Abstract
Metachromatic leukodystrophy (MLD) is an autosomal recessive lysosomal disorder caused by the deficiency of arylsulfatase A (ASA), resulting in impaired degradation of sulfatide, an essential sphingolipid of myelin. The clinical manifestations of MLD are characterized by progressive demyelination and subsequent neurological symptoms resulting in severe debilitation. The availability of therapeutic options for treating MLD is limited but expanding with a number of early stage clinical trials already in progress. In the development of therapeutic approaches for MLD, scientists have been facing a number of challenges including blood–brain barrier (BBB) penetration, safety issues concerning therapies targeting the central nervous system, uncertainty regarding the ideal timing for intervention in the disease course, and the lack of more in-depth understanding of the molecular pathogenesis of MLD. Here, we discuss the current status of the different approaches to developing therapies for MLD. Hematopoietic stem cell transplantation has been used to treat MLD patients, utilizing both umbilical cord blood and bone marrow sources. Intrathecal enzyme replacement therapy and gene therapies, administered locally into the brain or by generating genetically modified hematopoietic stem cells, are emerging as novel strategies. In pre-clinical studies, different cell delivery systems including microencapsulated cells or selectively neural cells have shown encouraging results. Small molecules that are more likely to cross the BBB can be used as enzyme enhancers of diverse ASA mutants, either as pharmacological chaperones, or proteostasis regulators. Specific small molecules may also be used to reduce the biosynthesis of sulfatides, or target different affected downstream pathways secondary to the primary ASA deficiency. Given the progressive neurodegenerative aspects of MLD, also seen in other lysosomal diseases, current and future therapeutic strategies will be complementary, whether used in combination or separately at specific stages of the disease course, to produce better outcomes for patients afflicted with this devastating inherited disorder.
Introduction
Metachromatic leukodystrophy (MLD) is an autosomal recessive lysosomal storage disorder caused by the deficiency of arylsulfatase A (ASA; EC 3.1.6.8), a lysosomal sulfatase that hydrolyzes the 3-O ester bond from sulfatides including 3-O-sulfogalactosylceramide and 3-O-sulfolactosylceramide.Citation1,Citation2 These sulfatides are expressed in specific tissues and are mostly abundant in the central (CNS) and peripheral nervous systems (PNS). Sulfatides are important components of myelin, which is a crucial insulator sheath that surrounds axons as a bilayer membrane.Citation3 An intact myelin sheath is important for the proper conduction of nerve impulse in both CNS and PNS.Citation3 The inappropriate recycling of sulfatides results in alterations of the myelin structure, affecting the physiological transmission of electrical impulse between nerve cells.Citation4 This process of degeneration of the myelin sheath surrounding axons, mostly known as demyelination, is the hallmark of MLD. Patients afflicted with this condition present with a broad spectrum of neurological manifestations centered on the impairment of CNS and PNS, including progressive coordination and speech problems, seizures, and behavioral disturbances.Citation5 In the design and development of disease-specific therapies for MLD, the biochemical pathogenesis and secondary molecular events to the ASA deficiency are important to be considered.
Epidemiology and pseudodeficiency ARSA alleles
Inherited disorders affecting the lysosomes have a collective incidence of 1 in 2,300–7,000 live births, making them a prevalent class of inborn organelle disorders.Citation6,Citation7 Among different lysosomal disorders, the general prevalence of MLD varies from 1:40,000 to 1:100,000.Citation8 In the Polish population, its incidence was reported as 4:100,000.Citation9 MLD is a pan-ethnic lysosomal storage disease with affected patients described in several populations including European, Japanese, Jewish, Lebanese, Muslim Arab, South African, Iranian, Indian, Polynesian, Algerian, Habbanite Jew, Navajo Indian, Alaskan Eskimo, and Christian Arab, with ranging from mild to severe forms of MLD.Citation8 The deficiency of ASA is caused by mutations in the ARSA gene in homo- or heterozygosity. Over 150 mutations have been reported in MLD patients.Citation10 Some specific alterations in the ARSA gene sequence result in an enzymatic activity of 10%–15% of the normal (wild-type) ASA, which is sufficient to physiologically hydrolyze sulfatides, maintaining their recycling process and avoiding their accumulation in the lysosomal compartment. These non-deleterious ARSA gene alterations resulting in reduction of ASA enzymatic activity are known as pseudodeficiency alleles. These ARSA alleles are considered polymorphisms, with no disease-associated symptoms either in hetero or homozygous states. Two most common ARSA pseudodeficiency alleles are c.1049A>G/p.N350S and c.*96A>G. As they occur in cistrans, they are described as c.[1049A>G; c.*96A>G].Citation11,Citation12 The c.1049A>G/p.N350S results in modification of one of the N-glycosylation sites of ASA, affecting the structural stability of ASA and its targeting to the lysosomes.Citation12 The c.*96A>G is found in the 3′ non-translated region that alters the signaling of the polyadenylation of mRNA, substantially reducing the amount of ASA produced.Citation13 The frequency of these ARSA pseudodeficiency alleles is up to 5% in the European population.Citation13–Citation17 The carrier frequency of the ARSA pseudodeficiency alleles in Australia is estimated to be 20%.Citation16 Its correlation with occurrence of other neurodegenerative disorders that occur later in life remains debatable.Citation14 The existence of the ARSA pseudodeficiency alleles demonstrates that the sulfatide degradation can occur normally in the presence of ASA variants functioning at 10–15% of the wild type ASA enzymatic activity. This biochemical observation has crucial implications in the development of therapeutic strategies for MLD.
Biochemistry and molecular genetics of MLD
As with other lysosomal hydrolases, ASA is synthesized in the rough endoplasmic reticulum (ER) and co-translationally transported to the lumen of ER.Citation18–Citation20 Once properly folded, ASA is targeted to the trans-Golgi network where it becomes a substrate of uridine diphosphate (UDP)-N-Acetylglucosamine-1 phosphotransferase (EC 2.7.8.17) and N-acetylglucosamine-1-phosphodiester alpha-N-acetylglucosaminidase (EC 3.1.4.45). These two enzymes catalyze the binding and formation of mannose-6-phosphate residue, the recognition marker of lysosomal hydrolases. This modification is important to allow ASA to interact with the mannose-6-phosphate receptor, and be targeted to the lysosomal compartment.Citation21 In terms of its molecular structure, 2.1-Å resolution crystal X-ray structure showed that ASA is a homo-octamer composed of a tetramer of dimers (α2)4.Citation22 In fact, the formation of ASA homodimers (α2) occurs in the ER. Subsequently, ASA only assumes the homo-octomeric conformation at the acidic pH of the lysosomal compartment.Citation22 The identification of the c.1277T>G/p.P426L mutation showed it preventing the formation of homo-octomers, as the P426 residue locates proximal to the E424 residue, whose protonation is necessary for the octomerization process.Citation23 The mutant ASA-P426L is degraded by cathepsin-L, a lysosomal protease, which recognizes a cleavage site in the ASA homo-dimer interface that is normally protected by the octomerization conformation seen in wild-type ASA.Citation23,Citation24 Functionally, ASA catalyzes the desulfation of 3-O-sulfogalactosyl-bound sphingolipids. Sulfatides, or 3-O-galactosylceramides, are the main sphingolipids containing sulfate residues. Sulfatides are abundant in both the CNS and PNS. These glycosphingolipids are the main components of the myelin sheath. In the CNS, oligodendrocytes synthesize sulfatides, which are located in the myelin surrounding neural axons. In the PNS, the Schwann cells are the myelin-producing cells. Interestingly, in the gall bladder of MLD-affected patients, sulfatides accumulate in the epithelium cells, resulting in sludge, gallstones, hemobilia, cholecystitis, polyposis, papillomatosis, and small or enlarged gallbladder.Citation25–Citation29 Sulfatides are also present, but at low levels, in the respiratory tract,Citation30 gastric mucosa,Citation31 and uterine endometrium.Citation32 The 3-O-sulfatelactosyl ceramide, which is another sulfatide substrate of ASA, is found in the renal tubules and liver.Citation33 Another deacylated sulfatide, known as lysosulfatide or psychosine sulfatide, is present in the CNS and renal tubules.Citation34,Citation35 The presence of sulfatides in the kidney is the cornerstone of the main diagnostic tool for MLD, which is based on measuring urinary sulfatides.Citation36 ASA is also responsible for catalyzing the desulfation of glycerol-containing sulfated sphingolipids, which have a sulfate bond to the galactose C-3 hydroxyl residue. These 3-O-sulfagalactosyl sphingolipids are present in the mammalian testes and sperm, which are mostly known as seminolipids.Citation37 These sphingolipids are produced by spermatocytes, and their roles in different stages of the spermatogenesis are still to be determined.Citation37
In MLD, the deficiency of ASA results in sulfatide accumulation in the myelin-producing cells: oligodendrocytes in CNS, and Schwann cells in PNS. With the progressive accumulation of sulfatides, the lysosomal-endosomal system becomes dysfunctional, and other secondary pathogenic cascades occur, ultimately resulting in apoptosis.Citation38 This results in progressive demyelination in both CNS and PNS, which correlates with the major clinical manifestations of MLD.Citation39 However, cellular inclusions called membranous cytoplasmic bodies (MCBs), which can correspond to accumulation of sulfatides, are observed in pyramidal neurons and Purkinje cells in the brain of MLD patients.Citation40
The deficiency of ASA is caused by mutations in the ARSA gene. Amongst the over 150 mutations reported in the ARSA gene, more than 70% are missense mutations.Citation10 The most common mutation is the splice donor site mutation of the exon2/intron2 border, IVS459+1G>A, which occurs in 15%–43% of MLD alleles, and is associated with the late infantile clinical form of MLD.Citation41 The second most common ARSA mutation is c.1277C>T/p.P426L, which, when in homozygosity or heterozygosity with an infantile mutation (eg, IVS459+1G>A), is associated with juvenile onset of MLD.Citation41 Each of these two common mutant alleles represents approximately 25% of all ARSA mutant alleles in MLD patients with European ancestry.Citation42 The third most prevalent ARSA mutation is c.536T>G/p.I179S, which, when in heterozygosity with the c.1277C>T/p.P426L mutation, is associated with adult onset of the disease.Citation41,Citation42 Interestingly, ASA-p.I179S presents the higher residual ASA enzymatic activity, and is correlated with later onset and more protracted MLD course.Citation41 The ARSA c.536T>G/p.I179S allele is present in 12%–13% of the total European population of mutant alleles.Citation41,Citation42 Though several missense mutations have been identified, few of them have been biochemically characterized.Citation10,Citation43 Most missense mutations result in misfolded mutant ASA that is early selected for the ER-associated degradation (ERAD) pathway and targeted to the ubiquitin-proteasome system.Citation44 Therefore, small amounts of partially folded and/or misfolded mutant ASA pass the ER post-translational quality control and ultimately reach the lysosomal compartment, resulting in the residual ASA enzymatic activity.Citation45 Such molecular characterizations, and their correlation with the MLD pathogenesis, may assist in delineating the molecular mechanism to correct the lysosomal enzymatic deficiencies at genetic and biochemical levels.
Clinical presentations
MLD is a neurological lysosomal storage disorder with a broad and heterogeneous clinical spectrum characterized by CNS and PNS demyelination resulting in a progressive demyelination and encephalopathy. The clinical presentation and disease course of MLD can be classified into three clinical forms. The three clinical forms are determined on the basis of the age of onset of first symptoms, the rate of progression of the neurological symptoms, and their severity. The late infantile form is the most severe form, in which patients develop first symptoms within a few months to 2 years of age.Citation46 The juvenile MLD clinical form includes patients who have the onset of symptoms occur between 3 and 16 years of age. Patients with the adult MLD clinical form have the onset of disease when older than 16 years. The two late-onset forms of MLD, juvenile and adult, sometimes overlap, and they usually present with a more insidious manifestation of a wide range of neurological symptoms, offering ample opportunity for possible therapeutic interventions.
Patients with the late infantile form of MLD present with delays in psychomotor development characterized by impairment of speech, gross, and fine motor development. Additionally, patients can present with muscular hypotonia, hyporreflexia, lately followed by pyramidal signs characterized by spasticity, and hypereflexia. Ataxia, spastic paresis, and optic dystrophy were also seen. The juvenile form of MLD is characterized by later onset of the above symptoms, which are usually accompanied by poor school performance, and some behavioral disturbance and peripheral neuropathies. Later on, MLD patients become debilitated, losing gait independence and speech. As disease progresses, seizures and recurrent pulmonary infections occur frequently.Citation8,Citation43 The adult form of MLD is characterized by gradual impairment of intellectual function, emotional instability, and behavioral/psychiatric disturbances. Polyneuropathy occurs later and does not usually present symptoms in this clinical form of MLD. In general, the course of disease is slower than the juvenile and late infantile clinical forms.Citation8
Pathogenesis
Sulfatide formation and its role in myelin
Sulfatides are membrane sphingolipids that show a cell type-specific expression.Citation3 They are expressed in the kidney and bile duct, in addition to brain tissue. Sulfatides are the most abundant sphingolipid in myelin, accounting for 4% of its composition. The stability of the myelin structure is one of its distinct features, as demonstrated in the intact myelin found in the 5,000-year old Tyrolean Iceman.Citation47 Two main factors confer stability of the myelin structure. First, the hydrophobic components of myelin work to repel the hydrophilic environment around its structure, represented by cytosol and extracellular fluids. Second, the same hydrophobic structures have strong attractive forces among them, making myelin a compact structure.Citation3 The sulfatide and galactosylceramide structures are major contributors to these attractive forces through formation of hydrogen bonds.Citation48 In MLD, the excess of sulfatides and deficiency of cerebrosides can lead to abnormal myelin composition and possibly affect the formation of a stable lipid bilayer.Citation49 Further, the sulfatide metabolite galactosylsphingosine can also cause cytotoxicity and lead to death and dysfunction of neuronal cells.Citation50
Sulfatides are formed by the action of a series of enzymes, as shown involved in the biosynthesis and degradation of sulfatides in . The importance of different myelin components has been investigated in specific mutant murine models with deficiencies in genes encoding the enzymes in the sulfatide biosynthetic pathway. UDP-galactose:ceramide galactosyltransferase (CGT) knockout mice (cgt −/−) were unable to synthesize the galactosylceramide or sulfatide structures.Citation51 Though the myelin structure seems normal, the cgt −/− mice develop progressive hind limb paralysis and extensive vacuolation of the ventral region of the spinal cord as they age.Citation2,Citation51,Citation52 Thus, these results indicate that the sulfatides are important in myelin stability and its physiological function. In the absence of CGT, the cgt −/− mice synthesize 2-OH glucocerebroside as the main component of myelin, a glycosphingolipid that is absent in the normal myelin composition. The 2-OH glucocerebroside gives a seemingly normal structure to myelin, and was hypothesized to prevent the severe effects of the loss of hydroxylated fatty acids such as galactosylceramide.Citation52,Citation53 To validate this hypothesis, a CGT/FA2H double knockout mouse (cgt −/−/fa2h −/−) was generated with an additional deletion of the FA2H gene encoding the fatty acid 2-hydroxylase.Citation54 These mice showed myelin comprised of non-hydroxylated sphingomyelin.Citation54 No phenotypic differences were observed between cgt −/−/fa2h −/− and cgt −/− knockout mice.Citation54 Thus, these results suggest that the presence of glucosylceramide and hydroxylated sphingomyelin in cgt −/− mice does not functionally compensate for the loss of hydroxylated fatty acid galactosylceramide.Citation54 On the other hand the fa2h −/− mice with intact CGT showed absence of 2-OH galactosylceramide and sulfatide in normal myelin.Citation48 However, myelin sheath degeneration was observed with aging.Citation48,Citation55 These studies of different murine knockout models deprived of specific myelin sphingolipids demonstrate that myelin biogenesis in the nervous system remains intact, likely due to the redundancies of these myelin components. The murine model with ceramide synthase 2 (CerS2) also showed interesting observations with regard to the importance of galactosylceramide and sulfatides in myelin.Citation56 The CerS2 knockout mice showed decreased levels of galactosylceramide, sulfatide, and very long-chain fatty acids.Citation56 From early adulthood the CerS2 null mice showed progressive myelin instability, with 50% loss of the compacted myelin pattern and ~80% loss of myelin-basic protein.Citation56 In a different study of the hepatic biochemical aspects of CerS2-null mice, the biophysical properties of lipid extracts isolated from liver microsomes were shown to be altered indicating higher membrane fluidity and morphological changes.Citation57 Taken as a whole, these observations on Cer2S knockout mice reiterate the role of the sulfatides in the maintenance of myelin stability, which seems to be of crucial importance during the aging process.
Figure 1 Sulfatide metabolic pathway.
Abbreviations: ASA, arylsulfatase A; EET, enzyme enhancement therapy; ERT, enzyme replacement therapy; Gal, galactose; HSCT, hematopoietic stem cell transplantation; Glc, glucose; SGal-Glc-cer, sulfo-galactose-glucosylceramide; MLD, metachromatic leukodystrophy; SRT, substrate reduction therapy.
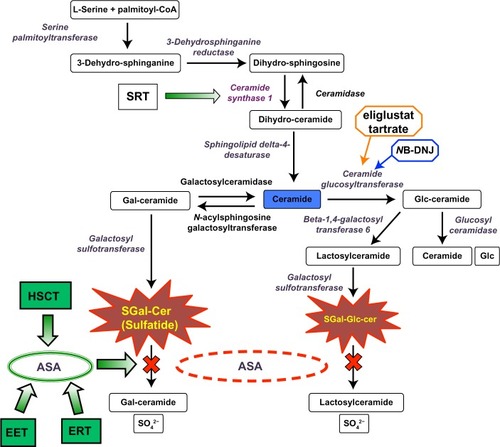
Mechanism of leukodystrophy
In MLD, ASA deficiency causes impairment of sulfatide degradation, and its accumulation mostly in nervous system (). Sulfatide accumulation does not impair the function of the renal tubular, respiratory epithelium, or endometrium as it does in the CNS and PNS.Citation58 Of note is that some degree of accumulation of sulfatides in the mucosa of the biliary tract is observed in MLD, resulting in mild to moderate clinical manifestations.Citation26–Citation29 This observation is explained because, physiologically, the levels of sulfatides present in the nervous system are much higher, reflecting its crucial function in the composition of myelin. In the arsa −/− MLD mouse model generated by Hess et al, mild neurological symptoms are only observed by the end of a normal life span.Citation59 As the arsa −/− mice fail to show sulfatide accumulation, no signs of demyelination are observed.Citation59 However, the arsa −/− mice showed neuromotor incoordination, suggesting that such neurological signs might occur before demyelination.Citation59 The lack of a complete recapitulation of the clinical and pathological MLD phenotype led to the generation of the arsa −/− mice with neural cells overexpressing the sulfatide synthesizing enzymes, including UDP-galactose:ceramide galactosyltransferase (CGT) and cerebroside sulfotransferase (CST).Citation60 These CGT/arsa −/− and CST/arsa −/− mice showed increased sulfatide storage in myelin-forming cells, resulting in axonal degeneration and development of neurological symptoms similar to MLD.Citation60 The hallmark of the disease is progressive demyelination with resulting neurological manifestations. The precise pathogenic mechanism of the demyelination is still not clearly understood.Citation43
Pathogenic cascades
In lysosome storage disorders (LSDs), several pathogenic cascades have been described secondary to the substrate accumulation in the lysosomal-endosomal system.Citation38,Citation61,Citation62 These cascades include calcium signaling, altered lipid trafficking, oxidative stress, ER stress/unfolded protein response, autophagy, and inflammation.Citation38 The role of these different pathogenic cascades in MLD is largely uncharacterized, but the cascades in some lysosomal disorders presenting with neurodegenerative processes are well studied. Calcium signaling is essential for cell motility and myelination of neurons. Therefore, perturbations of calcium signaling are likely to contribute to the demyelination observed in MLD.Citation63,Citation64 Studies in brain tissue from Gaucher disease patients indicate that defective calcium homeostasis may be a mechanism that leads to neuropathophysiology in the acute and chronic neuronopathic forms of Gaucher disease.Citation65 Studies in mice models of mucopolysaccharidosis type III showed that neurodegeneration proceeds independently of the signaling cascade associated with heparin sulfate.Citation66 Signaling cascades may direct cells towards autophagy, as observed in a lysosomal disease characterized by neuromuscular presentation known as Pompe disease. This inborn organelle disease is caused by deficiency of lysosomal acid α-glucosidase (GAA). In the fibroblasts from Pompe disease patients carrying c.546G>T mutation in the GAA gene, the misfolded mutant GAA was shown to induce ER stress, and subsequently elicit autophagy through activation of the p38 mitogen-activated protein kinase pathway.Citation67 Autophagy is the pathway through which a cell can recycle defective proteins in the system. Thus, autophagy is a key cleansing and regenerating pathway, in which products of protein breakdown can make amino acids available to the cell again. Also, autophagy and apoptosis have been shown previously to be inversely related in hematopoietic stem cells.Citation68 In another study in cultured Pompe patient fibroblasts, the inactivation of Akt can lead to autophagy, in a ER stress-independent manner, via the suppression of mTOR signaling.Citation69 In the absence of ER stress in Pompe disease fibroblasts, the inactivated Akt signaling cascade drives the cells to autophagy, which can be reversed with insulin, which activates Akt signaling.Citation69 In Niemann-Pick disease type C (NPC), sphingosine storage inhibits calcium uptake into the lysosomal acidic compartment, leading to insufficient calcium release for function of the endocytic pathway.Citation70 In this lysosomal disease, the altered lipid trafficking was demonstrated using fluorescent analogues of different lipids. Fluorescent dye-labeled sulfatides were shown to accumulate in lysosomes from fibroblasts obtained from MLD patients.Citation71 Studies have shown evidence that altered chain length of the lipids can affect its trafficking along the endocytic pathway.Citation72 Further, the storage of such metabolites in lysosomes is linked with defective autophagosome-lysosome fusion,Citation73 ultimately resulting in impaired autophagy. The processes of autophagy and apoptosis have been shown to be complementary in some cell types.Citation68 Thus, the impaired autophagy pathway in lysosomal disorders can lead to apoptosis. Inflammation is associated with the development of neurodegenerative diseases like multiple sulfatase deficiency (MSD).Citation74 MSD is pathogenically similar to MLD, since in both disorders sulfatide accumulation is noted. In MSD, inflammation was observed in the mouse model with null mutation in the SUMF1 gene,Citation75 which encodes the formylglycine-generating enzyme that is responsible for the oxidation of cysteine and formation of formylglycine (alanine-semialdehyde), the critical catalytic residue present in the active site of eukaryotic sulfatases.Citation76 The sumf1 −/− mice demonstrated complete loss of sulfatase activities and, consequently, early death, growth retardation, and neurological defects.Citation75 These mice showed inflammatory response characterized by the presence of vacuolated macrophages and activation of microglia in different regions of the brain.Citation75 In sumf1 −/ − mice between 4 and 6 months of age, elevated expression of inflammatory cytokines and apoptotic markers were observed in the CNS and liver, indicating that inflammation and apoptosis occur later in the disease and play an important role in the neurological phenotype.Citation75 Thus, arresting the disease after these processes occur becomes very difficult as, by this point in time, permanent and irreversible neural damage is already established in the CNS.Citation77
Challenges in development of treatment options
Blood-brain barrier
The blood-brain barrier (BBB) controls the migration of components from circulating blood to brain tissue. The BBB is formed by several types of cells, including endothelial cells with tight junctions, pericytes, astrocytes with foot processes, and perivascular neurons. Thus, the arterial capillaries in the CNS form a permeability barrier distinct from any other tissue, which harbors endothelial fenestrations between cells. The tight junctions found in the BBB generate high transendothelial electrical resistances of 1,500–2,000 Ω cm2. As a comparison, in tissues other than brain, the transendothelial electrical resistance ranges from 3–33 Ω cm2.78 Thus, the BBB contributes with the essential protection of neural structures preventing cellular damage from extra-cellular insults.
However, from the standpoint of the therapeutic development for inherited neurological disorders such as MLD, the BBB represents “a problem standing before the major problem”. In other words, BBB constitutes the major obstacle to novel therapeutic agents that require reaching the affected neural tissue to treat the neurological manifestations of the disease. Amongst the drugs in the current medicinal chemistry database, only a few – approximately 5%–6% – cross the BBB and are able to produce CNS effects.Citation79 These are small molecules with high Log P coefficient of lipophilicity, and molecular weight ranging from 400 to 450 D.Citation79 These drugs include antipsychotics, antidepressants, and hypnotics.Citation79 The BBB imposes the main obstacle to delivery of several of the novel therapies developed for MLD, including the recombinant enzyme, which is a large molecular weight therapeutic agent (~52 kD). Additionally, certain cells of hematopoietic origin, including macrophages, activated T-cells, and microglial cells, are capable of crossing the BBB and migration to the CNS.Citation80 Thus, both specific strategies, utilizing small molecules and specific cells, constitute potential novel avenues to deliver CNS disease-modifying agents for MLD patients.
Incomplete understanding of MLD pathogenesis
Sulfatides account for 4%–6% of the lipids forming the myelin and, as above, are essential sphingolipids in the myelin sheath surrounding the axons. They modulate the differentiation of myelinating cells, and are required for axon-glial interactions. Sulfatides are extracted from the myelin by saposin B (SapB), a heat-stable sphingolipid activator protein that is necessary to solubilize sulfatides and make them accessible to ASA (). Consequently, the molecular and clinical manifestations of MLD can also be resultant from defects in SapB, which was the first saposin described.Citation81 The arsa −/− mouse model showed a similar pattern of accumulation of sulfatides in the CNS and other organs.Citation59 However, the levels of sulfatide accumulation were lower, which resulted in mild phenotype with no myelin loss until the second year of mouse life, when progressive astrogliosis and microgliosis in the CNS and peripheral nerves were noticeable.Citation59 In the arsa −/− mouse, reduction of the levels of membrane raft-associated sulfatide-binding myelin and myelin and lymphocyte (MAL) protein were observed in the oligodendrocyte-generated myelin. This phenomenon indicates the presence of a compensatory mechanism to limit MAL-mediated delivery of sulfatide to the myelin membrane.Citation82 The sulfatide in the mice CNS has a long halflife (>6 months). Therefore, a short lifespan of the MLD mice model can limit the extent of sulfatide accumulation. How the accumulation of sulfatides causes the disruption of myelin sheath resulting in apoptosis of the myelin-forming cells, as observed in older arsa −/− mice, is still unclear.Citation59 In ultrastructural transmission electron microscopy studies of neurons from the arsa −/− mice, several MCBs were noticeable, indicating that sulfatides are likely accumulated in these cells.Citation83 The accumulation of sulfatides is possible in oligodendrocytes and Schwann cells of affected MLD patients, resulting in failure to produce and maintain myelin in axons located in the CNS and PNS, respectively. Thus, the complete inhibition of sulfatide biosynthesis is not an option in treatment, considering its importance as a myelin component. In addition, the biological effects of sulfatide accumulation in neural cells other than oligodendrocytes and Schwann cells are still unknown.Citation83 The arsa −/− mouse model of MLD showed incoordination signs in the absence of pathological signs of demyelinationCitation59, alluding that other pathogenic process may occur that can result in neurological symptoms, beyond demyelination.Citation83 Therefore, a better understanding of the molecular pathogenesis of MLD is a substantial challenge in developing therapies for MLD.
Time of intervention in the course of disease
In case of MLD, as in others lysosomal disorders, the classification of patients in clinical forms such as late infantile, juvenile, and adult is based on the age of onset of first symptoms.Citation58 In general, the earlier the onset of the disease, the more rapid is the disease progression. The different treatment options, though currently limited, can produce a better outcome when started in the earlier stages of the disease.Citation84 Generally, the earlier the intervention, the better the responses to that specific therapy observed. The importance of earlier intervention can be noted in MLD patients who underwent hematopoietic stem cell transplantation (HSCT).Citation85 MLD patients who received HSCT earlier usually had a better outcome than those transplanted at later stages of the disease. This can considerably increase an already high risk of mortality strictly related to the procedure.Citation85–Citation89 Some studies have shown that lysosomal storage in neuronal cells can be reduced to some extent in different LSDs. However, the cellular changes occurring downstream of storage are usually not reversible.Citation90 Thus, it is necessary to arrest such a primary cellular event at earlier stages of the natural history of the disease. Hence, the time of intervention becomes an important parameter in determining the prognosis and outcome.
Current treatments for MLD
Since MLD is caused by defective ASA, most therapeutic approaches have tried to correct the biochemical defect by providing wild-type ASA (). The different methods and sources of wild-type ASA constitute distinct therapeutic approaches. Enzyme replacement therapy (ERT) and HSCT rely on providing normal ASA to the deficient cells, while gene therapy approaches are based on the overexpression of wild-type ASA in different cell types ().
Enzyme replacement therapy
ERT has been a therapeutic modality used with remarkable success in treating some LSDs. Its applicability to MLD is challenged by the BBB. Lysosomal ASA is a high molecular weight therapeutic agent, and consequently is unable to penetrate the BBB to reach the affected brain tissue. In mice models, some studies have shown positive results,Citation91 leading to early phase clinical trials in MLD-affected patients. The efficacy of this treatment modality is inversely related to the age of mice.Citation92 The ERT approach also faces the difficulty of producing the recombinant enzyme at a large scale and in high purity, which has been overcome already by several pharmaceuticals.Citation93 In this setting, the BBB limitation has been addressed with different routes of administration, including intracerebroventricular or intrathecal ERT agent delivery. These approaches have been carefully evaluated as modes of delivery for ERT. The current ongoing clinical trials using Metazym, (Shire Human Genetics Therapeutics, Inc., Lexington, MA, USA) administered intrathecally as an ERT agent, are listed in . The final results of these clinical studies will be important to determine the efficacy of this mode of ERT delivery for MLD and other lysosomal diseases.
Table 1 Current therapeutic modalities in metachromatic leukodystrophy (MLD)
Hematopoietic stem cell therapy
As described earlier, the BBB prevents the delivery of ASA enzyme to the affected neural cells. This sets the scene for the development of various strategies that focus on overcoming the BBB and making the enzyme available to CNS cells. HSCT is one such approach, where hematopoietic stem cells (HSCs) cross the BBB and differentiate to microglial cells.Citation94 In principle, the donor microglial cells are able to secrete the wild-type ASA enzyme, which is uptaken by the recipient neural cells that are ASA-deficient. Uptake of the secreted wild-type ASA occurs through the mannose-6-phosphate receptor system, which brings the enzyme directly to the lysosomes, where sulfatide is accumulated in the recipient ASA-deficient neural cells.Citation95 One of the limitations of HSCT is the rate of repopulation of microglia into the recipient CNS, which should be a sufficient cell number to provide an adequate amount of wild-type ASA for the affected neural cells of the recipient. HSCT was shown to be efficacious in the treatment of MLD as early as 1990.Citation84 However, HSCT failed to show efficacy in the late infantile clinical form of MLD.Citation96 Though HSCT is a well-established therapeutic approach for a few LSDs, including mucopolysaccharidosis type I and globoid-cell leukodystrophy (Krabbe disease), this treatment modality is limited only to MLD patients who are diagnosed at earlier stages of the disease course ().Citation97–Citation99 HSCT brings other potential factors to be considered, such as identification of matched related donors, the rate of repopulation of microglial cells, along with specific transplant-related complications including acute and chronic graft-versus host diseases, and the level of engraftment.Citation100,Citation101 An alternative approach to HSCT is using umbilical cord blood as a source of hematopoietic and mesenchymal stem cells, as reviewed elsewhere.Citation102 Recently, the outcomes of 27 patients with different clinical forms of MLD were reported.Citation85 In this study, an inverse relationship between time of intervention and outcome was observed. In addition, children with the juvenile form of MLD showed better outcomes than those with late infantile onset.Citation85
Gene therapy
Types of gene therapies have been studied for the treatment of MLD. Autologous HSCs obtained from the affected MLD patient can be genetically modified to overexpress the ARSA gene using retrovirus gene transfer.Citation103 This approach has been shown to be highly successful in the arsa −/− mice model, as it prevents the development of motor conduction impairment, learning and coordination deficits, and neuropathological abnormalities typical of the disease.Citation104 Thus, genetically modified HSCs can help prevent neurodegeneration or slow its rate of progression. The synthesized wild-type ASA secreted by these genetically modified HSCs is uptaken by the adjacent ASA-deficient cells via the mannose-6-phosphate receptor system.Citation95 The overexpression of ARSA gene by the autologous HSCs before transplant is performed using retro- or lentivirus vectors.Citation105,Citation106 Different studies using this strategy have been discussed elsewhere.Citation103 The advantage of this therapeutic approach is the reduction in risk of graft-versus host disease. The major limitation of this approach is safety consideration, because of unknown risk of mutagenesis of cancer-related genes or oncogenes.Citation107 Currently, a phase I/II clinical study is ongoing with MLD patients receiving autologous HSCT with genetically modified HSCs ().
Specific cell therapy
In addition to HSCT, various other cell-based approaches can be used to deliver ASA to affected cells.Citation102 Briefly, microencapsulated recombinant cells that overexpress the ARSA gene can be directly delivered. These cells offer substantial flexibility in terms of the dosage and control of delivery, since they can be easily retracted.Citation108,Citation109 Similarly, oligodendrocyte progenitor cellsCitation110 and neural progenitor cellsCitation111,Citation112 are other cell types that can be used for cell-based therapy. Embryonic stem cells constitute another cell source for ARSA overexpression and delivery. Studies in the arsa −/− mice, using embryonic stem cells, have demonstrated significant decreases in sulfatide accumulation.Citation113 These specific cell therapies are attractive options for the treatment of neurological manifestations of MLD. All these studies have demonstrated considerable clearance of the storage of sulfatides in animal models.Citation111–Citation113 However, their efficacy and (of utmost importance) safety and tolerability are still to be demonstrated in humans.
Potential novel therapies
Small molecule-based therapies
In MLD, no specific treatment has been shown to completely arrest disease progression. Therapies using small molecules (in general, <500 D) have hardly been explored in MLD. Small molecule-based therapies have shown encouraging results in the case of some lysosomal disorders like Gaucher disease and NPC. N-butyldeoxynojirimycin (NB-DNJ) miglustat (Zavesca®, Actelion Pharmaceuticals Ltd, Allschwil, Switzerland) was showed to reduce the biosynthesis of glucosylceramide by competitively inhibiting the UDP-glucose-N-acysphingosine D-glucosyltransferase (EC 2.4.1.80) or ceramide-specific glucosyltransferase.Citation114 The mechanism of action of miglustat was shown using an in cellulo model of Gaucher disease.Citation115 In studies using cultured human macrophages pre-exposed with conduritol-β-epoxide (CBE), miglustat was shown to reduce glucosylceramide storage, which is the main substrate accumulated in Gaucher disease.Citation115 CBE is an irreversible inhibitor of glucocerobrosidase, leading to accumulation of glucosylceramide in cultured cells.Citation116,Citation117 Ceramide-specific glucosyltransferase is the key enzyme on the biosynthetic pathway of several glycosphingolipids, including the ganglio- series (acidic glycosphingolipids), lacto(neo)- series (neutral glycosphingolipids), and globo-series ().Citation118 Based on this principle, miglustat was tested in the murine model for Tay-Sachs disease (hexa −/−), and was showed to reduce the accumulation of GM2 ganglioside.Citation119 However, the hexa −/− mouse lacks an overt Tay-Sachs disease phenotype, as mice can partially bypass the blocked catabolic pathway and escape disease.Citation119 In an early clinical trial with patients with the juvenile clinical form of GM2 gangliosidosis, miglustat showed adequate pharmacokinetics and safety profiles,Citation120 but failed to arrest some aspects of disease progression.Citation121 However, patients affected by NPC, a lysosomal disorder resulting in intra-cellular lipid trafficking defect and secondary glycosphingolipid storage, showed improvement and stabilization of a number of clinical endpoints, including swallowing capacity and ambulatory index, when receiving miglustat in a clinical trial.Citation122
Small molecule-based therapy can potentially overcome limitations of the current therapies for MLD and may also tackle, at multiple levels, different pathogenic and molecular mechanisms involved in MLD. Small molecules can easily cross the BBB, which is a major hindrance in the ERT strategy. Warfarin (coumadin) is a classical anti-coagulant that functions by competitively inhibiting the multi-unit vitamin K epoxide reductase complex, resulting in impairment of the activation of vitamin K-dependent coagulation factors II, VII, IX, and X. Based on microorganism studies in vitamin K, warfarin administration in mice showed to reduce the levels of sulfatides by 33%, along with cerebrosides and sphingomyelin in the brain.Citation123 Warfarin was then hypothesized as a potential substrate-reducing agent to be explored therapeutically for MLD. Recently, a clinical trial in a small cohort of MLD patients tested whether warfarin would control the progression of MLD.Citation124 During a 45-day treatment with warfarin at doses of 0.2 mg/kg/day, adjusted to maintain INR within the range 2–2.5, no beneficial effect of this small molecule was observed in four enrolled patients with the late infantile form of MLD ().Citation124 The urinary sulfatide levels of these patients remained elevated. Brain magnetic resonance spectroscopy showed that the levels of N-acetyl-aspartate and myo-Inositol remained unchanged after the treatment period.Citation124
Small molecules as enzyme enhancement agents
Lysosomal storage disorders can be caused by missense mutations in specific genes that encode misfolded mutant lysosomal enzymes (). Most of these misfolded mutant lysosomal enzymes are earlier degraded in the ERAD pathwaysCitation125 and, consequently, a very small amount of the mutant enzyme reaches the lysosomal compartment and is functionally active. The resultant residual enzymatic activity, in the case of ASA mutants, ranges between 0%–10% of the normal activity detected from the wild-type ASA.Citation126 This ASA enzymatic activity, above which the activity is sufficient to degrade the sulfatides and prevent their accumulation, is known as the critical threshold – which is ~10% of that measured from the wild-type ASA.Citation126 Interestingly, the levels of ASA enzymatic activity of the pseudodeficiency ARSA alleles are just above the critical threshold. Therefore, in ASA pseudodeficiency, the levels of enzyme activity between 10%–15% of wild-type ASA levels result in no sulfatide accumulation, and consequently no disease-related symptoms of MLD.Citation126
Figure 2 Schematic representation of the mechanism of action of enzyme enhancement agents including pharmacological chaperones (PCs) and proteostasis regulators (PRs).
Abbreviations: ASA, arylsulfatase A; ER, endoplasmic reticulum; ERAD, endoplasmic reticulum associate degradation; PC, pharmacological chaperones; PR, proteostasis regulators.
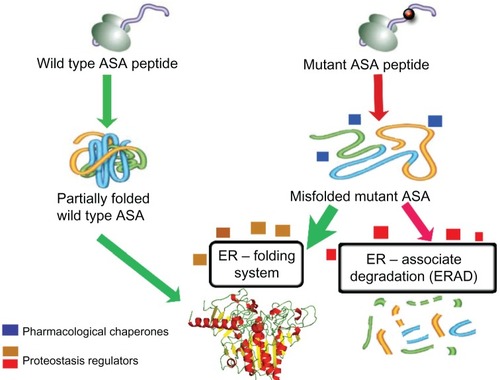
Specific small molecules are able to rescue misfolded proteins. These molecules are able to physically interact with a specific misfolded protein, assist its folding process, and prevent it from being targeted to the ERAD pathway and ultimately degraded by the ubiquitin-proteasomal system ().Citation127 Thus, by evading the ERAD pathway, and subsequently enhancing the level of the available mutant ASA to the lysosomal compartment, the residual ASA enzymatic activity will be increased and potentially overcome the 10% critical threshold.Citation126 In this regard, the ER folding system, with its numerous ER-resident chaperones, plays an important physiological role in promoting adequate and functional folding of several lysosomal enzymes, including ASA. The ER-resident chaperones are proteins that assist the folding or unfolding of nascent proteins synthesized in the ER that are not able to do so spontaneously.Citation127 Similarly, pharmacological chaperones (PCs) are small molecules that function like chaperones, enhancing the levels of the misfolded-prone mutant enzymes by assisting their ER folding and preventing their interaction with the ERAD pathway and subsequent degradation.Citation128–Citation130 Small molecules functioning as PCs were identified for misfolded mutant hexosaminidase A, the lysosomal enzyme deficient in GM2 gangliosidosis,Citation128 and for misfolded mutant glucocerebrosidase (Glc-ase), the lysosomal enzyme deficient in Gaucher disease.Citation129,Citation131 In addition to PCs, proteostasis regulators (PRs) are small molecules that improve the protein folding capacity of cells, resulting in the enhancement of folded mutant proteins.Citation132 PRs work by interacting with the ER folding components and eventually activating physiological cellular response mechanisms, including unfolded protein response and heat shock proteins (). PRs can also function by inhibiting the degradation of the misfolded mutant enzyme, by interacting with the ERAD components or HSC molecules ().Citation133–Citation135 The effect of PRs can be enhanced by simultaneously using PCs for assisting the folding of misfolded-prone mutant enzymes.Citation127 Interestingly, these two classes of small molecules can work together synergistically to guide the misfolded mutant enzyme to a folded state (assisted by PRs) and maintain its structural stability (assisted by PCs).Citation136 Some PRs have been identified for specific lysosomal mutant enzymes.Citation136,Citation137 Inhibitors of histone deacetylase (HDAC) are showed to reduce the acetylation of heat shock protein 90 (Hsp90)Citation138. Since Hsp90 plays an important role in the degradation pathway of the misfolded N370S and L444P Glc-ase mutants, HCAD inhibitors resulted in increased levels of mutant Glc-ase enzymatic activity.Citation138 Thus, these inhibitors act as PRs by preventing the degradation of mutant enzymes while interacting with components of the degradation pathway. MG-132 and celastrol are other examples of small molecules that were shown to function as PRs for some mutant Glc-ase and Hex A enzymes.Citation136 PRs can be potentially applied to a range of LSDs, since these small molecules target the cellular enzyme folding pathway and their effect can be enhanced using PCs. These specific small molecule-based therapies can work in conjunction with ERT by interacting with and, consequently, stabilizing the administered recombinant ASA, extending its half-life in either extra or intracellular compartments.
How can these therapeutic small molecules be identified? Small molecules that work as PCs or PRs for specific lysosomal mutant enzymes can be identified using high throughput screening (HTS) assays. Recently, the use of cell-based assays in the HTS of chemical compound collections has become increasingly common, as targets are presented in a more biologically relevant context than classical in vitro biochemical-based HTS assays.Citation139 In cell-based HTS assays, the identification of small molecules that penetrate cellular membranes and reach specific cellular organelles, along with their cytotoxicity information, can already be obtained at the primary screening stage.Citation140 However, in most cellular HTS assays, the cells utilized are usually not disease relevant. Thus, using patient-derived cell lines for HTS campaigns brings considerable advantages, compared to the traditional, single-target biochemical assays ().Citation141,Citation142 Cellular assays utilizing cells from affected patients provide a unique opportunity to assess a target protein and/or pathway in the context of potentially disrupted biochemical and/or signaling pathways secondary to the disease process. In addition, this pathogenic in cellulo setting permits the evaluation of multiple intervention points, which are potentially altered in the disease, as opposed to the commonly-used cell expression systems of a specific protein target, or a predefined step of a purified or recombinant protein-based assay (). Therefore, cell-based HTS can identify both PCs and PRs.Citation141,Citation142 A cell-based HTS assay was developed to identify small molecules for mutant ASAs.Citation141 Using a transformed fibroblast cell line from a MLD patient with measurable residual ASA activity, a fluorescence quench absorbance ASA assay was developed. This HTS assay showed robust signals in diverse multi-well plates, including 1,536-well plates.Citation141 The implementation of the cell-based HTS assay against larger and diverse small molecule libraries should allow the identification of novel potential drugs for MLD. This and other cell-based HTS assays open a novel opportunity to identify numerous small molecules, as several potential therapeutic targets are accessible in this disease-cellular environment.Citation141,Citation142
Figure 3 Cell-based high-throughput screening (HTS) assay.
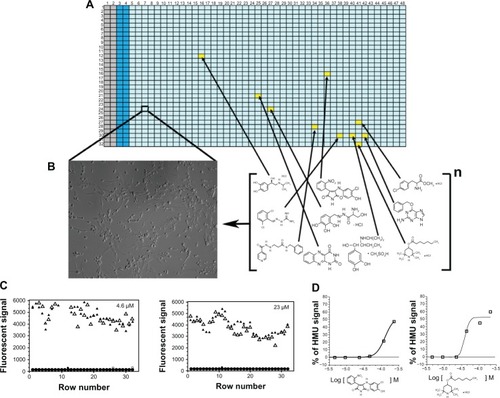
Small molecules as substrate reduction agents
Some small molecules can also have their point of action upstream to the biochemical defect: ie, sulfatide accumulation in MLD (). Miglustat is an example of a substrate reduction therapy (SRT) agent that functions as a competitive inhibitor of ceramide-specific transferase.Citation118 Similarly, the sulfatide synthesis pathway can be potentially targeted for substrate (sulfatide) reduction at multiple points in the pathway, as seen in . Specifically, ceramide synthase can be targeted using small molecules, which would lead to reduction of the biosynthesis of sulfatides (). Reducing ceramide synthase catalytic activity can result in the reduction of further accumulation of sulfatides. However, complete arrest of sulfatide biosynthesis may cause disruption of myelin stability, as shown in different murine models described above.Citation48,Citation51,Citation54 Thus, physiological levels of sulfatides are crucial, as these are main glycosphingolipids of the myelin sheath. Hence, the optimality of therapeutic dose of potential small molecules functioning as SRT agents is a prerequisite for the success of this therapeutic strategy. Therefore, SRT agent doses should be determined by the molecule concentrations capable of partially inhibiting the catalytic activity of a target enzyme in the biosynthetic pathway of sulfatides at intracellular levels (ie, in oligodendrocytes and Schwann cells) (). This will be crucial to avoid potential adverse events of SRT agents in the treatment of MLD.
Small molecules to control pathogenic cascades
Small molecules can also be used to manipulate downstream pathways that are initiated or disturbed secondary to the primary lysosomal enzymatic defect. Diverse pathways have been described to be impaired in lysosomal storage diseases. These are known as pathogenic cascades, which are characterized by disturbances in molecular signaling pathways occurring secondary to sulfatide accumulation, ultimately leading to apoptosis.
In a few lysosomal disorders, some small molecules have been identified to target specific pathogenic cascades. In studies of NPC patient fibroblasts and Npc1 −/− mice, sphingosine storage was shown to precede low calcium levels in the lysosomal compartment, which explains the profound block in late endosome to lysosome transport, resulting in the storage of cholesterol and glycosphingolipids – the cellular hallmark of this lysosomal disorder.Citation70 In the presence of curcumin, a small molecule that is a weak antagonist of the sarco (endo) plasmic reticulum ATPase (SERCA), the levels of calcium in the acidic compartments were normalized and clearance of lipid storage was noted, resulting in increased Npc1 −/− life expectancy.Citation70 SERCA is a pump that transports calcium ions from the cytoplasm into the sarco(endo)plasmic reticulum.Citation143 The inflammatory cascades can be targeted using non-steroidal and anti-inflammatory drugs, which have resulted in clinical benefits in other lysosomal diseases, including Sandhoff diseaseCitation144 and NPCCitation145. Most of these small molecules have already been characterized to interact with specific molecular targets. To discover further small molecules, cell-based HTS assays can be developed using as the readout signal a specific relevant target of a pathogenic cascade related to ASA deficiency. These HTS assays will identify potential therapeutic small molecules to tackle signaling pathways that are disturbed secondary to the ASA deficiency. These small molecules may produce synergistic effects along with the therapeutic modalities that tackle primarily the ASA deficiency, such as ERT, gene therapy, and cell-based therapies. By using combinations of therapies to specific targets, it is more likely that the arrest or even reversal of disease progression can be achieved.
Conclusion
Metachromatic leukodystrophy is a lysosomal disorder causing progressive neurological disability secondary to the accumulation of sulfatides and the resultant demyelination process that affects both CNS and PNS. The range of age of onset of symptoms and rate of disease progression are broad. The available therapeutic options are limited. In developing novel therapeutic approaches for MLD, BBB penetration, lack of a better understanding of the disease pathogenesis, uncertainty regarding the ideal time for intervention in the disease course, and safety issues related to specific therapies are the main challenges. Diverse therapeutic strategies have been developed and are now moving to clinical trials including cell-based therapies, gene therapy, ERT, and small molecule-based therapies. Most of these therapies tackle specific pathogenic aspects of the disease. Some of these strategies may result in better outcomes at specific stages of the natural disease course. These diverse strategies complement each other, either when applied in combination or in a stepwise manner. The determination of the more appropriate combinations and times of administration of these therapies in the disease course will certainly produce the best outcomes for patients afflicted with this devastating inherited neurological disorder.
Acknowledgments
We are grateful for our collaborators including Marc Ferrer PhD and his group at the NIH-Chemical Genomic Center (NCGC) for the instrumental assistance in the development of high-throughput screening cell-based assays targeting different lysosomal enzymes. We are also thankful for our collaborators who donated cell lines including Dr Hirokazu Furuya MD, PhD, neurologist from NHO Omuta Hospital, Omuta, Fukuoka, Japan.
Disclosure
Shilpa Patil is partially supported by National MPS Society. Gustavo Maegawa is currently supported by grants NIH-NIMH 1R03MH098689 and NIH-NINDS 1R01NS079655. Dr Maegawa also receives research grants from the National MPS Society and received research grants from the National Tay-Sachs Disease and Allied Diseases Association. Dr Maegawa receives research funds for clinical and educational studies from Genzyme Therapeutics Ltd, Biomarin Pharmaceutical Inc, Protalix Biotherapeutics Inc, and Shire HGT. The authors declare no other conflicts of interest in this work.
References
- Austin J McAfee D Armstrong D O’Rourke M Shearer L Bachhawat B Low sulfatase activities in metachromatic leukodystrophy (MLD). A controlled study of enzymes in 9 living and 4 autopsied patients with MLD Trans Am Neurol Assoc 1964 89 147 150 5828490
- Bosio A Binczek E Stoffel W Functional breakdown of the lipid bilayer of the myelin membrane in central and peripheral nervous system by disrupted galactocerebroside synthesis Proc Natl Acad Sci U S A 1996 12 93 23 13280 13285 8917582
- Aggarwal S Yurlova L Simons M Central nervous system myelin: structure, synthesis and assembly Trends Cell Biol 2011 21 10 585 593 21763137
- Faust PL Kaye EM Powers JM Myelin lesions associated with lysosomal and peroxisomal disorders Expert Rev Neurother 2010 10 9 1449 1466 20819015
- Gieselmann V Metachromatic leukodystrophy: genetics, pathogenesis and therapeutic options Acta Paediatr Suppl 2008 97 457 15 21 18339182
- Mechtler TP Stary S Metz TF Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria Lancet 2012 379 9813 335 341 22133539
- Meikle PJ Hopwood JJ Clague AE Carey WF Prevalence of lysosomal storage disorders. JAMA Jan 1999 281 3 249 254
- Von Figura K Metachromatic Leukodystrophy Scriver C Beaudet A Arthur L Sly WS Valle D Metabolic and Molecular Basis of Inherited Disease New York McGraw-Hill 2001 3 3696 3724
- Lugowska A Poninska J Krajewski P Broda G Ploski R Population carrier rates of pathogenic ARSA gene mutations: is metachromatic leukodystrophy underdiagnosed? PLoS One 2011 6 6 e20218 21695197
- Stenson PD Ball EV Howells K Phillips AD Mort M Cooper DN The Human Gene Mutation Database: providing a comprehensive central mutation database for molecular diagnostics and personalized genomics Hum Genomics 2009 4 2 69 72 20038494
- Gieselmann V An assay for the rapid detection of the arylsulfatase A pseudodeficiency allele facilitates diagnosis and genetic counseling for metachromatic leukodystrophy Hum Genet 1991 86 3 251 255 1671769
- Gieselmann V Polten A Kreysing J von Figura K Molecular genetics of metachromatic leukodystrophy J Inherit Metab Dis 1994 17 4 500 509 7967499
- Gort L Coll MJ Chabas A Identification of 12 novel mutations and two new polymorphisms in the arylsulfatase A gene: haplotype and genotype-phenotype correlation studies in Spanish metachromatic leukodystrophy patients Hum Mutat 1999 14 3 240 248 10477432
- Francis GS Bonni A Shen N Metachromatic leukodystrophy: multiple nonfunctional and pseudodeficiency alleles in a pedigree: problems with diagnosis and counseling Ann Neurol 1993 34 2 212 218 8101704
- Hohenschutz C Eich P Friedl W Waheed A Conzelmann E Propping P Pseudodeficiency of arylsulfatase A: a common genetic polymorphism with possible disease implications Hum Genet 1989 82 1 45 48 2565866
- Nelson PV Carey WF Morris CP Population frequency of the arylsulphatase A pseudo-deficiency allele Hum Genet 199 87 1 87 88
- Ott R Waye JS Chang PL Evolutionary origins of two tightly linked mutations in arylsulfatase-A pseudodeficiency Hum Genet 1997 101 2 135 140 9402957
- Sommerlade HJ Hille-Rehfeld A von Figura K Gieselmann V Four monoclonal antibodies inhibit the recognition of arylsulphatase A by the lysosomal enzyme phosphotransferase Biochem J 1994 297 Pt 1 123 130 7506530
- Fluharty AL Fluharty CB Bohne W von Figura K Gieselmann V Two new arylsulfatase A (ARSA) mutations in a juvenile metachromatic leukodystrophy (MLD) patient Am J Hum Genet 1991 49 6 1340 1350 1684088
- Harvey JS Nelson PV Carey WF Robertson EF Morris CP An arylsulfatase A (ARSA) missense mutation (T274M) causing late-infantile metachromatic leukodystrophy Hum Mutat 1993 2 4 261 267 8104633
- Ghosh P Dahms NM Kornfeld S Mannose 6-phosphate receptors: new twists in the tale Nat Rev Mol Cell Biol 2003 4 3 202 212 12612639
- Lukatela G Krauss N Theis K Crystal structure of human arylsulfatase A: the aldehyde function and the metal ion at the active site suggest a novel mechanism for sulfate ester hydrolysis Biochemistry 1998 37 11 3654 3664 9521684
- Von Bulow R Schmidt B Dierks T Defective oligomerization of arylsulfatase a as a cause of its instability in lysosomes and metachromatic leukodystrophy J Biol Chem 2002 277 11 9455 9461 11777924
- von Bulow R Schmidt B Dierks T von Figura K Uson I Crystal structure of an enzyme-substrate complex provides insight into the interaction between human arylsulfatase A and its substrates during catalysis J Mol Biol 2001 305 2 269 277 11124905
- Siegel EG Lucke H Schauer W Creutzfeldt W Repeated upper gastrointestinal hemorrhage caused by metachromatic leukodystrophy of the gall bladder Digestion 1992 51 2 121 124 1499874
- Ries M Deeg KH Polyposis of the gallbladder associated with metachromatic leukodystrophy Eur J Pediatr 1993 152 5 450 451 8319717
- Kim TS Kim IO Kim WS Choi YS Yeon KM Hwang YS Involvement of the gallbladder in childhood metachromatic leukodystrophy: ultrasonographic findings J Ultrasound Med 1996 15 12 821 825 8947856
- Oak S Rao S Karmarkar S Papillomatosis of the gallbladder in metachromatic leukodystrophy Pediatr Surg Int 1997 12 5–6 424 425 9244117
- Simanovsky N Ackerman Z Kiderman A Fields S Unusual gallbladder findings in two brothers with metachromatic leukodystrophy Pediatr Radiol 1998 28 9 706 708 9732500
- Krivan HC Olson LD Barile MF Ginsburg V Roberts DD Adhesion of Mycoplasma pneumoniae to sulfated glycolipids and inhibition by dextran sulfate J Biol Chem 1989 264 16 9283 9288 2470753
- Sugano K Tai T Kawashima I Localization of sulfatides in the epithelial lining of gastric mucosa: studies with a monoclonal antibody to sulfatides J Clin Gastroenterol 1995 21 Suppl 1 S98 S103 8774999
- Takamatsu K Phytosphingosine-containing neutral glycosphingolipids and sulfatides in the human female genital tract: their association in the cervical epithelium and the uterine endometrium and their dissociation in the mucosa of fallopian tube with the menstrual cycle Keio J Med 1992 41 3 161 167 1434311
- Malone MJ Stoffyn P A comparative study of brain and kidney glycolipids in metachromatic leucodystrophy J Neurochem 1966 13 11 1037 1045 5924656
- Toda K Kobayashi T Goto I Kurokawa T Ogomori K Accumulation of lysosulfatide (sulfogalactosylsphingosine) in tissues of a boy with metachromatic leukodystrophy Biochem Biophys Res Commun 1989 15 159 2 605 611 2539117
- Toda K Kobayashi T Goto I Lysosulfatide (sulfogalactosylsphingosine) accumulation in tissues from patients with metachromatic leukodystrophy J Neurochem 1990 55 5 1585 1591 1976756
- Natowicz MR Prence EM Chaturvedi P Newburg DS Urine sulfatides and the diagnosis of metachromatic leukodystrophy Clin Chem 1996 2 2 232 238 8595716
- Honke K Zhang Y Cheng X Kotani N Taniguchi N Biological roles of sulfoglycolipids and pathophysiology of their deficiency Glycoconj J 2004 21 1–2 59 62 15467400
- Vitner EB Platt FM Futerman AH Common and uncommon pathogenic cascades in lysosomal storage diseases J Biol Chem 2010 285 27 20423 20427 20430897
- Webster HD Schwann cell alterations in metachromatic leukodystrophy: preliminary phase and electron microscopic observations J Neuropathol Exp Neurol 1962 21 534 554 13999412
- Peng L Suzuki K Ultrastructural study of neurons in metachromatic leukodystrophy Clin Neuropathol 1987 6 5 224 230 2826059
- Berger J Loschl B Bernheimer H Occurrence, distribution, and phenotype of arylsulfatase A mutations in patients with metachromatic leukodystrophy Am J Med Genet 1997 69 3 335 340 9096767
- Polten A Fluharty AL Fluharty CB Kappler J von Figura K Gieselmann V Molecular basis of different forms of metachromatic leukodystrophy N Engl J Med 1991 24 1 18 22 1670590
- Gieselmann V Krageloh-Mann I Metachromatic leukodystrophy – an update Neuropediatrics 2010 41 1 1 6 20571983
- Poeppel P Habetha M Marcao A Bussow H Berna L Gieselmann V Missense mutations as a cause of metachromatic leukodystrophy. Degradation of arylsulfatase A in the endoplasmic reticulum FEBS J 2005 272 5 1179 1188 15720392
- Poeppel P Abouzied MM Volker C Gieselmann V Misfolded endoplasmic reticulum retained subunits cause degradation of wild-type subunits of arylsulfatase A heteromers FEBS J 2010 277 16 3404 3414 20646068
- Hagberg B Clinical aspects of globoid cell and metachromatic leukodystrophies. Birth Defects Orig Art Ser Feb 1971 7 1 103 112
- Hess MW Kirschning E Pfaller K Debbage PL Hohenberg H Klima G 5000-year-old myelin: uniquely intact in molecular configuration and fine structure Curr Biol 1998 8 15 R512 R513 9705943
- Zoller I Meixner M Hartmann D Absence of 2-hydroxylated sphingolipids is compatible with normal neural development but causes late-onset axon and myelin sheath degeneration J Neurosci 2008 24 28 39 9741 9754 18815260
- O’Brien JS Sampson EL Myelin Membrane: a molecular abnormality Science 1965 150 3703 1613 1614 5866661
- Tanaka K Nagara H Kobayashi T Goto I The twitcher mouse: accumulation of galactosylsphingosine and pathology of the central nervous system Brain Res 1989 482 2 347 350 2706492
- Coetzee T Li X Fujita N Molecular cloning, chromosomal mapping, and characterization of the mouse UDP-galactose:ceramide galactosyltransferase gene Genomics 1996 35 1 215 222 8661123
- Coetzee T Fujita N Dupree J Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability Cell 1996 86 2 209 219 8706126
- Coetzee T Suzuki K Popko B New perspectives on the function of myelin galactolipids Trends Neurosci 1998 21 3 126 130 9530920
- Meixner M Jungnickel J Grothe C Gieselmann V Eckhardt M Myelination in the absence of UDP-galactose:ceramide galactosyltransferase and fatty acid 2 -hydroxylase BMC Neurosci 2011 12 22 21366909
- Potter KA Kern MJ Fullbright G Central nervous system dysfunction in a mouse model of FA2H deficiency Glia 2011 59 7 1009 1021 21491498
- Imgrund S Hartmann D Farwanah H Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas J Biol Chem 2009 284 48 33549 33560 19801672
- Pewzner-Jung Y Park H Laviad EL A critical role for ceramide synthase 2 in liver homeostasis: I. alterations in lipid metabolic pathways The Journal of biological chemistry 2010 285 14 10902 10910 20110363
- von Figura K Metachromatic Leukodystrophy Scriver C Beaudet A Arthur L Sly WS Valle D Metabolic and Molecular Basis of Inherited Disease New York McGrw-Hill 2001 3 3696 3724
- Hess B Saftig P Hartmann D Phenotype of arylsulfatase A-deficient mice: relationship to human metachromatic leukodystrophy Proc Natl Acad Sci U S A 1996 93 25 14821 14826 8962139
- Eckhardt M Hedayati KK Pitsch J Sulfatide storage in neurons causes hyperexcitability and axonal degeneration in a mouse model of metachromatic leukodystrophy J Neurosci 2007 27 34 9009 9021 17715338
- Walkley SU Pathogenic mechanisms in lysosomal disease: a reappraisal of the role of the lysosome Acta Paediatr Suppl 2007 96 455 26 32 17391436
- Walkley SU Pathogenic cascades in lysosomal disease-Why so complex? J Inherit Metab Dis 2009 32 2 181 189 19130290
- Paez PM Fulton D Colwell CS Campagnoni AT Voltage-operated Ca(2+) and Na(+) channels in the oligodendrocyte lineage J Neurosci Res 2009 87 15 3259 3266 19021296
- Paez PM Fulton DJ Spreuer V Golli myelin basic proteins regulate oligodendroglial progenitor cell migration through voltagegated Ca2+ influx J Neurosci 2009 29 20 6663 6676 19458236
- Pelled D Trajkovic-Bodennec S Lloyd-Evans E Sidransky E Schiffmann R Futerman AH Enhanced calcium release in the acute neuronopathic form of Gaucher disease Neurobiol Dis 2005 18 1 83 88 15649698
- Ausseil J Desmaris N Bigou S Early neurodegeneration progresses independently of microglial activation by heparan sulfate in the brain of mucopolysaccharidosis IIIB mice PLoS One 2008 3 5 e2296 18509511
- Shimada Y Kobayashi H Kawagoe S Endoplasmic reticulum stress induces autophagy through activation of p38 MAPK in fibroblasts from Pompe disease patients carrying c.546G>T mutation Mol Genet Metab 2011 104 4 566 573 21982629
- Warr MR Binnewies M Flach J FOXO3A directs a protective autophagy program in haematopoietic stem cells Nature 2013 494 7437 323 327 23389440
- Nishiyama Y Shimada Y Yokoi T Akt inactivation induces endoplasmic reticulum stress-independent autophagy in fibroblasts from patients with Pompe disease Mol Genet Metab 2012 107 3 490 495 23041259
- Lloyd-Evans E Morgan AJ He X Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium Nat Med 2008 14 11 1247 1255 18953351
- Chen CS Patterson MC Wheatley CL O’Brien JF Pagano RE Broad screening test for sphingolipid-storage diseases Lancet 1999 354 9182 901 905 10489949
- Mukherjee S Soe TT Maxfield FR Endocytic sorting of lipid analogues differing solely in the chemistry of their hydrophobic tails J Cell Biol 1999 144 6 1271 1284 10087269
- Settembre C Fraldi A Jahreiss L A block of autophagy in lysosomal storage disorders Hum Mol Genet 2008 17 1 119 129 17913701
- Settembre C Arteaga-Solis E McKee MD Proteoglycan desulfation determines the efficiency of chondrocyte autophagy and the extent of FGF signaling during endochondral ossification Genes Dev 2008 22 19 2645 2650 18832069
- Settembre C Annunziata I Spampanato C Systemic inflammation and neurodegeneration in a mouse model of multiple sulfatase deficiency Proc Natl Acad Sci U S A 2007 104 11 4506 4511 17360554
- Dierks T Schmidt B Borissenko LV Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme Cell 2003 113 4 435 444 12757705
- Lo EH Degeneration and repair in central nervous system disease Nat Med 2010 16 11 1205 1209 21052074
- Pardridge WM The blood-brain barrier: bottleneck in brain drug development NeuroRx 2005 2 1 3 14 15717053
- Ghose AK Viswanadhan VN Wendoloski JJ A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases J Comb Chem 1999 1 1 55 68 10746014
- Priller J Flugel A Wehner T Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment Nat Med 2001 7 12 1356 1361 11726978
- Mehl E Jatzkewitz H A cerebrosidesulfatase from swine kidney Hoppe Seylers Z Physiol Chem 1964 339 1 260 276 5829234
- Saravanan K Schaeren-Wiemers N Klein D Specific downregulation and mistargeting of the lipid raft-associated protein MAL in a glycolipid storage disorder Neurobiol Dis 2004 16 2 396 406 15193296
- Wittke D Hartmann D Gieselmann V Lullmann-Rauch R Lysosomal sulfatide storage in the brain of arylsulfatase A-deficient mice: cellular alterations and topographic distribution Acta Neuropathol 2004 108 4 261 271 15322834
- Krivit W Shapiro E Kennedy W Treatment of late infantile metachromatic leukodystrophy by bone marrow transplantation N Engl J Med 1990 322 1 28 32 1967188
- Martin HR Poe MD Provenzale JM Kurtzberg J Mendizabal A Escolar ML Neurodevelopmental outcomes of umbilical cord blood transplantation in metachromatic leukodystrophy Biol Blood Marrow Transplant 2013 19 4 616 624 23348427
- Malm G Ringden O Winiarski J Clinical outcome in four children with metachromatic leukodystrophy treated by bone marrow transplantation Bone Marrow Transplant 1996 17 6 1003 1008 8807106
- Nagai T Kaneko T Shichijou H Karato T Maruyama A Tsuchiya Y Bone marrow transplantation for late infantile metachromatic leukodystrophy: pathogenic investigation for graft rejection Acta Paediatr Jpn 1993 35 5 404 408 8256624
- Navarro C Dominguez C Fernandez JM Fachal C Alvarez M Case report: four-year follow-up of bone-marrow transplantation in late juvenile metachromatic leukodystrophy J Inherit Metab Dis 1995 18 2 157 158 7564234
- Solders G Celsing G Hagenfeldt L Ljungman P Isberg B Ringden O Improved peripheral nerve conduction, EEG and verbal IQ after bone marrow transplantation for adult metachromatic leukodystrophy Bone Marrow Transplant 1998 22 11 1119 1122 9877277
- Walkley SU Wurzelmann S Siegel DA Ectopic axon hillockassociated neurite growth is maintained in metabolically reversed swainsonine-induced neuronal storage disease Brain Res 1987 410 1 89 96 3107757
- Matzner U Lullmann-Rauch R Stroobants S Enzyme replacement improves ataxic gait and central nervous system histopathology in a mouse model of metachromatic leukodystrophy Mol Ther 2009 17 4 600 606 19174759
- Matthes F Stroobants S Gerlach D Efficacy of enzyme replacement therapy in an aggravated mouse model of metachromatic leukodystrophy declines with age Hum Mol Genet 2012 21 11 2599 2609 22388935
- Martino S Consiglio A Cavalieri C Expression and purification of a human, soluble Arylsulfatase A for Metachromatic Leukodystrophy enzyme replacement therapy J Biotechnol 2005 117 3 243 251 15862354
- Asheuer M Pflumio F Benhamida S Human CD34+ cells differentiate into microglia and express recombinant therapeutic protein Proc Nat Acad Sci U S A 2004 101 10 3557 3562
- Saftig P Klumperman J Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function Nat Rev Mol Cell Biol 2009 10 9 623 635 19672277
- Bredius RG Laan LA Lankester AC Early marrow transplantation in a pre-symptomatic neonate with late infantile metachromatic leukodystrophy does not halt disease progression Bone Marrow Transplant 2007 39 5 309 310 17237829
- Krivit W Peters C Shapiro EG Bone marrow transplantation as effective treatment of central nervous system disease in globoid cell leukodystrophy, metachromatic leukodystrophy, adrenoleukodystrophy, mannosidosis, fucosidosis, aspartylglucosaminuria, Hurler, Maroteaux-Lamy, and Sly syndromes, and Gaucher disease type III Curr Opin Neurol 1999 12 2 167 176 10226749
- Malatack JJ Consolini DM Bayever E The status of hematopoietic stem cell transplantation in lysosomal storage disease Pediatr Neurol 2003 29 5 391 403 14684234
- Peters C Steward CG Hematopoietic cell transplantation for inherited metabolic diseases: an overview of outcomes and practice guidelines Bone Marrow Transplant 2003 31 4 229 239 12621457
- Biffi A Lucchini G Rovelli A Sessa M Metachromatic leukodystrophy: an overview of current and prospective treatments Bone Marrow Transplant 2008 42 Suppl 2 S2 S6 18978739
- Orchard PJ Tolar J Transplant outcomes in leukodystrophies Semin Hematol 2010 47 1 70 78 20109614
- Batzios S P Zafeiriou DI Developing treatment options for metachromatic leukodystrophy Mol Genet Metab 2012 105 1 56 63 22078456
- Matzner U Gieselmann V Gene therapy of metachromatic leukodystrophy Expert Opin Biol Ther 2005 5 1 55 65 15709909
- Biffi A De Palma M Quattrini A Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells J Clin Invest 2004 113 8 1118 1129 15085191
- Biffi A Capotondo A Fasano S Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice J Clin Invest 2006 116 11 3070 3082 17080200
- Matzner U Hartmann D Lullmann-Rauch R Bone marrow stem cell-based gene transfer in a mouse model for metachromatic leukodystrophy: effects on visceral and nervous system disease manifestations Gene Ther 2002 9 1 53 63 11850723
- Capotondo A Cesani M Pepe S Safety of arylsulfatase A overexpression for gene therapy of metachromatic leukodystrophy Hum Gene Ther 2007 18 9 821 836 17845130
- Consiglio A Martino S Dolcetta D Metabolic correction in oligodendrocytes derived from metachromatic leukodystrophy mouse model by using encapsulated recombinant myoblasts J Neurol Sci 2007 255 1–2 7 16 17341424
- Lagranha VL Baldo G de Carvalho TG In vitro correction of ARSA deficiency in human skin fibroblasts from metachromatic leukodystrophy patients after treatment with microencapsulated recombinant cells Metab Brain Dis 2008 23 4 469 484 18797988
- Givogri MI Galbiati F Fasano S Oligodendroglial progenitor cell therapy limits central neurological deficits in mice with metachromatic leukodystrophy J Neurosci 2006 26 12 3109 3119 16554462
- Kawabata K Migita M Mochizuki H Ex vivo cell-mediated gene therapy for metachromatic leukodystrophy using neurospheres Brain Res 2006 1094 1 13 23 16729983
- Givogri MI Bottai D Zhu HL Multipotential neural precursors transplanted into the metachromatic leukodystrophy brain fail to generate oligodendrocytes but contribute to limit brain dysfunction Dev Neurosci 2008 30 5 340 357 18667806
- Klein D Schmandt T Muth-Kohne E Embryonic stem cell-based reduction of central nervous system sulfatide storage in an animal model of metachromatic leukodystrophy Gene Ther 2006 13 24 1686 1695 16871228
- Zervas M Somers KL Thrall MA Walkley SU Critical role for glycosphingolipids in Niemann-Pick disease type C Curr Biol 200 11 16 1283 1287 11525744
- Platt FM Neises GR Dwek RA Butters TD N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis J Biol Chem 1994 269 11 8362 8365 8132559
- Grabowski GA Osiecki-Newman K Dinur T Human acid betaglucosidase. Use of conduritol B epoxide derivatives to investigate the catalytically active normal and Gaucher disease enzymes J Biol Chem 1986 261 18 8263 8269 3087971
- Adachi M Volk BW Gaucher disease in mice induced by conduritol-B-epoxide: morphologic features Arch Pathol Lab Med 1977 101 5 255 259 576797
- Platt FM Jeyakumar M Substrate reduction therapy Acta Paediatr Suppl 2008 97 457 88 93 18339196
- Platt FM Neises GR Reinkensmeier G Prevention of lysosomal storage in Tay-Sachs mice treated with N-butyldeoxynojirimycin Science 1997 276 5311 428 431 9103204
- Maegawa GH van Giersbergen PL Yang S Pharmacokinetics, safety and tolerability of miglustat in the treatment of pediatric patients with GM2 gangliosidosis Mol Genet Metab 2009 97 4 284 291 19447653
- Maegawa GH Banwell BL Blaser S Substrate reduction therapy in juvenile GM2 gangliosidosis Mol Genet Metab 2009 98 1–2 215 224 19595619
- Patterson MC Vecchio D Prady H Abel L Wraith JE Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study Lancet Neurol 2007 6 9 765 772 17689147
- Sundaram KS Lev M Warfarin administration reduces synthesis of sulfatides and other sphingolipids in mouse brain J Lipid Res 11 1988 29 11 1475 1479 3241123
- Assadi M Wang DJ Anderson K Carran M Bilaniuk L Leone P Vitamin k antagonist warfarin for palliative treatment of metachromatic leukodystrophy, a compassionate study of four subjects J Cent Nerv Syst Dis 2012 4 73 79 23650469
- Bernier V Lagace M Bichet DG Bouvier M Pharmacological chaperones: potential treatment for conformational diseases Trends Endocrinol Metab 2004 15 5 222 228 15223052
- Leinekugel P Michel S Conzelmann E Sandhoff K Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease Hum Genet 1992 88 5 513 523 1348043
- Wiseman RL Powers ET Buxbaum JN Kelly JW Balch WE An adaptable standard for protein export from the endoplasmic reticulum Cell 2007 131 4 809 821 18022373
- Maegawa GH Tropak M Buttner J Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis J Biol Chem 2007 282 12 9150 9161 17237499
- Maegawa GH Tropak MB Buttner JD Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease J Biol Chem 2009 284 35 23502 23516 19578116
- Beck M Emerging drugs for lysosomal storage diseases Expert Opin Emerg Drugs 2010 15 3 495 507 20557271
- Fan JQ A counterintuitive approach to treat enzyme deficiencies: use of enzyme inhibitors for restoring mutant enzyme activity Biol Chem 2008 389 1 1 11 18095864
- Balch WE Morimoto RI Dillin A Kelly JW Adapting proteostasis for disease intervention Science 2008 319 5865 916 919 18276881
- Harbut MB Patel BA Yeung BK Targeting the ERAD pathway via inhibition of signal peptide peptidase for antiparasitic therapeutic design Proc Natl Acad Sci U S A 2012 109 52 21486 21491 23236186
- Elfrink HL Zwart R Baas F Scheper W Inhibition of endoplasmic reticulum associated degradation reduces endoplasmic reticulum stress and alters lysosomal morphology and distribution Mol Cells 2013 35 4 291 297 23515578
- Wang F Segatori L Remodeling the proteostasis network to rescue glucocerebrosidase variants by inhibiting ER-associated degradation and enhancing ER folding PLoS One 2013 8 4 e61418 23620750
- Mu TW Ong DS Wang YJ Chemical and biological approaches synergize to ameliorate protein-folding diseases Cell 2008 134 5 769 781 18775310
- Mu TW Fowler DM Kelly JW Partial restoration of mutant enzyme homeostasis in three distinct lysosomal storage disease cell lines by altering calcium homeostasis PLoS Biol 2008 6 2 e26 18254660
- Yang C Rahimpour S Lu J Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones Proc Natl Acad Sci U S A 2013 110 3 966 971 23277556
- An WF Tolliday N Cell-based assays for high-throughput screening Mol Biotechnol 2010 45 2 180 186 20151227
- Hampton SL Kinnaird AI Genetic interventions in mammalian cells; applications and uses in high-throughput screening and drug discovery Cell Biol Toxicol 2010 26 1 43 55 19904619
- Geng H Whiteley G Ribbens J Novel patient cell-based HTS assay for identification of small molecules for a lysosomal storage disease PLoS One 2011 6 12 e29504 22216298
- Ribbens J Whiteley G Furuya H A high-throughput screening assay using Krabbe disease patient cells Anal Biochem 2012 434 1 15 25 23138179
- Pelled D Lloyd-Evans E Riebeling C Jeyakumar M Platt FM Futerman AH Inhibition of calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase in a mouse model of Sandhoff disease and prevention by treatment with N-butyldeoxynojirimycin J Biol Chem 2003 278 32 29496 29501 12756243
- Jeyakumar M Smith DA Williams IM NSAIDs increase survival in the Sandhoff disease mouse: synergy with N-butyldeoxynojirimycin Ann Neurol 2004 56 5 642 649 15505823
- Smith D Wallom K-L Williams IM Jeyakumar M Platt FM Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1 Neurobiol Dis 2009 36 2 242 251 19632328
- Inglese J Auld DS Jadhav A Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries Proc Natl Acad Sci U S A 2006 103 31 11473 11478 16864780
- National Center for Research Resources (NCRR) Stem cell transplantation (SCT) for genetic diseases Available from http://clinicaltrials.gov/ct2/show/NCT00004378?term=NCT00004378&rank=1. NLM identifier NCT00004378Accessed July 8, 2013
- Masonic Cancer Center, University of Minnesota Stem cell transplant for inborn errors of metabolism Available from http://clinicaltrials.gov/ct2/show/NCT00176904?term=NCT00176904&rank=1. /NLM identifier NCT00176904Accessed July 8, 2013
- Shire Human Genetic Therapies, Inc Long-term Metazym treatment of patients with late infantile metachromatic leukodystrophy (MLD) Available from http://clinicaltrials.gov/ct2/show/NCT00633139?term=NCT00633139&rank=1. NLM identifier NCT00633139Accessed July 8, 2013
- Shire Human Genetic Therapies, Inc Multicenter study of HGT-1110 enzyme replacement therapy administered intrathecally in children with metachromatic leukodystrophy (IDEAMLD) Available from http://clinicaltrials.gov/ct2/show/NCT01510028?term=NCT01510028&rank=1. NLM identifier NCT0150039Accessed July 8, 2013
- Assistance Publique – Hôpitaux de Paris Efficacy of Metazym for the treatment of metachromatic leukodystrophy treated with hematopoietic stem cell transplantation (Azylis) Available at http://clinicaltrials.gov/ct2/show/NCT01303146?term=NCT01303146&rank=1. NLM identifier NCT 01303146Accessed July 8, 2013
- Shire Human Genetic Therapies, Inc Open-label extension study of recombinant human arylsulfatase A (HGT-1111) in late infantile MLD Available at http://clinicaltrials.gov/ct2/show/NCT00681811?term=NCT00681811&rank=1. NLM identifier NCT 00681811Accessed July 22, 2013
- Shire Human Genetic Therapies, Inc Metazym for the treatment of patients with late infantile metachromatic leukodystrophy Available from http://clinicaltrials.gov/ct2/show/NCT00418561?term=NCT00418561&rank=1. NLM identifier NCT00418561Accessed July 8, 2013
- IRCCS San Raffaele Gene therapy for metachromatic leukodystrophy (TIGET-MLD Available from http://clinicaltrials.gov/ct2/show/NCT01560182?term=NCT01560182&rank=1. NLM identifier NCT01560182Accessed July 8, 2013
- Institut National de la Santé Et de la Recherche Médicale, France Intracerebral gene therapy for children with early onset forms of metachromatic leukodystrophy (TG-MLD) Available from http://clinicaltrials.gov/ct2/show/NCT01801709?term=NCT01801709&rank=1. NLM identifier NCT 01801709Accessed July 8, 2013
- The Cooper Health System Effect of warfarin in the treatment of metachromatic leukodystrophy Available at http://clinicaltrials.gov/ct2/show/NCT00683189?term=NCT00683189&rank=1. NLM identifer NCT00683189Accessed July 8, 2013