
Abstract
Purpose
As a novel antidepressant drug, agomelatine has good therapeutic effect on the mood disorder and insomnia in Alzheimer’s disease (AD). Recent studies have shown the neuroprotective function of agomelatine, including anti-oxidative and anti-apoptosis effect. However, it remains unclear whether agomelatine exerts neuroprotection in AD. Thus, the neuroprotective effect of agomelatine against amyloid beta 25–35 (Aβ25–35)-induced toxicity in PC12 cells was evaluated in this study.
Methods
The concentration of malondialdehyde (MDA), LDH, and ROS was investigated to evaluate oxidative damage. The expression of P-tau, tau, PTEN, P-Akt, Akt, P-GSK3β, and GSK3β proteins was assessed by Western blotting. Our results demonstrated that Aβ25–35 significantly increased the content of MDA, LDH, and ROS. Meanwhile, Aβ25–35 upregulated the expression of P-tau and PTEN as well as downregulated P-Akt and P-GSK3β expression. These effects could be blocked by agomelatine pretreatment. Furthermore, luzindole, the melatonin receptor (MT) antagonist, could reverse the neuroprotective effect of agomelatine.
Conclusion
The results demonstrated that antidepressant agomelatine might prevent the tau protein phosphorylation and oxidative damage induced by Aβ25–35 in PC12 cells by activating MT-PTEN/Akt/GSK3β signaling. This study provided a novel therapeutic target for AD in the future.
Introduction
It has been accepted that the extracellular deposition of amyloid beta (Aβ) plaques and the accumulation of intracellular tau neurofibrillary tangles (NFT) are the most important pathophysiology of AD.Citation1 Tau, a microtubule-associated protein, is the main component of the intracellular filamentous inclusions, which is essential for the regulation of microtubule structure and axonal transport by binding to the microtubule. In the pathological state, tau protein hyperphosphorylation has been reported to drive tau aggregation and enhance tau-mediated neurotoxicity,2 leading to spine collapse and dendritic injury,Citation3 and aggravate neurodegeneration, which is involved in several neurodegenerative diseases, including AD and frontotemporal dementia with parkinsonism-17 (FTDP-17).
Oxidative stress is defined as an imbalance between oxidants and antioxidants, resulting in excessive generation of toxic molecules such as ROS.Citation4 When the concentration of reactive species is beyond the control of internal protective mechanisms, oxidative damage occurs to proteins, lipids, and DNA, leading to cytotoxicity.Citation5 The level of malondialdehyde (MDA), a marker of lipid peroxidation index, reflects the extent of lipid peroxidation, which is considered as crucial factor in AD.Citation6 Besides, glycolytic enzyme LDH increases along with plasma membrane damage, which is often used as an indicator of necrotic cell death caused by a plethora of external stress factors. Oxidative damage to neurons and loss of cholinergic neurons in the forebrain region are observed in AD,Citation7 and several investigations have revealed that oxidative stress plays an important role in the pathogenesis of AD.Citation8 Furthermore, several anti-oxidative and anti-tau protein hyperphosphorylation therapeutic strategies show great potential in treating AD.Citation9,Citation10
AD often expresses with multiple comorbidities such as depression. AD and depression share some common etiology, including oxidative stress and nitrosative stress;Citation11 therefore, more and more evidence demonstrated that antidepressant exerted neuroprotective effect in the development of AD.Citation12,Citation13 As a novel antidepressant drug, agomelatine, widely applied in clinic, is a receptor agonist that affects both MT1 and MT2 melatonin receptors and an antagonist that affects 5-hydroxytyriptamine (5HT) 2C receptor. Agomelatine was quite effective not only for insomnia but also for anxiety and depressive symptoms. Recent studies have shown that agomelatine provided neuroprotective effect in multiple disease models, such as ischemic stroke animal modelCitation14 and depression animal model,Citation15 by anti-oxidative injury,Citation15 anti-apoptosis, and by promoting neural restoration.Citation16,Citation17 However, it remains unclear whether agomelatine exerts neuroprotection in AD.
In the present study, the PC12 cell line was used and aimed to explore 1) effect of agomelatine on tau protein phosphorylation and oxidative damage induced by Aβ25–35 and 2) the neuroprotective mechanism of agomelatine. This study aimed to provide new insights in the therapy of AD.
Materials and methods
Materials
Aβ25–35 (#A4559), agomelatine (#A1362), luzindole (#L2407), the primary antibodies against phosphotau (Ser396) (#SAB4504557), tau (#SAB4501830), PTEN (#SAB1406331), GAPDH (#SAB2701826), goat antirab-bit IgG (#A3687), and antibody antimouse IgG (#M8770) were purchased from Sigma-Aldrich Co., St Louis, MO, USA. The primary antibodies against phospho-Akt (Ser473) (#4060) and Akt (#4691) were purchased from Cell Signaling Technology, Danvers, MA, USA. The primary antibodies against phospho-GSK3β (Ser9) (Ab131097) and GSK3β (Ab93926) were purchased from Abcam, Cambridge, UK. Cell counting kit-8 (CCK-8) (#E606335-0500) and ROS assay kit (#50101ES01) were obtained from Sango Biotech (Shanghai, China). Cell MDA assay kit (#A003-4) and LDH assay kit (#A020-2) were purchased from Nanjing Jiancheng Bioengineering Institute (China).
Cell culture
PC12 cells were purchased from Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM basal culture medium (Thermo Fisher Scientific, Waltham, MA, USA) with 10% FBS (HyClone, Logan, UT, USA) and 1% penicillin–streptomycin at 37°C in 5% CO2 incubator. In agomelatine pretreatment group, after agomelatine pretreatment at different concentration for 24 hours, PC12 cells were then exposed to Aβ25–35 for 24 hours. In agomelatine posttreatment group, after Aβ25–35 pretreatment for 24 hours, PC12 cells were then exposed to agomelatine for 24 hours. Besides, in luzindole treatment group, PC12 cells were first treated with agomelatine in the presence of luzindole treatment for 24 hours, then exposed to Aβ25–35 for 24 hours.
Cell viability assay
The cell viability was assessed by CCK-8 assay. PC12 cells were first plated into 96-well plates and incubated in DMEM medium at 37°C for 24 hours. After treatment, CCK-8 assay kit was used to determine cell viability, according to the manufacturer’s instructions. The assays were performed in triplicate.
Measurement of malondialdehyde (MDA) production
To evaluate lipid peroxidation reactions, intracellular MDA generation was measured using the MDA assay kit. The cells were lysed by lysate buffer, and the lysate was used to measure the level of MDA according to the manufacturer’s instructions. The results were expressed as OD values by determining the absorbance at 530 nm. The assays were performed in triplicate.
Lactate dehydrogenase (LDH) measurements
To assess oxidative stress-induced cell injury, LDH activity in the supernatant of PC12 cells was measured by a commercial kit. At the end of the different treatments, the experiment was performed following the manufacturer’s instructions. The total absorbance was measured at 490 nm.
Measurement of ROS production
Dichlorodihydrofluorescein diacetate (DCFH-DA), indicator of ROS, was used to analyze intracellular accumulation of ROS production. After staining with DCFH-DA for 30 minutes at 37°C, the cells were harvested by trypsinization, centrifuged, and suspended in 1 mL PBS buffer, as described in the ROS assay kit. The intracellular accumulation of ROS production was measured by flow cytometry (BD, Franklin Lakes, NJ, USA).
Western blot analysis
After designated treatment, lysates were generated by RIPA lysis buffer, and then Bradford assay was performed to determine total protein concentrations. Subsequently, samples were prepared in sample buffer and heated to 95°C for 5 minutes. Equal amounts of lysates were fractionated using 10% SDS-PAGE and electrotransferred onto nitrocellulose membranes. Gels were run at a constant current (10–15 mA) for 3–4 hours for maximum separation. Wet transfer was performed for 1 hour at constant current (300 mA) using poly-vinylidenedifluoride membrane presoaked in methanol. The membrane was blocked in 5% milk in 0.2% Phosphate Buffered Saline Tween-20. The membrane was then washed in Phosphate Buffered Saline Tween-20 ×3 for 15 minutes each. After overnight incubation at 4°C with the primary antibodies (P-tau 1:1,000, tau 1:1,000, PTEN 1:1,000, P-Akt 1:1,000, Akt 1:1,000, P-GSK3β 1:1,000, GSK3β 1:1,000, GAPDH 1:5,000), the blots were washed and exposed for 1 hour to corresponding horseradish peroxidase-conjugated secondary antibodies. Chemiluminescent (Bio-Rad, Hercules, CA, USA) detection was then used to detect expression of each protein; GAPDH levels served as internal loading controls.
Statistical analysis
Statistical analysis was performed using SPSS 16.0 statistical software. All data were expressed as mean ± SEM from at least three independent experiments. P-values were determined using one-way ANOVA. Significance was defined as P<0.05.
Results
Agomelatine prevents Aβ25–35-induced oxidative injury in PC12 cells
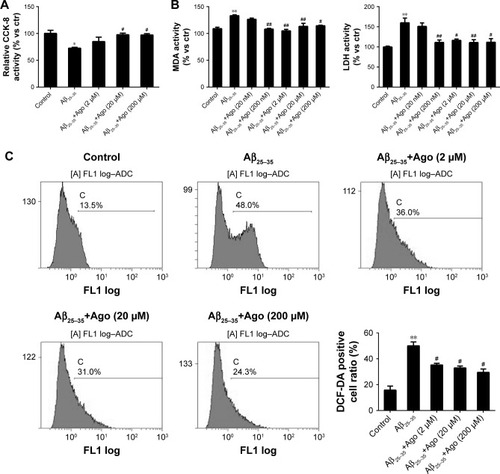
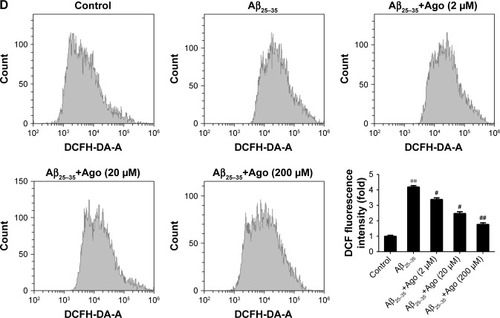
To assess Aβ25–35-induced oxidative damage in PC12 cells, the pretreatment study was conducted first. The concentration of MDA and LDH was determined. Then ROS level was analyzed via flow cytometry analysis. As shown in , the result of CCK-8 assay showed that cell viability was significantly reduced by Aβ25–35 treatment (20 µM) (P<0.05), and the injury effect was remarkably attenuated by agomelatine with different concentrations (20, 200 µM). Besides, as shown in , Aβ25–35 (20 µM) could obviously increase the concentration of MDA, LDH, and ROS in PC12 cells (P<0.01), and agomelatine pretreatment could protect against the oxidative damage (P<0.05). Second, the effect of agomelatine postinsult was investigated. As shown in , after PC cells were already stressed with Aβ25–35 treatment, agomelatine posttreatment could obviously reverse the increase in ROS level induced by Aβ25–35. The results demonstrated that agomelatine provided protective effect on Aβ-induced oxidative injury in vitro.
Figure 1 Effects of agomelatine on oxidative stress induced by Aβ25–35 in PC12 cells.
Abbreviations: Aβ25–35, amyloid beta 25–35; Ago, agomelatine; CCK-8, cell counting kit-8; DCFH-DA, dichlorodihydrofluorescein diacetate; LDH, lactate dehydrogenase; MDA, malondialdehyde.
Agomelatine protects against Aβ25–35-induced tau phosphorylation in PC12 cells
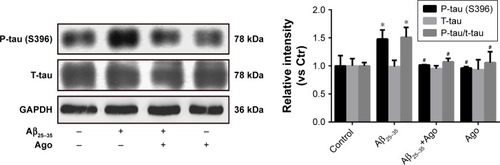
To assess the extent of tau phosphorylation, Western blot analysis was explored. As shown in , as compared to control, the protein expression of P-tau and P-tau/tau ratio were significantly upregulated in Aβ25–35-treated group (20 µM) and indicated that Aβ25–35 could promote tau phosphorylation in PC12 cells. Furthermore, agomelatine pretreatment could downregulate increased P-tau expression and P-tau/tau ratio induced by Aβ25–35. Therefore, we concluded that agomelatine could attenuate tau protein hyperphosphorylation induced by Aβ25–35 in AD.
Figure 2 Effects of agomelatine on tau phosphorylation induced by Aβ25–35 in PC12 cells.
Abbreviations: Aβ25–35, amyloid beta 25–35; Ago, agomelatine.
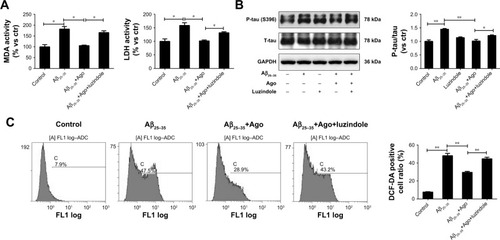
To explore the potential molecular mechanism contributing to the neuroprotective activity of agomelatine, the melatonin receptor antagonist luzindole was used. First, oxidative stress response was evaluated. As shown in , Aβ25–35 (20 µM) obviously promoted the generation of MDA, LDH, and ROS, and agomelatine (20 µM) pretreatment significantly blocked Aβ25–35-induced increase in MDA, LDH, and ROS production (P<0.05). Moreover, when cotreated with luzindole (1 µM), this effect could be reversed (P<0.05). Second, tau protein phosphorylation was determined by Western blot analysis. As shown in , Aβ25–35 (20 µM) remarkably upregulated the expression of P-tau protein (P<0.01), and Aβ25–35-induced tau phosphorylation could be blocked by agomelatine (20 µM). When compared with agomelatine group, P-tau/tau ratio obviously increased by luzindole (1 µM) cotreatment. According to the results, we found that the anti-oxidative and anti-tau protein phosphorylation effect of agomelatine was prevented by melatonin receptor antagonist luzindole. Therefore, agomelatine may provide neuroprotective function by activating melatonin receptor.
Figure 3 Effect of agomelatine and luzindole on oxidative stress and tau phosphorylation induced by Aβ25–35 in PC12 cells.
Abbreviations: Aβ25–35, amyloid beta 25–35; Ago, agomelatine; DCFH-DA, dichlorodihydrofluorescein diacetate; LDH, lactate dehydrogenase; MDA, malondialdehyde.
The signaling pathway involved in the neuroprotective effect of agomelatine
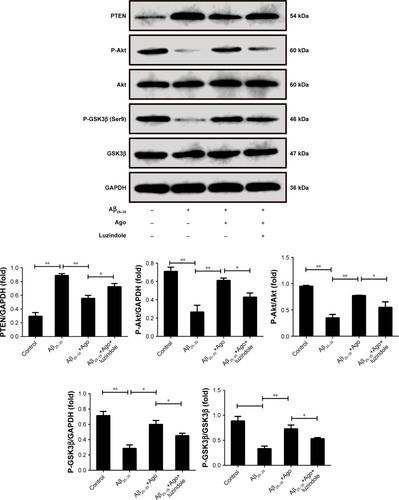
PTEN/Akt/GSK3β signaling pathway plays an important role in the process of oxidative stress and tau protein hyperphosphorylation. Thus, Western blot analysis was explored to detect the expression of PTEN, P-Akt, Akt, P-GSK3β, and GSK3β. As shown in , Aβ25–35 (20 µM) obviously promoted the upregulation of PTEN expression and decreased P-Akt and P-GSK3β expression (P<0.01). Furthermore, agomelatine pretreatment could partly inhibit the effect of Aβ25–35. When cotreated with luzindole, agomelatine-induced PTEN/Akt/GSK3β pathway inhibition was obviously reversed. According to the results, we concluded that agomelatine may exert anti-oxidative effect and prevent tau hyperphosphorylation through activating MT-PTEN/Akt/GSK3β signaling.
Figure 4 The signaling involved in the neuroprotective effect of agomelatine. Measurement of PTEN, P-Akt, Akt, P-GSK3β (Ser9), and GSK3β expression by Western blot in PC12 cells.
Abbreviations: Aβ25–35, amyloid beta 25–35; Ago, agomelatine.
Discussion
As the most prevalent neurodegenerative disease, the etiology of AD is not well understood.Citation18,Citation19 The extracellular deposition of Aβ plaques and the accumulation of intracellular NFT are considered to be the most crucial pathophysiology of AD.Citation1 Tau protein plays an important role in maintenance of the integrity of genomic DNA,Citation20 regulation of neuronal activity,Citation21 and neurogenesis.Citation22 Generally, to maintain physiological functions of tau, an appropriate extent of phosphorylation is necessary, modulated by various protein kinases and phosphatases, including proline-directed protein kinases (PDPKs), non-PDPKs, and tyrosine kinases.Citation23 In the pathological state, tau protein hyperphosphorylation promoted microtubule instability,Citation24 axon transport impairment,Citation25 and altered its truncation,Citation26 eventually leading to neurodegeneration. Tau phosphorylation is involved in several neurodegenerative diseases, including AD and FTDP-17. In the progression of AD, tau protein hyperphosphorylation has been reported to drive tau aggregation, and tau aggregation is essential for tau-induced toxicity.2 Several experiments indicated that Aβ exposure could aggravate tau-mediated neurotoxicity by facilitating tau phosphorylation, leading to spine collapse and dendritic degeneration.Citation3 In our study, we found that Aβ could obviously promote tau protein phosphorylation by upregulation of P-tau expression and P-tau/tau ratio in PC12 cells, which suggested that interaction between Aβ and tau might facilitate the development of AD.
A lot of evidence revealed that oxidative stress is involved in the development and progression of AD. It has been widely accepted that oxidative stress damage contributes to initiation and progression of AD,Citation27 through several pathways, including 1) aggravation of Aβ production, secretion, and aggregation;Citation28–Citation30 2) causing mitochondrial dysfunction;Citation31 and 3) promoting hyperphosphorylation of tau proteins, tau-mediated toxicity, and intracellular NFT formation.Citation32 Our results showed that Aβ25–35 treatment could significantly enhance oxidative stress response by increasing the concentration of MDA, LDH, and ROS, which indicate that oxidative stress may play an important role in Aβ-induced toxicity and pathological process of AD.
Furthermore, several anti-oxidative and anti-tau protein hyperphosphorylation therapeutic strategies show great potential in treating AD.Citation9,Citation10 In addition, AD often manifests with multiple comorbidities such as depression. More and more evidence demonstrated that antidepressants exert a neuroprotective effect in the development of AD.Citation12 As a novel antidepressant drug, agomelatine, widely applied in clinic, could act on the SCN, hippocampus, frontal cortex, and striatum. Agomelatine was quite helpful not only for insomnia through improvement of sleep duration and restoration of the circadian rhythmCitation33 but also for anxiety and depressive symptoms and has no adverse effects on cognition during the treatment of patients, especially the elderly.Citation34 Agomelatine is effective in the therapy of mood disorder in AD. Furthermore, animal studies have demonstrated that agomelatine treatment could increase generation of brain-derived neurotrophic factor levels and promote hippocampal and prefrontal cortex neogenesis.Citation17 On the other hand, agomelatine was found to provide potent antioxidative effect in multiple disease models. It was shown that agomelatine potentially helps reducing testicular damage by decreasing oxidative stress level in STZ-induced type I diabetic rats.Citation35 In ischemic stroke animal model, agomelatine treatment could protect the brain from cerebral I/R injury by anti-apoptosis and antioxidant properties.Citation14 In rats with chronic mild stress-induced depression, agomelatine exhibited protective effects against brain, kidney, and liver oxidative damage by regulation of the glutathione concentrations and glutathione peroxidase activity.Citation15 However, it remains unclear whether agomelatine exerts neuroprotection in AD. In our study, we first reported that agomelatine could significantly prevent Aβ25–35-induced increase in P-tau expression and P-tau/tau ratio. Moreover, as compared to Aβ25–35 group, agomelatine pretreatment could alleviate oxidative stress damage caused by Aβ25–35 by decreasing the generation of MDA, LDH, and ROS. Our results indicated that agomelatine had great potential for inhibiting tau protein hyperphosphorylation and oxidative damage in AD.
Then the neuroprotective mechanism of agomelatine was investigated. Agomelatine is a receptor agonist that affects both MT1 and MT2 melatonin receptors and an antagonist that affects 5HT 2C receptors. Previous studies demonstrated that MT receptors are obviously decreased in multiple brain areas such as the pineal gland, pyramidal and cortical layers, and hippocampus in AD.Citation36,Citation37 MT receptors regulate the neurotoxicity and clearance of amyloid-β,Citation38,Citation39 the stabilization of synapses,Citation40 the generation of neurofibrillary tangles, and level of oxidative stress in AD.Citation41 In our study, after luzindole pretreatment, we found the novel results that the decrease in P-tau/tau ratio and oxidative production by agomelatine was obviously upregulated. The study revealed that luzindole, antagonist of melatonin receptor, could reverse the anti-tau phosphorylation and anti-oxidative stress effect provided by agomelatine in Aβ-treated PC12 cells; therefore, we speculated that agomelatine may prevent the pathological injury by activating melatonin receptor in AD.
On the other hand, PTEN/Akt/GSK3 signaling pathway is closely related to the onset and progression of AD. As a serine/threonine protein kinase, GSK3 exists in two isoforms, GSK3α and GSK3β, expressed in brain abundantly. GSK3 activity could be modulated by the site-specific phosphory-lation of GSK3β, for example, ser9 of GSK3β inhibits its activity.Citation42 GSK3 signaling is involved in a number of cellular pathological and physiological process, including glucose regulation, inflammation, oxidative stress, and apoptosis.Citation43,Citation44 A lot of evidence has shown that both GSK3 isoforms were abnormally activated in the brains of postmortem AD samples and AD animal model,Citation45,Citation46 resulting in excessive Aβ formation and aggregation.Citation47 Besides, GSK3 activation in AD significantly promotes tau protein hyperphosphorylation, leading to the formation of NFTs and neuronal death.Citation48,Citation49 Therefore, GSK3 is widely known as the key kinase responsible for the hyperphosphorylation of tau protein, and GSK3 inhibitor provides potential neuroprotective function in the development of AD.Citation48 GSK3 activity is regulated by several upstream kinases, such as PTEN (also named MMAC1/TEP1). PTEN is considered as a dual phosphatase with both protein and lipid phosphatase activities, modulating cellular growth, survival, and metabolism.Citation50 It is now well established that PTEN could inhibit a major cell growth and survival signaling pathway, including PI3K/AKT signaling pathway,51 and upregulate the expression and activity of GSK3.Citation52–Citation54 Furthermore, targeted suppression of PTEN activity could protect against oxidative stress damage, neurotoxicity,Citation55,Citation56 endoplasmic reticulum stress,Citation57 and tau hyperphosphorylationCitation56 in the brain of patients with AD. However, little is known about the link between agomelatine, MT receptors, and PTEN/Akt/GSK3β signaling in AD. According to our study, we found the novel results that Aβ25–35 obviously activated PTEN/Akt/GSK3β signaling by increasing PTEN expression and decreasing P-Akt and P-GSK3β expression. Moreover, agomelatine could prevent the effect of Aβ25–35 through inhibition of PTEN/Akt/GSK3β axis, which was reversed by melatonin receptor antagonist luzindole. Our study demonstrated that the neuroprotective effect provided by agomelatine was via activating MT-PTEN/Akt/GSK3β signaling pathway. The results have not been published before and revealed the novel neuroprotective mechanism of agomelatine in AD.
Conclusion
Our data presented that antidepressant agomelatine could prevent the tau protein phosphorylation and oxidative damage induced by Aβ25–35 in PC12 cells. Furthermore, agomelatine might exert protective effect by activating MT-PTEN/Akt/GSK3β signaling pathway. Importantly, the neuroprotective effect of agomelatine must be discussed in vivo in AD animal models in the future.Citation58,Citation59 The data above revealed the neuroprotective function of agomelatine in AD-associated pathological injury and provided new insights in the therapy of AD.
Acknowledgments
The authors thank Heng-bing Zu (Department of Neurology, Jinshan Hospital Affiliated to Fudan University, Shanghai, China) for excellent technical assistance in this study. The study was supported by research grants from Research Project of Jinshan District Health and Family Planning Commission (No JSKJ-KTMS-2018-19), Qi Hang project of Jinshan Hospital (No 2018-JSYYQH-06).
Disclosure
The authors report no conflicts of interest in this work.
References
- QuerfurthHWLaferlaFMAlzheimer’s diseaseN Engl J Med Overseas Ed20103624329344
- AhmedTVander Jeugd ABlumDCognition and hippocampal synaptic plasticity in mice with a homozygous tau deletionNeurobiol Aging201435112474247824913895
- De FeliceFGWuDLambertMPAlzheimer’s disease-type neuronal tau hyperphosphorylation induced by Aβ oligomersNeurobiol Aging20082991334134717403556
- SiesHBerndtCJonesDPOxidative stressEncyclopedia Stress200762104548
- GagnéFOxidative stressBiochemical Ecotoxicology: Principles and MethodsAmsterdamElsevier2014103115
- GaoHMZhouHHongJSNADPH oxidases: novel therapeutic targets for neurodegenerative diseasesTrends Pharmacol Sci201233629530322503440
- LimónIDDíazAMendietaLAmyloid-beta (25–35) impairs memory and increases NO in the temporal cortex of ratsNeurosci Res200963212913719084561
- WangXWangWLiLPerryGLeeHGZhuXOxidative stress and mitochondrial dysfunction in Alzheimer’s diseaseBiochim Biophys Acta18422014812401247
- GräffJTsaiLHThe potential of HDAC inhibitors as cognitive enhancersAnnu Rev Pharmacol Toxicol201353131133023294310
- KanninenKMalmTMJyrkkänenHKNuclear factor erythroid 2-related factor 2 protects against beta amyloidMol Cell Neurosci200839330231318706502
- PlotnikoffNP2007Cytokines: stress and immunityBoca Raton. FLCRC Press405
- ManjiHKMooreGJChenGClinical and preclinical evidence for the neurotrophic effects of mood stabilizers: implications for the patho-physiology and treatment of manic-depressive illnessBiol Psychiatry200048874075411063971
- NobleWPlanelEZehrCInhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivoProc Natl Acad Sci U S A2005102196990699515867159
- ChumboatongWThummayotSGovitrapongPTocharusCJittiwatJTocharusJNeuroprotection of agomelatine against cerebral ischemia/reperfusion injury through an antiapoptotic pathway in ratNeurochem Int201710211412228012846
- DemirdaşANazıroğluMÜnalGÖAgomelatine reduces brain, kidney and liver oxidative stress but increases plasma cytokine production in the rats with chronic mild stress-induced depressionMetab Brain Dis20163161445145327438049
- YucelAYucelNOzkanlarSEffect of agomelatine on adult hippocampus apoptosis and neurogenesis using the stress model of ratsActa Histochem2016118329930426970810
- PompiliMSerafiniGInnamoratiMAgomelatine, a novel intriguing antidepressant option enhancing neuroplasticity: a critical reviewWorld J Biol Psychiatry201314641243123530731
- RosenbergRNLambracht-WashingtonDYuGXiaWGenomics of Alzheimer disease: a reviewJAMA Neurol201673786727135718
- KillinLOStarrJMShiueIJRussTCEnvironmental risk factors for dementia: a systematic reviewBMC Geriatr201616117527729011
- VioletMDelattreLTardivelMA major role for tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditionsFront Cell Neurosci2014868424672431
- GheyaraALPonnusamyRDjukicBTau reduction prevents disease in a mouse model of Dravet syndromeAnn Neurol201476344345625042160
- HongXPPengCXWeiWEssential role of tau phosphorylation in adult hippocampal neurogenesisHippocampus201020121339134919816983
- ChungSHAberrant phosphorylation in the pathogenesis of Alzheimer’s diseaseBMB Rep200942846747419712581
- HangerDPAndertonBHNobleWTau phosphorylation: the therapeutic challenge for neurodegenerative diseaseTrends Mol Med200915311211919246243
- HooverBRReedMNSuJTau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegenerationNeuron20106861067108121172610
- DickeyCAKamalALundgrenKThe high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteinsJ Clin Invest2007117364865817304350
- LiuZZhouTZieglerACDimitrionPZuoLOxidative stress in neurodegenerative diseases: from molecular mechanisms to clinical applicationsOxid Med Cell Longev201720174111
- SherringtonRRogaevEILiangYCloning of a gene bearing missense mutations in early-onset familial Alzheimer’s diseaseNature199537565347547607596406
- BondaDJWangXPerryGOxidative stress in Alzheimer disease: a possibility for preventionNeuropharmacology2010594–529029420394761
- AseervathamGSSivasudhaTJeyadeviRArul AnanthDEnvironmental factors and unhealthy lifestyle influence oxidative stress in humans – an overviewEnviron Sci Pollut Res Int20132074356436923636598
- AbramovAYBerezhnovAVFedotovaEIZinchenkoVPDolgachevaLPInteraction of misfolded proteins and mitochondria in neurodegenerative disordersBiochem Soc Trans201745410251033
- GiraldoELloretAFuchsbergerTViñaJAβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: protective role of vitamin ERedox Biol2014287387725061569
- LaudonMFrydman-MaromATherapeutic effects of melatonin receptor agonists on sleep and comorbid disordersInt J Mol Sci2014159159241595025207602
- PlesničarBKEfficacy and tolerability of agomelatine in the treatment of depressionPatient Prefer Adherence2014860361224833894
- YigitturkGAcaraACErbasOThe antioxidant role of agomelatine and gallic acid on oxidative stress in STZ induced type I diabetic rat testesBiomed Pharmacother20178724024628061407
- BrunnerPSözer-TopcularNJockersRPineal and cortical melatonin receptors MT1 and MT2 are decreased in Alzheimer’s diseaseEur J Histochem200650431131617213040
- SavaskanEAyoubMARavidRReduced hippocampal MT2 melatonin receptor expression in Alzheimer’s diseaseJ Pineal Res2005381101615617532
- ChinchalongpornVShuklaMGovitrapongPMelatonin ameliorates Aβ42-induced alteration of βAPP-processing secretases via the melatonin receptor through the Pin1/GSK3β/NF-κB pathway in SH-SY5Y cellsJ Pineal Res2018644e1247029352484
- PappollaMAMatsubaraEVidalRMelatonin treatment enhances Aβ lymphatic clearance in a transgenic mouse model of amyloidosisCurr Alzheimer Res201815763764229637859
- ShiYFangYYWeiYPMelatonin in synaptic impairments of Alzheimer’s diseaseJ Alzheimers Dis201863311629578489
- CaballeroBVega-NaredoISierraVFavorable effects of a prolonged treatment with melatonin on the level of oxidative damage and neurodegeneration in senescence-accelerated miceJ Pineal Res200845330231118410310
- DajaniRFraserERoeSMCrystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibitionCell2001105672173211440715
- RanaAKSinghDTargeting glycogen synthase kinase-3 for oxidative stress and neuroinflammation: opportunities, challenges and future directions for cerebral stroke managementNeuropharmacology201813912413630017999
- GeXHShaoLZhuGJOxymatrine attenuates brain hypoxic-ischemic injury from apoptosis and oxidative stress: role of p-Akt/GSK3beta/ HO-1/Nrf-2 signaling pathwayMetab Brain Dis20183361869187530032345
- TerwelDMuyllaertDDewachterIAmyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic miceAm J Pathol2008172378679818258852
- LeroyKBoutajangoutAAutheletMWoodgettJRAndertonBHBrionJPThe active form of glycogen synthase kinase-3β is associated with granulovacuolar degeneration in neurons in Alzheimer’s diseaseActa Neuropathol20021032919911810173
- HooperCKillickRLovestoneSThe GSK3 hypothesis of Alzheimer’s diseaseJ Neurochem200810461433143918088381
- MaqboolMMobashirMHodaNPivotal role of glycogen synthase kinase-3: a therapeutic target for Alzheimer’s diseaseEur J Med Chem2016107638126562543
- MorrisGBerkMMaesMPuriBKCould Alzheimer’s disease originate in the periphery and if so how so?Mol Neurobiol Epub2018429
- WorbyCADixonJEPTENAnnu Rev Biochem201483164166924905788
- RobbinsHLHagueAThe PI3K/Akt pathway in tumors of endocrine tissuesFront Endocrinol20166377–387188
- MaJGuoXZhangJPTEN gene induces cell invasion and migration via regulating Akt/GSK-3β/β-catenin signaling pathway in human gastric cancerDig Dis Sci201762123415342529030742
- ShenHLiLYangSMicroRNA-29a contributes to drug-resistance of breast cancer cells to adriamycin through PTEN/AKT/ GSK3β signaling pathwayGene20165931849027523474
- HeEPanFLiGLiJFractionated ionizing radiation promotes epithelial-mesenchymal transition in human esophageal cancer cells through PTEN deficiency-mediated Akt activationPLoS One2015105e012614926000878
- LiuXYZhangLJChenZLiuLBThe PTEN inhibitor bpV (pic) promotes neuroprotection against amyloid β-peptide (25–35)-induced oxidative stress and neurotoxicityNeurol Res201739875876528436304
- Pinto-AlmazánRSegura-UribeJJSoriano-UrsúaMAFarfán-GarcíaEDGallardoJMGuerra-AraizaCEffect of tibolone pretreatment on kinases and phosphatases that regulate the expression and phosphorylation of tau in the hippocampus of rats exposed to ozoneNeural Regen Res201813344029623928
- CuiWWangSWangZWangZSunCZhangYInhibition of PTEN attenuates endoplasmic reticulum stress and apoptosis via activation of PI3K/Akt pathway in Alzheimer’s diseaseNeurochem Res201742113052306028819903
- LaferlaFMGreenKNAnimal models of Alzheimer diseaseCold Spring Harb Perspect Med2012211a006320a00632923002015
- GötzJIttnerLMAnimal models of Alzheimer’s disease and frontotemporal dementiaNat Rev Neurosci20089753254418568014