Abstract
This case report presents a newly diagnosed congenital factor VII deficiency treated with recombinant activated factor VII (rFVIIa). Congenital factor VII deficiency is a rare autosomal-recessive bleeding disorder that occurs in fewer than 1/500,000 persons. Its presentation can vary from epistaxis to hemarthroses and severe central nervous system bleeding, and correlates poorly with factor VII levels. Our patient had not had a significant hemostatic challenge prior to his presentation and therefore never had any symptomatology suggestive of this disease. He was treated with rFVIIa, and was able to undergo repair of his fractures without bleeding.
Case report
A 19-year-old African-American male presented to the emergency room after an altercation that resulted in significant trauma. He sustained bilateral mandibular angle fractures and orbital floor fractures, requiring urgent surgical correction. On initial evaluation, he was noted to have a prolonged prothrombin time of 40.1 seconds, with an International Normalized Ratio of 4.0, a normal activated partial thromboplastin time of 29.9 seconds, and a platelet count of 241. After receiving vitamin K and fresh frozen plasma, he was taken to the operating room for a temporary rigid maxillomandibular fixation. A 1:1 mixing study with normal plasma corrected the prothrombin time (decreasing from 40.7 to 14.7 seconds) and a factor VII assay revealed 5% of the normal factor VII level. The patient was diagnosed with congenital factor VII deficiency. Due to his coagulopathy and the extensive surgical correction needed, rFVIIa was administered and surgery was accomplished without hemorrhagic sequelae.
Conclusion
This case report and review describes a rare congenital disease, the history of rFVIIa use, and its mechanism. rFVIIA use in our patient provided a treatment option that allowed the necessary surgical correction, but further prospective studies on dose optimization would ensure adequate dosing with minimal risk of severe side effects.
Introduction
The first coagulation cascade discoveries were made in the 1940s. Since that time, research has provided a better understanding of its mechanism as well as the effects on hemostasis when deficiencies occur within the system. Some of the initial research leads to understanding of the causality of bleeding in hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency) patients. These patients often develop severe bleeding episodes that can now be treated with replacement of the missing factor. However, over time, they may develop antibodies, primarily immunoglobulin G, known as inhibitors, which are directed against the specific deficient factor replacement. When this occurs, replacement of their own deficient factor will not control bleeding episodes and alternative treatment must be used. Recombinant activated factor VII (rFVIIa, NovoSeven®) was originally developed for the treatment of cases of hemophilia A and hemophilia B that had inhibitors or acquired inhibitors to replacement factor VIII and factor IX. Its uses as approved by the US Food and Drug Administration have now been expanded, and it is also indicated for prevention of bleeding in hemophilia and congenital factor VII deficiency during surgical procedures, and treatment of bleeding episodes in patients with factor VII deficiency. In addition, recent research has supported the use of rFVIIa in other hemorrhagic conditions, such as Glanzmann’s thrombasthenia, von Willebrand disease, Bernard–Soulier syndrome, and factor XI deficiency.Citation1–Citation4 Despite its known use for controlling bleeding in these abnormal states of coagulation, use of NovoSeven is not without risk of side effects. Due to its mechanism as a potent thrombin generator, it can cause serious thrombosis. A case of newly diagnosed congenital factor VII deficiency is presented, and utilization of rFVIIa and its effects on the coagulation cascade with regard to prevention of bleeding complications, as well as the risk of thrombosis, is described.
Case report
A 19-year-old African-American male, with no notable past medical history, was admitted to the hospital for significant head and facial trauma. The patient had been in his usual state of health until the evening of admission when he sustained significant facial trauma, secondary to a physical assault with an unknown blunt object. The patient’s multiple facial injuries included bilateral mandibular angle fractures, bilateral orbital floor fractures, and bilateral nasal bone fractures requiring urgent surgical correction. On initial physical examination, the only abnormalities noted were the above-mentioned fractures and associated contusions, all of which were felt to be appropriate for the known mechanism of injury. Specifically, the patient was without abnormal bleeding, petechiae, purpura, or hepatosplenomegaly. A computerized tomographic scan of the head revealed small focal collections of subarachnoid hemorrhage in the left frontal and parietal lobes. Standard preoperative laboratory tests were obtained, and he was found to have an elevated International Normalized Ratio (INR) of 4.0 (reference range 0.9–1.1), a prolonged prothrombin time of 40.1 seconds (reference range 11.6–14.7) with a normal activated partial thromboplastin time of 29.9 seconds (reference range 26.0–42.0) and a platelet count of 241 (reference range 150–450). He was initially given intravenous vitamin K to attempt correction of his coagulopathy; however, it persisted until he was transfused with multiple units of fresh frozen plasma and given prothrombin complex concentrate. The patient was taken to surgery following this temporizing therapy, and a rigid maxillomandibular fixation was completed. Repair of his bilateral orbital floor fractures was deferred until a thorough workup could be completed and further recommendations for treatment could be delineated by hematology.
Upon further evaluation, the patient specifically denied any history of abnormal bleeding, bruising, epistaxis, or hemarthroses. However, he also denied any previous surgical procedures or major trauma. The patient had no significant past medical history and his only medications included recent postoperative pain medications. He had no known allergies to medications and he and his family specifically denied known family history of bleeding disorders or problems. Review of systems was essentially negative aside from the pain associated with the recent trauma and surgical procedure.
On physical examination, he was found to be hemodynamically stable. His weight was 89.2 kg and his height was 6 feet and 1.5 inches. He was awake, alert, and oriented in person, place, time, and situation, and was resting comfortably, in no acute distress. However, communication was limited secondary to rigid fixation of his mandible. He had some residual edema to his face, but was otherwise without significant ecchymoses. His respiratory, cardiovascular, abdominal, and neurological examinations were all within normal limits.
A mixing study revealed correction of the prothrombin time from 40.7 to 14.7 seconds (within normal limits) when the patient’s plasma was mixed 1:1 with normal control plasma. Given the patient’s prolonged prothrombin time and INR with a normal activated partial thromboplastin time, a factor VII activity assay was performed on the patient’s plasma. It revealed a 5% level of normal factor VII and the patient was diagnosed with congenital factor VII deficiency. In preparation for repair of his bilateral orbital floor fractures, it was recommended that the patient receive rFVIIa prior to and following his surgery. Based on the available dosing recommendations of 15–30 μg/kg for patients with congenital factor VII deficiency undergoing elective surgery, 2000 μg (22 μg/kg) were given immediately prior to surgery. This allowed normalization of his INR and created a window during which he could undergo repair of his orbital floor fractures without complication. He remained in the hospital postoperatively for continued observation, monitoring of his INR, and infusion of recombinant factor VII, with continued dosing every 4 hours for the first 24 hours postoperatively then every 6 hours for the subsequent 48 hours.
His postoperative course was complicated by an episode of subconjunctival emphysema related to the patient forcefully blowing his nose. However, he had no problems with excess bleeding or bruising. The patient’s subconjunctival emphysema spontaneously improved, and on postoperative day 3 he was tolerating a no-chew diet, the rFVIIa was discontinued, and he was discharged home.
Discussion
Factor VII and coagulation cascade
The coagulation cascade and our understanding of this pathway have evolved tremendously over the past 70 years. In the 1940s came the initial discoveries that led to elucidation of the coagulation cascade that is well known today. Specifically in 1949, Alexander et al described a factor in the serum that facilitated conversion of prothrombin to thrombin, which at that time was termed serum prothrombin conversion accelerator (SPCA).Citation5–Citation7 In 1951, Koller et al published on factor VII as a newly discovered clotting factor.Citation8 It was later determined that SPCA and factor VII as described separately by these individuals were in fact the same entity.
It is now known that factor VII plays a pivotal role in the coagulation cascade, and actually initiates the process of coagulation. Tissue factor (TF, factor III) is a transmembrane protein found on endothelium. Upon vascular injury, it is exposed to the blood, binds to circulating factor VII and activates factor VII (FVIIa), with the aid of factor IXa, factor Xa, factor XIIa, thrombin, and plasmin.Citation9,Citation10 It then creates a complex referred to as TF:FVIIa. This complex then converts factor X and factor IX into FXa and FIXa, respectively, which provides feedback amplification once activatedCitation9–Citation12 ( and ).
Figure 1 Factor VII is activated by tissue factor to VIIa, much the way a match is lit when struck (initiation).
Abbreviations: VII, Factor VII; TF, tissue factor; VIIa, Activated Factor VII; VIIIa, Activated Factor VIII; IXa, Activated Factor IX; Va, Activated Factor V; Xa, Activated Factor X; II, Factor II; IIa, Activated Factor II.
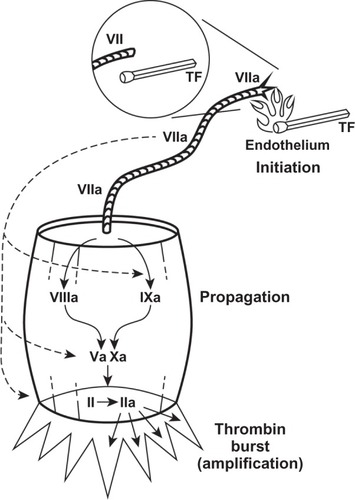
Figure 2 Coagulation cascade and the effects of recombinant activated Factor Vll (rFVlla).
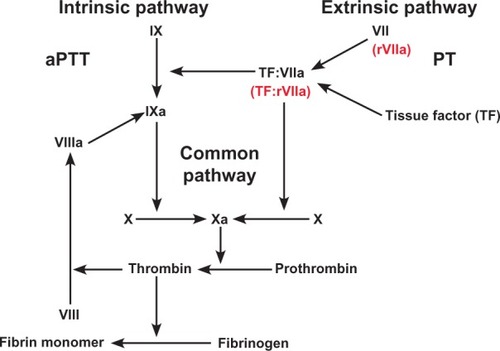
Creation of the TF:FVIIa complex at the site of injury is the initiating event that leads to hemostasis by formation of a fibrin clot, as a result of the conversion of prothrombin to thrombin and fibrinogen to fibrin. It is thrombin that activates factors V, VIII, and XI, and platelets, and activates thrombin-activatable fibrinolysis inhibitor which, in turn, protects the fibrin clot from lysis.Citation9 In addition, TF:FVIIa also activates factor VIII, factor IX, and factor X (). Although factors VIII and IX are considered part of the intrinsic pathway, it is the complex direct and indirect effects of TF:FVIIa on them that supports the view that the deficiencies in factor VIII and IX (hemophilia A and B, respectively) are actually an abnormality of the TF:FVIIa pathway ().Citation13 Due to limited production of the TF:FVIIa complex and via the above-described process, there is minimal formation of a fibrin plug and is one of the primary reasons why bleeding is observed in hemophiliacs and other bleeding disorders.Citation14
Congenital factor VII deficiency
Soon after SPCA (Factor VII) was described, Alexander et al subsequently went on to report a disease process referred to as congenital SPCA deficiency, which was later known to be the first reported case of congenital factor VII deficiency.Citation5–Citation7,Citation15
Congenital factor VII deficiency is a rare autosomalrecessive bleeding disorder, with a reported incidence of 1/500,000.Citation16,Citation17 Although rare, it is the most common among the recessively inherited coagulation disorders.Citation18 These patients may have clinical presentations that vary greatly. Patients may be asymptomatic or present with symptoms and complications that range from mild to lethal. Symptoms may be mild and include epistaxis, easy bruising, hematuria, and bleeding gums, or be more severe, with symptoms such as muscle hematomas, hemarthroses, and postoperative bleeding. Even lethal symptoms may occur, such as central nervous system bleeding.Citation12,Citation18
In vivo studies using knockout mice that were completely factor VII-deficient showed normal fetal development, but death soon after birth due to severe perinatal bleeding, revealing that complete deficiency of factor VII is incompatible with life.Citation19 Further studies have shown that there are numerous polymorphisms that result in factor VII deficiency, which explains the great heterogeneity observed with this disease.Citation17,Citation20,Citation21 Also, because of this variation in phenotype, patients can present and be diagnosed at various stages in their life, and may not be diagnosed until a significant hemostatic challenge is experienced.
Usual laboratory data reveal discordance between a prolonged prothrombin time and a normal activated partial thromboplastin time, depending on plasma FVII levels.Citation13,Citation18 The prolonged prothrombin time corrects on performing a 1:1 mixing study with normal plasma.Citation12 Confirmatory factor VII levels can confirm a diagnosis after preliminary laboratory data are consistent with factor VII deficiency. Patients with ≤1% normal factor VII levels typically present with severe symptoms that are often difficult to distinguish from hemophilia A or B, and often suffer from life-threatening intracranial hemorrhage and hemarthroses.Citation13,Citation16 However, patients with >5% normal factor VII levels often have minimal bleeding, such as epistaxis, gingival bleeding, menorrhagia, and easy bruising.Citation13 Despite this observation, there is no definitive correlation between factor VII levels and the prevalence of bleeding episodes and their severity.Citation12,Citation16 There are only a few reported cases in the literature of patients who have <1% of normal factor VII levels and undergo surgery without replacement therapy and experience no bleeding.Citation22–Citation25 This makes bleeding prophylaxis and prevention complicated in patients with factor VII deficiency, because it is not completely understood who will be at greatest risk for bleeding and what procedures may be the most dangerous.
Due to the rarity of factor VII deficiency, guidelines for the management of these patients have not been established.Citation12 Supportive care has historically included administration of fresh frozen plasma, prothrombin complex concentrates, factor VII concentrates, and, recently, rFVIIa, which enables an alternative approach to treatment, without exposing patients to the volume overload or risk of blood-borne infection associated with administering plasma-based products.Citation16
Recombinant activated factor VII
Recombinant FVIIa is a vitamin-K dependent glycoprotein that is structurally similar to human plasma-derived factor VIIa. No human serum or proteins are used in the production of rFVIIa. It is manufactured by cloning of the human factor VII gene and is expressed in baby hamster kidney cells, then cultured in bovine albumin.Citation26,Citation27 This process eliminates the risk of human blood-borne infection.Citation26 Recombinant FVIIa is a recombinant human coagulation factor that is intended for promoting hemostasis through activation of the extrinsic pathway of the coagulation cascade.Citation28 It does this by bypassing the coagulation cascade and creating a “thrombin burst”, thereby leading to a stable fibrin clot ().Citation26
There are numerous theories about the exact mechanism of rFVIIa in vivo. In hemophilia A and B (factor VIII and IX deficiencies), it has been shown that factor VIIa will bind to platelets and activate factor X to factor Xa, resulting in thrombin generation, despite deficiencies in the intrinsic pathway.Citation14 There has also been shown to be a dose-response relationship between the amount of rFVIIa and thrombin formation, so increasing doses of rFVIIa can increase thrombin generation even in the absence of factor VIII and IX.Citation14 In addition to upregulation of thrombin formation, it has also been shown that rFVIIa inhibits fibrinolysis by activation of thrombin activatable fibrinolytic inhibitor in factor VIII-deficient plasma.Citation29
As mentioned, rFVIIa was originally developed as a bypassing agent for the treatment of bleeding in hemophilia A and hemophilia B patients with inhibitors to replacement factor VIII and factor IX.Citation30 However, recent research has supported its use in numerous other hematological conditions.Citation1–Citation4,Citation26 Its safety and efficacy in congenital factor VII deficiency has been shown, and it has been used as prophylaxis for bleeding associated with surgery as well as used in the management of acute bleeding episodes.Citation30 Despite this, exact dosing preoperatively has been questioned, because it appears that more than just factor VII levels play a role in bleeding.Citation22–Citation25
It has been reported that in patients with congenital factor VII deficiency and hemophilia, the half-life of rFVIIa is 2.96 (2.82–3.11) and 2.3 (1.7–2.7) hours, respectively, and therefore it needs to be dosed frequently.Citation27 According to current recommendations, when used in patients with factor VII deficiency, the doses range from 15 to 30 μg/kg every 4–6 hours, but higher doses may be required for life-threatening hemorrhage.Citation27,Citation30 Effective treatment requires at least three doses to be administered for adequate hemostasis. These dosage recommendations vary dramatically from the dosing guidelines for patients with hemophilia, in which rFVIIa is currently recommended at 90 μg/kg every 2 hours until hemostasis is achieved.Citation27 It has been suggested by Tran et al that continuous infusion of rFVIIa during surgery results in the desired hemostasis as well as requiring a significantly reduced amount of rFVIIa, when compared with bolus injections.Citation31 Such infusional dosing has not become regarded as standard and therefore bolus dosing remains recommended. In addition, rFVIIa is listed as pregnancy category C, and it is unknown if it is excreted in breast milk.Citation27 Therefore, caution should be maintained in administration to these subsets of patients, and the risks versus benefits should be considered.
There have been several case reports which address safety concerns and administration both in the setting of bleeding prophylaxis as well as the complications that may occur. Tokgoz et al reported a case in which an infant with congenital factor VII deficiency had recurrent intracranial hemorrhage and was therefore placed on rFVIIa three times weekly, and had no further intracranial hemorrhage.Citation32 In addition, Jahromi and Karimi reported a case series of five patients with recurrent intracranial hemorrhages, discussing the dosing and duration of rFVIIa, also resulting in no further intracranial hemorrhage.Citation33 These cases, among others, reveal that there can be extreme benefit from prophylactic utilization of rFVIIa, but strong evidence of optimal dosing is lacking. Although bleeding improved in all patients in the previous cases, there are also reports of rFVIIa causing significant side effects. Due to the fact that rFVIIa is a potent thrombin generator, thrombosis resulting in serious events should be of concern to the prescribing physician. Review of the literature reveals reports of cerebral infarction, cerebral ischemia, angina pectoris, myocardial infarction, pulmonary embolism, and deep vein thrombosis.Citation27 In 2011, Girolami et al reported a series of patients who developed significant thrombotic events after factor replacement,Citation34 emphasizing that although these patients have severe clotting defects, there is still a risk of thrombosis. The Cochrane Collaboration published a large review and meta-analysis of multiple trials to evaluate the efficacy and safety of rFVIIa. This review included 29 randomized controlled trials with over 4290 patients. The data support utilization of rFVIIa, but due to the severe side effects observed, off-label use is cautioned against, but is observed and reported quite frequently in the literature.Citation35 This further supports that dosing is difficult in these patients, and additional prospective studies on administration of rFVIIa are mandatory.
Conclusion
Prophylactic administration of recombinant FVIIa in our patient was used empirically in light of the severity of his facial trauma as well as the location and extensiveness of the surgery. During administration, factor VII levels were not monitored, because our goal was not to reach a certain factor VII level, but to maintain a normal INR and prothrombin time and prevent any hematological complications that could have rendered this young patient blind. This case report describes the presentation of a rare congenital disease and demonstrates the prophylactic clinical use of recombinant FVIIa to prevent bleeding during urgent surgical correction of significant facial trauma. Further studies should be performed to assess optimal dosing schedules in this patient population. These studies may allow for smaller doses and less frequent administration when used in clinical situations such as ours.
Disclosure
The authors report no conflicts of interest in this work.
References
- d’OironRMenartCTrzeciakMCUse of recombinant factor VIIa in 3 patients with inherited type I Glanzmann’s thrombasthenia undergoing invasive proceduresThromb Haemost20008364464710823254
- KubiszPStaskoJRecombinant activated factor VII in patients at high risk of bleedingHematology2004931733215763970
- PoonMCDemersCJobinFWuJWRecombinant factor VIIa is effective for bleeding and surgery in patient with Glazmann thrombastheniaBlood1999943951395310572113
- PetersMHeijboerHTreatment of a patient with Bernard-Soulier syndrome and recurrent nosebleeds with recombinant factor VIIaThromb Haemost1998803529716174
- De VriesAAlexanderBGoldsteinRA factor in serum which accelerates the conversion of prothrombin to thrombin; its determination and some physiologic and biochemical propertiesBlood1949424725818110687
- AlexanderBdeVriesAGoldsteinRA factor in the serum which accelerates the conversion of prothrombin to thrombin; its evolution with special reference to the influence of conditions which affect blood coagulationBlood1949473974618129759
- AlexanderBde VriesAA factor in the serum which accelerates the conversion of prothrombin to thrombin; its relationship to the coagulation defect of thrombocytopenic bloodBlood1949474775118129760
- KollerFLoeligerADuckertFExperiments on a new clotting factor (Factor VII)Acta Haematol1951611814856604
- DahlbackBBlood coagulationLancet20003551627163210821379
- GolinoPThe inhibitors of the tissue factor: factor VII pathwayThromb Res2002106V257V26512356487
- RaoLVRapaportSIActivation of factor VII bound to tissue factor: a key early step in the tissue factor pathway of blood coagulationProc Natl Acad Sci USA198885668766913261869
- IngerslevJKristensenHLClinical picture and treatment strategies in factor VII deficiencyHaemophilia199846896969873815
- KaushanskyKWilliamsWJWilliams Hematology8th edNew York, NYMcGraw-Hill2010
- RobertsHRMonroeDMWhiteGCThe use of recombinant factor VIIa in the treatment of bleeding disordersBlood20041043858386415328151
- AlexanderBGoldsteinRLandwehrGCookCDCongenital SPCA deficiency: a hitherto unrecognized coagulation defect with hemorrhage rectified by serum and serum fractionsJ Clin Invest19513059660814841260
- HunaultMBauerKARecombinant factor VIIa for the treatment of congenital factor VII deficiencySemin Thromb Hemost20002640140511092215
- MarianiGHermannFHDolceAClinical phenotypes and factor VII genotype in congenital factor VII deficiencyThromb Haemost20059348148715735798
- MarianiGBernardiFFactor VII deficiencySemin Thromb Hemost20093540040619598068
- RosenEDChanJCIdusogieEMice lacking factor VII develop normally but suffer fatal perinatal bleedingNature19973902902949384381
- BernardiFMarchettiGPinottiMFactor VII gene polymorphisms contribute about one third of the factor VII level variation in plasmaArterioscler Thromb Vasc Biol19961672768548429
- TriplettDABrandtJTBatardMADixonJLFairDSHereditary factor VII deficiency: heterogeneity defined by combined functional and immunochemical analysisBlood198566128412874063521
- YorkeAJMantMJFactor VII deficiency and surgery. Is preoperative replacement therapy necessary?JAMA1977238424425577560
- Giansily-BlaizotMBiron-AndreaniCAguilar-MartinezPInherited factor VII deficiency and surgery: clinical data are the best criteria to predict the risk of bleedingBr J Hematol2002117172175
- StraussHSSurgery in patients with congenital factor VII deficiency (congenital hypoproconvertinemia): experience with one case and review of the literatureBlood19652532533414263207
- Giansily-BlaizotMSchvedJFPotential predictors of bleeding risk in inherited factor VII deficiency. Clinical, biological and molecular criteriaThromb Haemost20059490190616363229
- AgarwalMBPatnaikMRecombinant activated factor VII (rFVIIa, NovoSeven)J Assoc Physicians India20055371772016398083
- NovoSevenRTCoagulation factor VIIa (recombinant) [package insert] Available from: http://www.novo-pi.com/novosevenrt.pdfAccessed January 5, 2013
- RobertsHRThoughts on the mechanism of action of action of FVIIaPresented at the Second Symposium on New Aspects of Hemophilia TreatmentCopenhagen, DenmarkJan 1991
- LismanTMosnierLOLambertTInhibition of fibrinolysis by recombinant factor VIIa in plasma from patients with severe hemophilia ABlood20029917517911756168
- TchengWYDonkinJKonzalSWongWYRecombinant factor VIIa prophylaxis in a patient with severe congenital factor VII deficiencyHaemophilia20041029529815086331
- TranHTTTjonnfjordGEPausAHomeArFVIIa administered by continuous infusion during surgery in patients with severe congenital FVII deficiencyHaemophilia20111776477021707871
- TokgozHCaliskanULavigne-LissaldeGGiansily-BlaizotMSuccessful prophylactic use of recombinant activated factor VII (rFVIIa) in a patient with congenital FVII deficiency and inhibitors to FVIIHaemophilia201218e25e2721973016
- JahromiBJKarimiMLong-term follow-up of prophylaxis with recombinant activated factor VII in patients with congenital factor VII deficiencyHaemophilia20111771371521299748
- GirolamiABertozziIRigoniIMuzzolonRVettoreSCongenital FVII deficiency and thrombotic events after replacement therapyJ Thromb Thrombolysis20113236236721681394
- SimpsonELinYStanworthSBirchallJDoreeCHydeCRecombinant factor VIIa for the prevention and treatment of bleeding in patients without haemophiliaCochrane Database Syst Rev20123CD00501122419303