Ca2+ level in the endoplasmic reticulum restores abnormal endogenous F508del-CFTR trafficking in airway epithelial cellsTraffic20067556257316643279
- EganMEPearsonMWeinerSACurcumin, a major constituent of turmeric, corrects cystic fibrosis defectsScience2004304567060060215105504
- SongYSonawaneNDSalinasDEvidence against the rescue of defective ΔF508-CFTR cellular processing by curcumin in cell culture and mouse modelsJ Biol Chem200427939406294063315280357
- GrubbBRGabrielSEMengosASERCA pump inhibitors do not correct biosynthetic arrest of ΔF508 CFTR in cystic fibrosisAm J Respir Cell Mol Biol200634335536316284361
- YangSYuBSuiYCFTR chloride channel is a molecular target of the natural cancer preventive agent resveratrolPharmazie201368977277624147347
- IllekBLizarzaburuMELeeVNantzMHKurthMJFischerHStructural determinants for activation and block of CFTR-mediated chloride currents by apigeninAm J Physiol Cell Physiol20002796C1838C184611078699
- AlexanderNSHatchNZhangSResveratrol has salutary effects on mucociliary transport and inflammation in sinonasal epitheliumLaryngoscope201112161313131921480283
- DhoogheBBouckaertCCapronAWallemacqPLealTNoelSResveratrol increases F508del-CFTR dependent salivary secretion in cystic fibrosis miceBiol Open20154792993626092868
- WoodworthBAResveratrol ameliorates abnormalities of fluid and electrolyte secretion in a hypoxia-induced model of acquired CFTR deficiencyLaryngoscope2015125suppl 7S1S1325946147
- HamdaouiNBaudoin-LegrosMKellyMResveratrol rescues cAMP-dependent anionic transport in the cystic fibrosis pancreatic cell line CFPAC1Br J Pharmacol2011163487688621366549
- JaiYShahKBridgesRJBradburyNAEvidence against resveratrol as a viable therapy for the rescue of defective ΔF508 CFTRBiochim Biophys Acta20151850112377238426342647
- Hay MeleBCitroVAndreottiGCubellisMVDrug repositioning can accelerate discovery of pharmacological chaperonesOrphanet J Rare Dis2015105525947946
- SawkarARChengWCBeutlerEWongCHBalchWEKellyJWChemical chaperones increase the cellular activity of N370S β-glucosidase: a therapeutic strategy for Gaucher diseaseProc Natl Acad Sci U S A20029924154281543312434014
- NoëlSWilkeMBotAGMDe JongeHRBecqFParallel improvement of sodium and chloride transport defects by miglustat (n-butyldeoxynojyrimicin) in cystic fibrosis epithelial cellsJ Pharmacol Exp Ther200832531016102318309088
- NorezCNoelSWilkeMRescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the α-glucosidase inhibitor miglustatFEBS Lett200658082081208616546175
- LubambaBLebacqJLebecquePAirway delivery of low-dose miglustat normalizes nasal potential difference in F508del cystic fibrosis miceAm J Respir Crit Care Med2009179111022102819299496
- LeonardALebecquePDingemanseJLealTA randomized placebo-controlled trial of miglustat in cystic fibrosis based on nasal potential differenceJ Cyst Fibros201211323123622281182
- PeakTCYafiFASangkumPHellstromWJGEmerging drugs for the treatment of erectile dysfunctionExpert Opin Emerg Drugs201520226327525740087
- HamblyNGrantonJRiociguat for the treatment of pulmonary hypertensionExpert Rev Respir Med20159667969526599488
- LeierGBangel-RulandNSobczakKKnieperYWeberWMSildenafil acts as potentiator and corrector of CFTR but might be not suitable for the treatment of CF lung diseaseCell Physiol Biochem2012295–677579022613978
- DormerRLHarrisCMClarkZSildenafil (Viagra) corrects ΔF508-CFTR location in nasal epithelial cells from patients with cystic fibrosisThorax2005601555915618584
- DhoogheBNoëlSBouzinCBehets-WydemansGLealTCorrection of chloride transport and mislocalization of CFTR protein by vardenafil in the gastrointestinal tract of cystic fibrosis micePLoS One2013810e7731424204804
- LubambaBLecourtHLebacqJPreclinical evidence that sildenafil and vardenafil activate chloride transport in cystic fibrosisAm J Respir Crit Care Med2008177550651518006891
- Taylor-CousarJLWileyCFeltonLAPharmacokinetics and tolerability of oral sildenafil in adults with cystic fibrosis lung diseaseJ Cyst Fibros201514222823625466700
- GhofraniHAGalièNGrimmingerFRiociguat for the treatment of pulmonary arterial hypertensionN Engl J Med2013369433034023883378
- MittendorfJWeigandSAlonso-AlijaCDiscovery of riociguat (BAY 63-2521): a potent, oral stimulator of soluble guanylate cyclase for the treatment of pulmonary hypertensionChemMedChem20094585386519263460
- KonstanMWSchluchterMDXueWDavisPBClinical use of ibuprofen is associated with slower FEV(1) decline in children with cystic fibrosisAm J Respir Crit Care Med2007176111084108917872492
- CarlileGWRobertRGoeppJIbuprofen rescues mutant cystic fibrosis transmembrane conductance regulator traffickingJ Cyst Fibros2015141162524974227
- GahlWAEarly oral cysteamine therapy for nephropathic cystinosisEur J Pediatr20031621S38S4114610675
- LucianiAVillellaVREspositoSDefective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibitionNat Cell Biol201012986387520711182
- AbdulrahmanBAKhweekAAAkhterAAutophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosisAutophagy20117111359137021997369
- LucianiAVillellaVREspositoSTargeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on ΔF508 cystic fibrosis transmembrane conductance regulatorAutophagy20128111657167222874563
- StefanoDDVillellaVREspositoSRestoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutationAutophagy201410112053207425350163
- ToscoADe GregorioFEspositoSA novel treatment of cystic fibrosis acting on-target: cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTRCell Death Differ Epub2016722
- SirtoriCRAescin: pharmacology, pharmacokinetics and therapeutic profilePharmacol Res200144318319311529685
- MutyamVDuMXueXDiscovery of clinically approved agents that promote suppression of CFTR nonsense mutationsAm J Respir Crit Care Med Epub2016422
- MaTVetrivelLYangHHigh-affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high-throughput screeningJ Biol Chem200227740372353724112161441
- PedemonteNLukacsGLDuKSmall-molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screeningJ Clin Invest200511592564257116127463
- Van GoorFStraleyKSCaoDRescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small moleculesAm J Physiol Lung Cell Mol Physiol20062906L1117L113016443646
- Sermet-GaudelusIRenouilMFajacAIn vitro prediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot studyBMC Med200755517394637
- HowardMFrizzellRABedwellDMAminoglycoside antibiotics restore CFTR function by overcoming premature stop mutationsNat Med1996244674698597960
- BedwellDMKaenjakABenosDJSuppression of a CFTR premature stop mutation in a bronchial epithelial cell lineNat Med1997311128012849359706
- WilschanskiMYahavYYaacovYGentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutationsN Engl J Med2003349151433144114534336
- WilschanskiMFaminiCBlauHA pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutationsAm J Respir Crit Care Med2000161386086510712334
- ClancyJPBebökZRuizFEvidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosisAm J Respir Crit Care Med200116371683169211401894
- ClancyJPRoweSMBebokZNo detectable improvements in cystic fibrosis transmembrane conductance regulator by nasal aminoglycosides in patients with cystic fibrosis with stop mutationsAm J Respir Cell Mol Biol2007371576617347447
- NudelmanIRebibo-SabbahACherniavskyMDevelopment of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutationsJ Med Chem20095292836284519309154
- SabbavarapuNMShavitMDeganiYSmolkinBBelakhovVBaasovTDesign of novel aminoglycoside derivatives with enhanced suppression of diseases-causing nonsense mutationsACS Med Chem Lett20167441842327096052
- XueXMutyamVTangLSynthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftorAm J Respir Cell Mol Biol201450480581624251786
- RoweSMSloanePTangLPSuppression of CFTR premature termination codons and rescue of CFTR protein and function by the synthetic aminoglycoside NB54J Mol Med (Berl)201189111149116121779978
- DuMJonesJRLanierJAminoglycoside suppression of a premature stop mutation in a Cftr−/− mouse carrying a human CFTR-G542X transgeneJ Mol Med (Berl)200280959560412226741
- HamedSDrug evaluation: PTC-124: a potential treatment of cystic fibrosis and Duchenne muscular dystrophyIDrugs200691178378917096300
- KeremEHirawatSArmoniSEffectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trialLancet2008372964071972718722008
- Sermet-GaudelusIDe BoeckKCasimirGJAtaluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosisAm J Respir Crit Care Med2010182101262127220622033
- KeremEKonstanMWDe BoeckKCystic Fibrosis Ataluren Study GroupAtaluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trialLancet Respir Med20142753954724836205
- BrownCRHong-BrownLQBiwersiJVerkmanASWelchWJChemical chaperones correct the mutant phenotype of the ΔF508 cystic fibrosis transmembrane conductance regulator proteinCell Stress Chaperones1996121171259222597
- SatoSWardCLKrouseMEWineJJKopitoRRGlycerol reverses the misfolding phenotype of the most common cystic fibrosis mutationJ Biol Chem199627126356388557666
- MoyerBDLoffing-CueniDLoffingJReynoldsDStantonBAButyrate increases apical membrane CFTR but reduces chloride secretion in MDCK cellsAm J Physiol19992772F271F27610444582
- RubensteinRCEganMEZeitlinPLIn vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTRJ Clin Invest199710010245724659366560
- RubensteinRCZeitlinPLSodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of ΔF508-CFTRAm J Physiol Cell Physiol20002782C259C26710666020
- RubensteinRZeitlinPA pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in Δ F508-homozygous cystic fibrosis patientsAm J Respir Crit Care Med199815724844909476862
- LooTBartlettMClarkeDCorrectors promote folding of the CFTR in the endoplasmic reticulumBiochem J20084131293618361776
- LooTBartlettMWangYClarkeDThe chemical chaperone CFcor-325 repairs folding defects in the transmembrane domains of CFTR-processing mutantsBiochem J2006395pt 353754216417523
- HuttDMHermanDRodriguesAPCReduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosisNat Chem Biol201061253319966789
- SampsonHRobertRLiaoJIdentification of a NBD1-binding pharmacological chaperone that corrects the trafficking defect of F508del-CFTRChem Biol201118223124221338920
- RenHYGroveDEDe La RosaOVX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1Mol Biol Cell201324193016302423924900
- Van GoorFHadidaSGrootenhuisPDJCorrection of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809Proc Natl Acad Sci U S A201110846188431884821976485
- ClancyJPRoweSMAccursoFJResults of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutationThorax2012671121821825083
- BoyleMPBellSCKonstanMWVX09-809-102 Study GroupA CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trialLancet Respir Med20142752753824973281
- WainwrightCEElbornJSRamseyBWLumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTRN Engl J Med2015373322023125981758
- JonesAMBarryPJLumacaftor/ivacaftor for patients homozygous for Phe508del-CFTR: should we curb our enthusiasm?Thorax201570761561626071414
- OrensteinDMO’SullivanBPQuintonPMCystic fibrosis: breakthrough drugs at break-the-bank pricesGlob Adv Health Med20154685726659555
- CholonDMQuinneyNLFulcherMLPotentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosisSci Transl Med20146246246ra296
- VeitGAvramescuRGPerdomoDSome gating potentiators, including VX-770, diminish ΔF508-CFTR functional expressionSci Transl Med20146246246ra297
- PilewskiJMCookeJLekstrom-HimesJDonaldsonSWS01.4 VX-661 in combination with ivacaftor in patients with cystic fibrosis and the F508del-CFTR mutationJ Cyst Fibros201514S1
- BeumerWSwildensJHenigNWS01.2 QR-010, an RNA therapy, restores CFTR function using in vitro and in vivo models of delF508 CFTRJ Cyst Fibros201514S1
- HenigNBeumerWAnthonijszHQR-010, an RNA Therapy, Restores CFTR Function in the Saliva Secretion AssayA37. It Won’t be Long: Advances in Adult Cystic FibrosisNew York, NYAmerican Thoracic Society2015A1449A1449
- Van GoorFHadidaSGrootenhuisPDJRescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770Proc Natl Acad Sci U S A200910644188251883019846789
- AccursoFJRoweSMClancyJPEffect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutationN Engl J Med2010363211991200321083385
- DaviesJSheridanHBellNAssessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trialLancet Respir Med20131863063824461666
- RamseyBWDaviesJMcElvaneyNGVX08-770-102 Study GroupA CFTR potentiator in patients with cystic fibrosis and the G551D mutationN Engl J Med2011365181663167222047557
- De BoeckKMunckAWalkerSEfficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutationJ Cyst Fibros201413667468025266159
- DaviesJCCunninghamSHarrisWTSafety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm studyLancet Respir Med20164210711526803277
- FlumePALiouTGBorowitzDSVX 08-770-104 Study GroupIvacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutationChest2012142371872422383668
- UttamsinehVPiljaLGrotbeckBWS13.6 CTP-656 tablet confirmed superiority of pharmacokinetic profile relative to Kalydeco® in phase I clinical studiesJ Cyst Fibros201615S1S22
- ShamsahKJoseALaurieDQBW251 is a safe and efficacious CFTR potentiator for patients with cystic fibrosisAm J Respir Crit Care Med2016193A7789
- VanhoutteFPGouyMHaazenWSafety, tolerability and pharmacokinetics of a novel CFTR potentiator GPLG1837 in healthy volunteersPediatr Pulmonol201550S41S289
- MarozkinaNVYemenSBorowitzMHsp 70/Hsp 90 organizing protein as a nitrosylation target in cystic fibrosis therapyProc Natl Acad Sci U S A201010725113931139820534503
- ZamanKSawczakVZaidiAAugmentation of CFTR maturation by S-nitrosoglutathione reductaseAm J Physiol Lung Cell Mol Physiol20163103L263L27026637637
- ZamanKCarraroSDohertyJS-nitrosylating agents: a novel class of compounds that increase cystic fibrosis transmembrane conductance regulator expression and maturation in epithelial cellsMol Pharmacol20067041435144216857740
- Taylor-CousarJZemanickESolomonGThe pharmacokinetics of N91115, an inhibitor of S-nitrosoglutathione reductase in cystic fibrosis patientsPediatr Pulmonol201550S41S285S286
- MillerJDrewLGreenOAmplifiers are a new class of CFTR modulators that increase the abundance of CFTR protein and combined with potentiators and correctors enhance chloride transport activityPediatr Pulmonol201550S41S265
- DhoogheBHaafJBNoelSLealTStrategies in early clinical development for the treatment of basic defects of cystic fibrosisExpert Opin Investig Drugs2016254423436