Abstract
Epidermal growth factor receptor (EGFR) tyrosine inhibitors were first approved for the treatment of non-small cell lung cancer (NSCLC) in 2003 in the US. Activating EGFR mutations were subsequently discovered in 2004, and heralded the era of molecular targeted therapy in NSCLC. The discovery of anaplastic lymphoma kinase (ALK) rearrangement in NSCLC in 2007 by two independent groups not only represents the first time ALK rearrangement has been discovered in common solid tumors but also represents another important milestone in the era of molecular targeted therapy in NSCLC. Crizotinib, a mesenchymal-epithelial transition (MET)/ALK multi-targeted receptor tyrosine kinase inhibitor went into early Phase I clinical development in 2007. Using the knowledge that NSCLC patients with activating EGFR mutations benefited from EGFR tyrosine kinase inhibitors, crizotinib was rapidly and successfully developed as an inhibitor in ALK-rearranged NSCLC, based on a break apart fluorescence in situ hybridization assay, developed by two of the crizotinib Phase I sites. It cumulated in the conditional approval of crizotinib by the US Food and Drug Administration on August 26, 2011 for the treatment of ALK-rearranged NSCLC. The conditional approval was based on response rates of 50% and 61% from 255 ALK-rearranged NSCLC patients enrolled in two single-arm trials. Common adverse events of crizotinib include mild transient visual disorders, mild gastrointestinal toxicities, fatigue, rare alanine transaminase elevations, and even rarer pneumonitis (1.6%). Confirmatory trials comparing crizotinib with standard chemotherapy are ongoing. It took an unprecedented four years from the discovery of ALK rearrangement in NSCLC to the approval of crizotinib, the first ever ALK inhibitor, for the treatment of ALK-rearranged NSCLC.
Introduction
During the past few years, the seminal discovery of activating mutations in the kinase domain of the epidermal growth factor receptor (EGFR) gene has revolutionized the treatment of non-small cell lung cancer (NSCLC).Citation1–Citation3 While the first EGFR tyrosine kinase inhibitors (gefitinib, erlotinib) were approved for use in NSCLC prior to the knowledge of activating EGFR mutations,Citation4,Citation5 the clinical utility of EGFR tyrosine kinase inhibitors has been optimized after the discovery of EGFR mutations and heralded the era of molecular targeted therapy in NSCLC. Retrospective analysis of IPASS (Iressa Pan-Asia Study) clearly demonstrated that the presence or absence of EGFR mutations in Asian never-smokers/light former smokers significantly determines the presence or lack of response to EGFR tyrosine kinase inhibitors, respectively.Citation6,Citation7 Several prospective randomized trials have now confirmed the use of EGFR tyrosine kinase inhibitors in patients with advanced treatment-naïve NSCLC with EGFR mutations significantly improved the response rate and progression-free survival compared with standard platinum-based chemotherapy.Citation8–Citation11 The characterization of NSCLC patients with activating EGFR mutations provided the bulk of the molecular under-pinning of the seminal observation that NSCLC in neversmokers (<100 cigarettes lifetime) is a distinct clinical entity (higher proportion of adenocarcinoma, female, Asian, better survival).Citation12 However, as demonstrated by IPASS, even among a clinically defined NSCLC patient cohort (Asian, female, adenocarcinoma, never-smokers) only slightly more than half of these patients harbored activating EGFR mutations and that other “driver mutations “ remained to be discovered in NSCLC.Citation6,Citation7
Anaplastic lymphoma kinase (ALK) is thus named because it was first discovered to be translocated in anaplastic large cell lymphoma.Citation13 Since the late 1980s, alterations in the ALK gene have been well recognized as playing a key role in the pathogenesis of anaplastic large cell lymphoma, a subset of B cell non-Hodgkin’s lymphoma, inflammatory myofibro-blastic tumors, and in neuroblastoma.Citation14 However, perturbations in the ALK gene had not been found in common solid tumors until two groups independently reported the discovery of ALK rearrangement in NSCLC in 2007.Citation15,Citation16 Soda et al screened a cDNA library derived from adenocarcinoma of the lung of a 62-year-old male Japanese smoker for transforming activity.Citation15 This fusion arises from an intrachromosomal inversion on the short arm of chromosome 2 [Inv (2)(p21p23)] that joins exons 1–13 of the echinoderm microtubule-associated protein-like 4 gene (EML4) to exons 20–29 of ALK. The resulting chimeric protein, EML4-ALK, contains an N terminus derived from EML4 and a C terminus containing the entire intracellular tyrosine kinase domain of ALK. Since the initial discovery of this fusion, multiple other variants of EML4-ALK have been reported, all of which encode the same cytoplasmic portion of ALK but contain different truncations of EML4.Citation17,Citation18 Additionally, other fusion partners with ALK have been described (TFG and KIF5B), but these fusion variants are much less common than EML4-ALK.Citation17,Citation18 The various fusion partners of ALK mediate ligand-independent dimerization of ALK resulting in constitutive kinase activity. EML4-ALK possesses potent oncogenic activity in cell cultures. Citation15 In transgenic mouse models, lung-specific expression of EML4-ALK leads to development of numerous lung adenocarcinoma.Citation19 Treatment of EML4-ALK transgenic mice with ALK inhibitors also results in tumor regression.Citation19 Contemporaneously, Rikova et al independently discovered the same EML4-ALK translocation in NSCLC while searching for candidate tyrosine kinases in NSCLC by screening for phosphotyrosine activation in 150 NSCLC tumors as well as 41 NSCLC cell lines.Citation16 They identified kinases known to have a dominant role in NSCLC pathogenesis, such as EGFR and mesenchymal-epithelial transition (MET) receptor tyrosine kinase, as well as others not previously implicated in NSCLC, including platelet-derived growth factor receptor-α and ROS. The samples with ALK hyperphosphorylation were shown to harbor EML4-ALK (three cases) or TFG-ALK (one case).Citation16
ALK belongs to the leukocyte tyrosine kinase receptor superfamily. ALK is a single-chain transmembrane receptor. The extracellular domain contains an N-terminal signal peptide sequence and is the ligand-binding site for the activating ligands of ALK, pleiotrophin, and midkine. This is followed by the transmembrane and juxtamembrane region which contains a binding site for phosphotyrosine-dependent interaction with insulin receptor substrate-1. The final section has an intracellular tyrosine kinase domain with three phosphorylation sites (Y1278, Y1282, and Y1283), followed by the C-terminal domain with interaction sites for phospholipase C-gamma and Src homology 2 domain-containing SHC. The signaling pathways involving ALK have recently been the subject of an expert review.Citation20
Simultaneous with the discovery of ALK-rearranged NSCLC, crizotinib, a multitargeted receptor tyrosine kinase inhibitor, entered early Phase I clinical development primarily as a MET inhibitor. With the ability of a few Phase I clinical sites to develop and standardize a breakapart fluorescence in situ hybridization (FISH) assay for ALK-rearranged NSCLC, there was a concerted switch in the focus of the Phase I crizotinib trial when the first two ALK-rearranged NSCLC patients derived clinical benefit from crizotinib.
This review summarizes the preclinical and clinical data that led to conditional approval of crizotinib by the US Food and Drug Administration (FDA). It will discuss the advantages and limitations of the breakapart FISH assay as the companion diagnostic approved by the FDA for the detection of ALK-rearranged NSCLC that accompanied the approval of crizotinib. This review will also examine newer agents (second-generation ALK inhibitors, heat shock protein [Hsp] 90 inhibitors) that can potentially overcome the resistance to crizotinib that has already been discovered, and touches upon other tumors and receptor tyrosine kinases that may be inhibited by crizotinib.
Chemistry and pharmacology
Crizotinib (previously known as PF02341066, marketed as XalkoriTM, Pfizer, New York, NY) was synthesized primarily as a MET inhibitor. It was engineered based on interactions of PHA-665752, a potent MET inhibitor, at the ATP-binding sites of the MET kinase domain obtained from the cocrystal structure of PHA-665752 binding in the MET kinase domain. Binding of PHA-665752 to the MET kinase domain results in displacement of the kinase activation loop that interferes with ATP and substrate binding to the MET receptor tyrosine kinase. PHA-665752 is a powerful dose-dependent MET inhibitor in cell lines and in xenograft models. However, the low solubility of PHA-665752 at pH 7.4, its high meta-bolic clearance, and its poor permeability limit its ability as a clinical MET inhibitor. Crizotinib was designed using a structure-based drug design program to be less lipophilic and have a small hinge binder to allow better interaction in the kinase pocket ().Citation21
Figure 1 Synthesis and structure of crizotinib. The hinge binder 2 aminopyridine in crizotinib is highlighted in red. The molecular formula for crizotinib is C21H22Cl2FN5O. The molecular weight is 450.34 Da. The chemical name for crizotinib is (R)-3-[1-(2,6-Dichloro-3-fluorophenyl)-ethoxy]-5-[(1-piperidin-4-yl)-1H-pyrazol-4-yl]pyridin-2-amine. Figure provided by Dr Jean Cui.
![Figure 1 Synthesis and structure of crizotinib. The hinge binder 2 aminopyridine in crizotinib is highlighted in red. The molecular formula for crizotinib is C21H22Cl2FN5O. The molecular weight is 450.34 Da. The chemical name for crizotinib is (R)-3-[1-(2,6-Dichloro-3-fluorophenyl)-ethoxy]-5-[(1-piperidin-4-yl)-1H-pyrazol-4-yl]pyridin-2-amine. Figure provided by Dr Jean Cui.](/cms/asset/62850966-d0b3-4d39-a864-10f40ee301cd/dddt_a_19045_f0001_c.jpg)
Crizotinib was evaluated against a panel of more than 120 kinases in biochemical assays and twelve cell-based phosphorylation assays, and was determined to be nearly 20-fold more selective for ALK and MET compared with other kinases evaluated. Crizotinib inhibited nucleophosmin (NPM)-ALK phosphorylation in Karpas299 or SU-DHL-1 anaplastic large cell lymphoma cells (mean IC50 value, 24 nmol/L), and inhibited cell proliferation and induced apoptosis in NPM-ALK-dependent Karpas299 or SU-DHL-1 cell lines. Furthermore, crizotinib also inhibited ALK phosphorylation and growth in a NPM-ALK-dependent Karpas299 tumor xenograft model. Crizotinib potently inhibited cell proliferation, which was associated with G1-S phase cell cycle arrest and induction of apoptosis in ALK-positive anaplastic large cell lymphoma cells (IC50 values, about 30 nmol/L). Oral administration of crizotinib to severe combined immunodeficient-beige mice bearing Karpas299 anaplastic large cell lymphoma tumor xenografts resulted in dose-dependent antitumor efficacy, with complete regression of all tumors at the 100 mg/kg/day dose within 15 days of initial administration of the compound. A strong correlation was observed between antitumor response and inhibition of NPM-ALK phosphorylation and induction of apoptosis in tumor tissue. In addition, inhibition of key NPM-ALK signaling mediators, including phospholipase C gamma, STAT3, and Akt, by crizotinib was observed at concentrations or dose levels that correlated with inhibition of NPM-ALK phosphorylation and function.Citation22
First in-human Phase I trial of crizotinib
The first in-human Phase I trial of crizotinib (A8081001, NCT00585195) trial was designed as an open-label, multi-center two-part trial that was activated in 2006. The first part was the standard dose-escalation finding schema to determine the maximal tolerated dose and the recommended Phase II dose. There was also a single day -7 dose to establish the single-dose pharmacokinetics. Crizotinib was first dosed at 50 mg orally once daily and subsequently escalated to 300 mg orally twice daily at which two patients experienced grade three fatigue. The dose of crizotinib was eventually reduced to 250 mg orally twice daily and was found to be tolerable and was determined to be the maximal tolerated dose and recommended Phase II dose.Citation23 There was also a substudy to investigate the effect of food on crizotinib level and a midazolam substudy to assess the effect of cytochrome P450 (CYP) 3A isozyme inhibition by crizotinib. In contrast with other Phase I trials, the efficacy data from prospective enrollment of ALK-rearranged NSCLC patients into this Phase I crizotinib trial was used in part to obtain conditional FDA approval. Therefore, a lot of data on pharmacokinetics, efficacy, and adverse events were generated from this one Phase I trial.
Pharmacokinetics
Eleven patients were enrolled into the food effect sub-study. There was no food effect on the pharmacokinetics of crizotinib. The absorption of a single 250 mg dose of crizotinib after a high-fat, high-calorie meal did not differ from the concentration of a single 250 mg dose of crizotinib taken on an empty stomach.Citation24 The peak plasma concentration was reached 4 hours after a single dose of crizotinib. Pharmacokinetics were linear from a 100 mg once daily dose to a 300 mg twice daily dose.Citation23,Citation24 A steady-state concentration of crizotinib was reached after 15 days of repeated administration of crizotinib 250 mg orally twice a day, with a half-life of around 43–51 hours.Citation24 The mean steady-state trough plasma level for crizotinib 250 mg taken twice a day (the recommended Phase II dose) was 274 ng/mL or 57 nM of free drug, which exceeded the target efficacy levels predicted for the inhibition of MET (about 13 nM) and ALK (about 26 nM) based on preclinical mouse models.Citation24
Thirteen patients were enrolled in the midazolam substudy. The pharmacokinetics of a single 2 mg midazolam dose was evaluated before and 28 days after crizotinib 250 mg administered orally twice a day. Midazolam is metabolized by CYP3A isozymes. There was a 3.6-fold increase in the midazolam level (90% confidence interval 2.7–4.9) after 28 days of 250 mg twice a day of crizotinib, indicating that crizotinib is a moderate CYP3A inhibitor.Citation24 Crizotinib is also predominantly metabolized by the CYP3A isozymes in human microsomes. The total clearance of crizotinib from plasma was lower after 250 mg twice a day than after a single 250 mg dose (100 L/hour), likely due to the autoinhibition of CYP3A.Citation24 The cerebral spinal fluid to plasma ratio of crizotinib was measured and reported to be 0.0026 in a ALK-rearranged NSCLC patient.Citation25 The low crizotinib level in the cerebral spinal fluid implies poor blood-brain barrier penetration of the drug. However, this is the only report of the level of crizotinib achieved in cerebral spinal fluid in one patient.
Clinical efficacy
Crizotinib was initially developed as a MET inhibitor, and the second part of the protocol called for screening of patients with tumors that harbor MET amplification (non-Barrett’s gastroesophageal junction cancer, gastric cancer,) or MET mutations (lung cancer in former/current smokers, squamous cell carcinoma of the head and neck, papillary renal cell carcinoma) to enroll into the molecularly enriched cohort part of the protocol. Other rare tumors that may also be involved in the MET signaling pathway, such as alveolar soft part sarcoma and alveolar rhabdomyosarcoma were also eligible upon histological confirmation. However, during the dose-escalation phase, ALK rearrangement in NSCLC was discovered and one of the clinical sites involved in A8081001 was in the process of developing a breakapart FISH assay to detect ALK rearrangement in tumors. The first EML4-ALK NSCLC patient enrolled in the trial was a 49-year-old male never-smoker enrolled onto the 300 orally twice a day cohort on December 26, 2007 at Massachusetts General Hospital, Boston, MA, barely 4 months after publication of the Soda et al paper. He had a dramatic clinical response within weeks, but developed liver enzyme elevation and had to be taken off treatment. The second ALK-rearranged NSCLC patient was a 30-year-old male never-smoker enrolled into the trial at Beth Israel Deaconess Medical Center, Boston, MA on June 5, 2008, onto the 200 mg orally twice a day cohort and achieved stable disease for several months. He eventually developed brain metastasis. After radiation to the brain, his crizotinib dose was increased to 250 mg orally twice a day but he had disease progression after 1 month.Citation25 Because both ALK-rearranged NSCLC patients had dramatic improvements in symptoms, this prompted the protocol to be amended to allow a large-scale prospective screening for ALK-rearranged NSCLC among the seven participating clinical sites and nine participating hospitals. Screening for patients with MET-amplified or MET-mutated tumors continued simultaneously. The main eligibility for the ALK molecular-enriched cohort was that patients had to be tumor-positive for ALK rearrangement by FISH. Other common eligibility criteria include Eastern Cooperative Oncology Group performance status 0–2. Patients with stable brain metastasis or patients on stable coagulation were eligible. There was no limit on the number of previous treatment regimens for any patient. Patients enrolled onto the ALK or MET enriched molecularly defined portion of the trial were evaluated for safety at least once every two weeks during the first two cycles (one cycle is 28 days) and then at least every four weeks thereafter. Radiological assessments were performed at baseline and generally after every two cycles of treatment using the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0. Adverse events were reported according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events version 3.0.
Initially, there was no screening strategy for ALK-rearranged NSCLC patients, but as more and more ALK-rearranged NSCLC patients were identified it became clear that ALK-rearranged NSCLC patients generally present with adenocarcinoma (with the presence of signet ring cell features within the adenocarcinoma), are never-smokers, are younger than the general NSCLC population, and harbor wild-type EGFR.Citation18 The efficacy results for crizotinib in the first 19 patients with ALK-rearranged NSCLC were presented in 2009.Citation23 There were ten responders out of the first 19 patients (overall response rate, 53%) and five additional patients had achieved stable disease, giving a disease control rate (overall response rate + stable disease) of 79%. Subsequent efficacy results for crizotinib in 82 ALK-rearranged NSCLC patients were published in 2010.Citation26 The overall confirmed response rate was 57% (1% complete response + 56% partial response) and an additional 33% of patients achieved stable disease for a disease control rate of 90%. Of note, 87% disease control rate was achieved at eight weeks (two cycles, ). All of the ALK-rearranged NSCLC patients also screened negative for MET amplification. The mean duration of treatment was 6.4 months with ongoing follow-up, and the estimated probability of progression-free survival at 6 months was estimated at 72%. In a subset of ALK-rearranged patients where the fusion partners were able to be identified by reverse transcription polymerase chain reaction (PCR), there was a difference in efficacy among the different EML4-ALK fusion product.Citation26 In 2011, results of crizotinib on 119 ALK-rearranged NSCLC patients were presented.Citation27 These 119 patients were combined with 136 ALK-rearranged NSCLC patients from another trial and constituted the efficacy data that formed the basis for the conditional approval of crizotinib. The clinicopathologic characteristics of these 255 patients were listed in . Among the 119 patients, the median age is 51 years and equally divided between female and male, 72% were never-smokers, and 97% had adenocarcinoma histology. The response rate is 61% () with an estimated progression-free survival of about 10 months (95% confidence interval 8.2–14.7) and median overall survival has not been reached as only 19% of the patients have died. The median duration of response is 48 weeks at the time of data cutoff.Citation27 Median time to response is 8 weeks which is essentially at the follow-up diagnostic scan. Indeed, some of the patients achieved symptomatic relief and significant tumor shrinkage within days of starting crizotinib ().Citation28 The efficacy of crizotinib has been remarkably consistent since the efficacy of the first 19 ALK-rearranged NSCLC patients was reported, and has cemented confidence among oncologists about the efficacy of crizotinib in ALK-rearranged NSCLC patients while the confirmatory randomized trials are being done.
Table 1 Clinicopathologic characteristics of 255 patients with ALK-rearranged NSCLC, the data for whom were used for conditional approval of crizotinibCitation27,Citation29
Table 2 Efficacy of 255 patients with ALK rearranged non-small cell lung cancer enrolled in A8081001 and PROFILE 1005Citation27,Citation29
Figure 2 Response of ALK-rearranged NSCLC after 2 months of crizotinib.
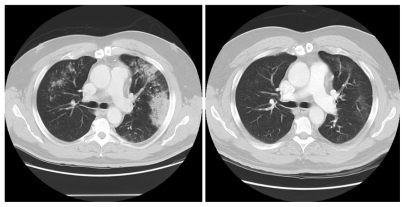
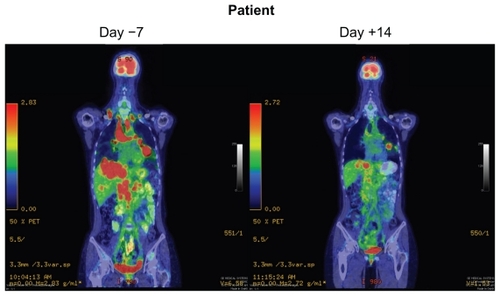
Figure 3 Dramatic response to crizotinib in a ALK-rearranged NSCLC patient.
Phase II trial of ALK-rearranged NSCLC
The Phase II trial of ALK-rearranged NSCLC (PROFILE 1005, NCT00932451) was originally designed as an open-label, single-arm study of the efficacy and safety of crizotinib in ALK-rearranged NSCLC who had failed more than two lines of chemotherapy. It serves as a compendium trial to the second-line randomized trial (PROFILE 1007) for patients who were randomized to and progressed on the chemotherapy arm of PROFILE 1007. PROFILE 1007 does not allow patients on the chemotherapy arm to crossover to crizotinib after disease progression. Instead, these patients are to enrol into PROFILE 1005.
In anticipation of the approval of crizotinib in the US, PROFILE 1007 was closed to accrual for patients in the US in order to protect the integrity of the trial because Pfizer anticipates patients on the chemotherapy arm may decide to take crizotinib off trial once it is approved. In conjunction with the closure of PROFILE 1007 in the US, major amendments were made to PROFILE 1005 to allow ALK-rearranged patients identified during the screening period of PROFILE 1007 to enroll without having progressed on chemotherapy. Other changes in the criteria for enrollment included patients with nonmeasurable disease who were allowed to enroll. However, the major change to the protocol was that patients who tested positive for ALK rearrangement other than by the breakapart FISH assay performed at the central laboratory were allowed to enroll on a “case by case” basis. Therefore, ALK rearrangement detected by immunohistochemistry (IHC), reverse transcription PCR, or breakapart FISH performed at commercial laboratories were potentially able to enroll into A8081005, essentially turning A8081005 into an expanded access program prior to the approval of crizotinib in August 2011.
The efficacy results for the first 136 ALK-rearranged NSCLC patients from PROFILE 1005 were presented in 2011.Citation29 The response rate was 50% at the time of data cutoff (). The efficacy data from these 136 patients together with the 119 ALK-rearranged NSCLC patients from A8081001 formed the basis of the FDA conditional approval of crizotinib in the US. The clinicopathologic characteristics of these 255 patients are listed in .
Phase III trials of ALK-rearranged NSCLC
Currently there are two ongoing randomized Phase III trials comparing crizotinib and chemotherapy. PROFILE 1014 (NCT01154140) is comparing crizotinib with platinum-pemetrexed combination chemotherapy in previously untreated patients with advanced ALK-rearranged adenocarcinoma of the lung. The primary endpoint is progression-free survival, and patients randomized to chemotherapy are allowed to cross over to crizotinib on disease progression. Breakapart FISH assay is the companion diagnostic test to detect ALK rearrangement. PROFILE 1007 (NCT00932451) is comparing crizotinib with pemetrexed or docetaxel in the second-line setting in patients with ALK-rearranged adenocarcinoma of the lung. The primary endpoint is progression-free survival. While the trial has been closed to accrual in the US as discussed above, accrual is still ongoing in the rest of the world.
Adverse events
Adverse events are tabulated together for the 255 ALK-rearranged NSCLC patients enrolled in A8081001 and PROFILE 1005 (). Most of the adverse events are mild (grade 1–2). Both trials are ongoing, and adverse events as well as efficacy data should be updated with further publications. The most common adverse event is a visual disorder consisting primarily of light flashes in the peripheral visual field during light transition (from dark to light or vice versa). These visual abnormalities are usually transient, lasting seconds and generally decreasing in frequency with prolonged crizotinib use. Repeat visual acuity and slit lamp examination were negative. The visual disorders disappeared after discontinuation of crizotinib. The vast majority of the patients were not bothered by these “visual disturbances”. PROFILE 1007 and 1014 are prospectively collecting data on these visual disturbances by questionnaire.
Table 3 Treatment-related adverse events in >10% of 255 patients with ALK rearranged non-small cell lung cancer treated on trials A8081001 and PROFILE 1005Citation27,Citation29
Other common adverse events were mild (grade 1–2) gastrointestinal in nature (nausea, vomiting, constipation, or diarrhea). Diarrhea was generally mild and on average not as severe as diarrhea from epithelial growth factor receptor (EGFR) tyrosine inhibitors. Nausea and vomiting can be mitigated by food so it is recommended that crizotinib should not be taken on an empty stomach. Food has a minimal effect on the level of crizotinib. Dysgeusia (taste change) occurred in 12% of the patients and is different from the usual lack of taste or metallic taste that are generally associated with other medications. Patients described a hypersensitivity to or accentuation of sweet or sour taste sensations while the taste for hot or spicy food is diminished.
Critical appraisal of adverse events
Three adverse events warranted “warnings and precautions” in the package insert, ie, pneumonitis, hepatic laboratory abnormalities, and QT interval prolongation.Citation30 Treatment-related and potential life-threatening pneumonitis occurred in four of 255 patients (1.6%) and all within two months of starting crizotinib. Citation30 It will be important to determine if there are risk factors for developing pneumonitis on crizotinib according to gender, ethnicity, comorbidities, smoking history, and prior radiation.
Serial electrocardiograms in triplicate two minutes apart were obtained following a single dose and then at steady-state to assess the QT interval prolongation potential of crizotinib in all patients (n = 308) who received 250 mg orally twice a day. Four of 308 (1.3%) were found to have a QTcF (corrected QT by Fridericia method: QTcF = QT/3√response rate) >500 msec.Citation30 However, the incidence of QTcF in the 255 patients with ALK-rearranged NSCLC have not been reported.Citation27,Citation29 Furthermore, QTcF was not associated with the dose level of crizotinib.Citation31
Grade 1–2 sinus bradycardia has been reported in 12 of 255 patients (5%) so far. Bradycardia is a pharmacodynamic effect of crizotinib. It has been reported that there was a decrease of heart rate of 2.5 beats per minute for every 100 mg/mL of crizotinib.Citation31 Indeed, several patients who received prolonged crizotinib developed asymptomatic but profound (heart rate ≤45) sinus bradycardia without an increase in the PR interval or QTc prolongation.Citation32 It will be important to determine the pharmacokinetics of crizotinib in patients who develop profound sinus bradycardia, to see if there is any correlation between overall response rate and the degree of bradycardia, given that bradycardia is a pharmacodynamic phenomenon with crizotinib. It will also be important to determine if there are any correlations between age, gender, length of treatment, ethnicity, and profound sinus bradycardia, given that Asian patients achieved, on average, a 40%–50% higher crizotinib level than Caucasian patients.Citation33
What is the best companion diagnostic test for ALK rearrangement?
The paradigm of molecularly targeted therapy in oncology depends critically on a validated test to provide a uniform platform for detecting the molecular alteration in question. The test has to be both highly sensitive and specific, yet relatively inexpensive and not highly technical to perform to enable widespread adoption and implementation of the paradigm, especially in developing countries so that the maximum number of patients can be identified to benefit from treatment. Breakapart FISH assay is the companion diagnostic test approved by the FDA to detect ALK-rearranged NSCLC in conjunction with conditional approval of crizotinib in the US. Two major criteria have to be satisfied for a sample to be positive for ALK rearrangement. First, more than 15% of the cells counted have to harbor signals that indicate ALK translocation, ie, separation of the 5′ (green color) and 3′ (red color) has to be greater than 2 signal diameter in length, OR the presence of’ signal (red color) alone. Second, at least 50 cells have to be counted (). These two criteria were developed by Dr Anthony John Iafrate’s laboratory at Massachusetts General Hospital and later validated by Dr Marileila Varella-Garcia at the University of Colorado Denver Cancer Center. Camidge et al reported that in ALK rearrangement-positive NSCLC, the mean percentage of positive cells for breakapart FISH was 53.8% (range 22.25%–86.62%), while in ALK rearrangement-negative NSCLC, the mean percentage of positive cells for breakapart FISH was 5.98% (range 3.51%–9.45%; P > 0.0001).Citation34 Furthermore, counting 45 nuclei will give >99% sensitivity and 100% specificity, while counting 60 cells will give 100% sensitivity and 100% specificity.Citation34
Table 4 Criteria for breakapart fluorescence in situ hybridization positivity for detection of ALK-rearranged non-small cell lung cancer
There are advantages and disadvantages to using the breakapart FISH assay. Breakapart FISH is the assay used to identify patients with ALK-rearranged NSCLC to enroll them prospectively into crizotinib trials and thus is the only assay validated to correspond with the response to crizotinib. Breakapart FISH does not depend on knowing all the fusion partners to ALK since some of the fusion partners to ALK in NSCLC may remain to be discovered. Breakapart FISH can be performed on formalin-fixed paraffin-embedded tissue, which is how the vast majority of lung cancer tissue is processed. On the other hand, the breakapart signal pattern resulting from intrachromosomal deletions and inversion events occurring in the setting of polysomy typical of lung cancer is subtle and can easily be missed. Thus, to interpret the ALK breakapart FISH assay requires experience, patience, precise measurements (>2 signal diameter separation), and technical expertise. Furthermore, breakapart FISH assay is expensive. Therefore, breakapart FISH is unlikely to be the preferred method to screen for ALK rearrangement of NSCLC in routine surgical pathology practice, thereby limiting the utility of breakapart FISH assay as a screening test.
A second assay to detect ALK rearrangement is reverse transcriptase-PCR.Citation35 Reverse transcriptase-PCR is relatively easy to perform in most laboratories worldwide. Second, the fusion partners to ALK must be known. Knowledge of the fusion partners will be important in the future to subdivide ALK-rearranged NSCLC into different fusion partners in terms of their clinicopathologic characteristics and natural history and potential response to ALK inhibitors. The disadvantage of reverse transcriptase-PCR is that RNA may not be well preserved in formalin-fixed paraffin-embedded tissues to allow a successful reverse transcriptase-PCR reaction. More importantly, in order not to miss any ALK rearrangement, all the know fusion partners for ALK have to be known in order for the PCR reaction to include primers for all the ALK fusion genes. Finally, reverse transcriptase-PCR requires more tumor tissue material than breakapart FISH and, unfortunately, in an era of molecularly targeted lung cancer treatment, the amount of tumor biopsied for diagnosis is still relatively small and frequently inadequate for molecular profiling.
A third screening test for ALK-rearranged NSCLC is immunohistochemistry (IHC). The ALK protein is generally not expressed in most normal tissues and its presence in a tumor specimen could be sufficiently abnormal as to indicate an oncogenic role in those cells. Currently, a commercially available ALK1 antibody (Dako, Glostrup, Denmark) is being used to detect ALK rearrangement in anaplastic large cell lymphoma. However, expression of ALK protein in ALK-rearranged NSCLC is at least fivefold lower than ALK protein expression in anaplastic large cell lymphoma, likely due to weaker transcriptional activity of the promoter-enhancer region of EML4 that drives the expression of EML4-ALK compared with that of the NPM promoter involved in the NPM-ALK fusion protein in anaplastic large cell lymphoma.Citation36 It has been demonstrated that in order to increase the sensitivity of ALK1 antibodies in detecting ALK rearrangement in NSCLC, the increased titer of ALK1 used leads to nonspecific staining of nontumor tissue, leading to decreased specificity (high false positive rate). Alternatively, in conditions that allow for 100% specificity (no false positives), the detection rate of ALK1 is greatly compromised (32%).Citation36 Several monoclonal antibodies are in development. D5F3 (Cell Signaling Technology, Beverly, MA), a newer and more specific antibody to ALK, was shown to have 100% sensitivity and 99% specificity, with a positive predictive value of 96% and negative predictive value of 100%, with excellent interobserver reproducibility of 94% (κ = 0.94).Citation36 In the same study, ALK1 only had a sensitivity of 67%, specificity of 97%, a positive predictive value of 78%, a negative predictive value of 95%, and κ = 0.74.Citation36
Two separate studies have compared IHC using ALK1Citation37 with another new antibody in development 5A4 (Novocastra, Newcastle, UK),Citation38 with the breakapart FISH assay (). The results indicated that while IHC 3+ or IHC 0 from both antibodies had perfect concordance with FISH, a few IHC 2+ or IHC 1+ patients were also positive for FISH. Thus, algorithms have to be developed in these intermediate IHC staining cases so that all ALK-positive patients can be identified because of the low prevalence of ALK rearrangement in NSCLC, even a few missed cases represent a significant proportion of all ALK rearranged NSCLC. Comparison of IHC, reverse transcription PCR, and FISH has been performed in anaplastic large cell lymphoma, and IHC with ALK performs similarly to FISH, while reverse transcription PCR is not as sensitive.Citation39 Thus, when these newer ALK antibodies become commercially available, they will likely allow mass screening for ALK rearrangement in NSCLC because IHC is easy to perform and interpret and is cost-effective. However, validation studies of IHC using the newer antibodies compared with FISH similar to what has been performed for anaplastic large cell lymphomaCitation39 will have to be performed before IHC is officially considered as a companion diagnostic assay for ALK-rearranged NSCLC.
Table 5 Comparison of immunohistochemistry using various ALK antibodies and fluorescence in situ hybridization
Ongoing clinical development of crizotinib
Phase I trials
COG-ADVL0912 (NCT00939770) is an ongoing Children’s Oncology Group Phase I/II study of crizotinib to determine the maximum dose that is safe and tolerable and to obtain preliminary information on clinical activity in pediatric patients with relapsed/refractory solid tumors, primarily central nervous system tumors and anaplastic large cell lymphoma. A8081002 (NCT00965731) is a combination study of erlotinib in combination with crizotinib in NSCLC with the intention of randomizing NSCLC patients to erlotinib ± crizotinib in advanced NSCLC once the recommended Phase II dose level of the erlotinib-crizotinib combination is achieved. A8081006 (NCT01121575) is a similarly designed trial combining crizotinib with dacomitinib (PF0299804), a pan HER inhibitor, in NSCLC.
Phase II
Crizotinib has been reported to result in excellent clinical benefit and response in inflammatory myofibroblastic tumorsCitation40 and anaplastic large cell lymphomaCitation41 in case report format. PROFILE 1013 (NCT01121588) is a Phase II trial that will further study the safety and potential clinical activity of crizotinib in patients with ALK rearrangement or mutation-positive tumors other than NSCLC, such as anaplastic large cell lymphoma, inflammatory myofibroblastic tumors, and neuroblastoma in a systemic manner. The primary endpoint of PROFILE 1013 is overall response rate.
Other targets of crizotinib
Crizotinib was initially developed as a MET inhibitor. Once the maximal tolerated dose has been reached and the recommended Phase II dose determined from the first part of A8081001, it is planned to enroll 25 patients with tumors that harbor MET amplification or mutations. Crizotinib has demonstrated clinical activity in MET-amplified NSCLC,Citation42 MET-amplified gastroesophageal carcinoma,Citation43 and MET-amplified glioblastoma.Citation44 Additionally, because of the evolutionary relatedness between ALK and ROS receptor tyrosine kinases,Citation45 crizotinib has also been shown in vitro to be a potential ROS inhibitor.Citation46 The current amendment to A8081001 allows for screening and testing of crizotinib in tumors with ROS rearrangement.
Future directions and newer agents
Resistance to tyrosine inhibitors invariably develops, and usually involves development of a gatekeeper mutation in the kinase domain, such as T315I in chronic myelogenous leukemia, and T790M in EGFR-mutated NSCLC.Citation47 Similarly, a patient developed resistance to crizotinib after 5 months of treatment and molecular analyses revealed the resistant tumor harbored two acquired but independent mutations within the kinase domain of EML4-ALK, C1156Y, and the gatekeeper mutation L1196M. BaF3 cells engineered to express these two EML4-ALK mutations indicated that these mutants were resistant to crizotinib.Citation48 In vitro assays have also identified the L1196M gatekeeper mutation to be the major mechanism of resistance to crizotinib.Citation49 Of note, amplification of translocation in ALK is the intermediate step of the resistance process based on the in vitro assays, but so far has not been observed from rebiopsy of tumors from patients who progressed on crizotinib.Citation47 A separate mutation, F1174L, has been described in a patient with an inflammatory myofibroblastic tumor and RANPB2-ALK translocation who progressed on crizotinib.Citation50
In light of the identification of the gatekeeper mutation before crizotinib is approved, many “second-generation” ALK inhibitors are now or soon to be entered into early clinical development to try to overcome crizotinib resistance, especially the L1196M gatekeeper mutation. One of the best characterized compounds is AP26113 (ARIAD Pharmaceuticals, Cambridge, MA), a small-molecule competitive inhibitor of the ALK tyrosine kinase domain. Of note, AP26113 is not only ten times more active than crizotinib in inhibiting wild-type ALK but is a hundred times more potent than crizotinib in inhibiting the L1196M gatekeeper mutation. Importantly, AP26113 inhibits ALK L1196M at the same subnanomolar concentration as it inhibits wild-type ALK kinase ().Citation49 Another second-generation ALK inhibitor, CH542802 (Chugai Pharmaceutical Co Ltd, Tokyo, Japan), is also reported to have strong inhibitory activity in both wild-type ALK and L1196M gatekeeper mutation-harboring cell lines.Citation51 However, to date, the number of patients reported with identified resistance mechanisms to crizotinib remains extremely small (less than four so far) and therefore it is premature to say that the L1196M gate-keeper mutation will be the most important mechanism of acquired resistance to crizotinib than other mechanisms that can bypass the ALK signaling pathway. Indeed, in vitro cell line experiments have demonstrated that activating the EGFR signaling pathway can confer resistance to crizotinib, and that a small subset of ALK-rearranged NSCLC also harbor activating EGFR mutations. This finding suggests that some patients with ALK-rearranged NSCLC may have intrinsic resistance to crizotinib.Citation52 Complicating the matter ever further, another study has shown that different ALK mutations confer differential resistance to structurally diverse ALK inhibitors. Therefore, it is of utmost importance to rebiopsy to allow repeat molecular profiling of ALK-rearranged tumors.Citation53
Table 6 Comparison of Ki between crizotinib and AP26113 for wild-type ALK and ALK with LII96M gate-keeper mutationCitation49
Another important pathway involved in EML4-ALK signaling is the Hsp family. EML4-ALK fusion protein is an aberrantly synthesized protein residing in the cytoplasm. Hsp are chaperone proteins involved in stabilizing the EML4-ALK fusion protein in the cytoplasm and sorting the substrates of EML4-ALK. Hsp90 is one of the major members of the Hsp family.Citation54 EML4-ALK is an Hsp90 client protein and coprecipitates with other members of the Hsp family, including Hsp90.Citation55 The association between Hsp90 and EML4-ALK can be disrupted by an Hsp90 inhibitor. Inhibition of Hsp90 leads to rapid degradation of EML4-ALK within 3 hours, together with downstream signaling pathways in ALK-dependent H3122 NSCLC cell lines.Citation55 Moreover, addition of Hsp90 inhibitors resulted in cell death in an ALK-dependent cell line, tumor regression in an EML4-ALK xenograft, and regression of lung adenocarcinoma in the EML4-ALK transgenic mouse. Most importantly, Hsp90 inhibitors lead to cell death in both crizotinib-sensitive and crizotinib-resistant ALK-dependent cell lines,Citation49,Citation55 including NSCLC cell lines harboring the ALK L1196M gatekeeper mutationCitation49 which provides another alternate pathway to overcome resistance to crizotinib. Indeed, Hsp90 inhibitors have demonstrated preliminary clinical activity in patients with ALK-rearranged NSCLC.Citation56,Citation57
Will crizotinib suffer the same fate as gefitinib in the US?
I raise this question not because crizotinib is an ineffective drug for patients with ALK-rearranged NSCLC. In oncologic drug development, to show clinical benefit, an investigational drug has to perform better in terms of survival or side effect profile than the current standard of care in order for the investigational drug to be approved. Crizotinib is one of the first oncogenic drugs approved for a specific molecular-enriched patient population based on its activity in that population. Crizotinib was conditionally approved in the US on August 26, 2011, based on response rates of 50% from PROFILE 1005 and 61% from the A8081001 trial. The approval language states “... This approval is based on response rate. There are no data available demonstrating improvement in patient reported outcomes or survival with XALKORI”.Citation30 This approval does not specify what line of crizotinib treatment should be used, meaning crizotinib could be used for first-line treatment of patients with advanced ALK-rearranged NSCLC. However, data from only 15 patients with ALK-rearranged NSCLC who received crizotinib as first-line treatment for their disease formed the clinical data submitted to the FDA (). Gefitinib, an EGFR tyrosine kinase inhibitor, was conditionally approved in the US in 2003 based also on response rates from two Phase II trials.Citation58 However, gefitinib did not achieve significant improvement in overall survival when compared with placebo in second-line treatment in the randomized Phase III ISEL (IRESSA® Survival Evaluation in Lung Cancer) study (hazard ratio [HR] 0.89, P = 0.087),Citation59 which resulted essentially in the withdrawal of gefitinib from the US market. In retrospect, the major reason why the ISEL trial failed is because knowledge of the optimal target of the EGFR tyrosine kinase inhibitor was not well understood at the time of design of the ISEL study. Indeed, subgroup analysis of ISEL revealed that gefitinib was beneficial in Asians, with significant improvement in overall survival when compared with placebo (HR 0.66; P = 0.010),Citation60 especially in never-smokers, females, and adenocarcinoma cases in the Asian subgroup, in which most patients are known to harbor activating EGFR mutations.
ALK-rearranged NSCLC is a molecularly defined sub-group of NSCLC, but remains quite heterogeneous because there are many EML4-ALK fusion proteins and other fusion partners for ALK.Citation17,Citation18 Secondly, there is now emerging evidence that ALK-rearranged NSCLC responds well to pemetrexed, which may be related to a lower level of thymidylate synthase, a target of pemetrexed, in the ALK-rearranged lung tumor.Citation61,Citation62 The frontline PROFILE 1014 (NCT01154140) trial is comparing pemetrexed-platinum combination therapy with crizotinib alone in untreated ALK-rearranged NSCLC. PROFILE 1014 is designed to detect a 50% improvement in progression-free survival from 6 to 9 months, with 85% power and a one-sided significance level of 0.025.
Maintenance pemetrexed is not allowed in PROFILE 1007, although the recently presented PARAMOUNT pemetrexed maintenance trial indicated that there is significant improvement in progression-free survival with continuous pemetrexed maintenance after four cycles of pemetrexed/cisplatin compared with best supportive care.Citation63 The progression-free survival from the start of cisplatin/pemetrexed to the end of pemetrexed maintenance was 6.9 months.Citation63 It is possible that patients with ALK-rearranged NSCLC may enjoy a long progression-free survival even without pemetrexed maintenance, and the trial may not have been sufficiently powered to detect a statistical significance in progression-free survival. Because PFS is chosen as the primary endpoint for the front line trial, RECIST criteria will be used to determine “progression”. Disease progression by RECIST is usually defined as either the appearance of new metastatic lesions or cumulative in tumor measurement > 20% from the smallest measurement achieved or a combination of both. I have listed three potential scenarios where cumulative tumor measurement increased by 33% (thus satisfying RECIST criteria for disease progression). Scenario one, where there is no response to treatment and the tumor continues to grow (). Scenario two, where there is some shrinkage of the tumor in response to treatment but not enough to stratify RECIST criteria for partial response and then the tumor grows back to a pre-treatment baseline (). The scenario is characteristic of a response to chemotherapy. In scenario three there is a dramatic response to treatment but then the tumor starts to slowly grow back. However, it is still much smaller than pre-treatment baseline (). Scenario three is characteristic of a highly effective targeted therapy such as crizotinib. Therefore, patients whose disease continues to progress while on crizotinib may still continue to derive clinical benefit and thus using progression-free survival and RECIST criteria may not truly reflect the beneficial effect of a highly effective targeted therapy.
Figure 4 Three scenarios where there is a 33% progression (as defined by Response Evaluation in Solid Tumor [RECIST]) can occur when the tumor is still significantly smaller than from baseline, especially with molecular targeted therapy.
![Figure 4 Three scenarios where there is a 33% progression (as defined by Response Evaluation in Solid Tumor [RECIST]) can occur when the tumor is still significantly smaller than from baseline, especially with molecular targeted therapy.](/cms/asset/cef246f3-7334-4e00-990e-8dd298fca7a8/dddt_a_19045_f0004_c.jpg)
Conclusion
The identification of activating EGFR mutations in NSCLC heralded the molecularly targeted therapy era in NSCLC and led to successful demonstration of the superiority of EGFR tyrosine inhibitors over standard platinum-doublet chemotherapy as the first line treatment of NSCLC patients with EGFR activating mutations. The identification of resistance mechanisms of first-generation EGFR tyrosine inhibitors several years later led to the rational development of second-generation EGFR tyrosine inhibitors.Citation47 These principles have guided the development of crizotinib ever since ALK rearrangement was first reported in NSCLC in August 2007, and have led to the identification of a gatekeeper mutation in EML4-ALK that preceded the US approval of crizotinib by months. The discovery of ALK mutations is now guiding the rapid development of second-generation ALK inhibitors and Hsp90 inhibitors. This paradigm will likely guide the development of molecularly targeted therapy in NSCLC patients against other potential driver mutations, such as the V600E BRAF mutation.Citation64,Citation65
Finally, one of the important findings in the era of molecular targeted therapy is that the tumor underwent rapid and dramatic shrinkage within a short period of time.Citation28 The current RECIST criteria used in all oncology trials was designed in the era of chemotherapy when massive tumor shrinkage was not common. However, one of the definitions of progression by RECIST is a 20% increase in cumulative tumor diameter from the smallest aggregate measurement. Thus, the greater the amount of tumor shrinkage a targeted therapy achieves, the less the tumor has to regrow to be defined as disease progression by RECIST. Thus, in many cases of molecularly targeted therapy at the time of progression, the tumor is still significantly smaller than baseline (). Therefore, the RECIST criteria may not be the optimal criteria to assess a highly efficacious targeted therapy when progression-free survival is the primary endpoint.
In the future, clinical studies of molecularly targeted therapy should allow patients to continue on treatment if there is “continual clinical benefit” and the pace of progression is measured, which was the case in A8081001 and PROFILE 1005. Indeed, at least 15 patients in the A8081001 study continued on treatment after progression defined by RECIST because of “continual clinical benefit”.Citation27
Disclosures
The author’s institution, University of California Irvine, received research support from Pfizer for conducting the A8081001 and PROFILE 1005 trials. The author has received honoraria from Pfizer for serving on crizotinib advisory boards.
References
- LynchTJBellDWSordellaRActivating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinibN Engl J Med20043502129213915118073
- PaezJGJannePALeeJCEGFR mutations in lung cancer: correlation with clinical response to gefitinib therapyScience20043041497150015118125
- PaoWMillerVZakowskiMEGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinibProc Natl Acad Sci U S A2004101133061331115329413
- CohenMHWilliamsGASridharaRUnited States Food and Drug Administration drug approval summary: gefitinib (ZD1839; Iressa) tabletsClin Cancer Res2004101212121814977817
- JohnsonJRCohenMSridharaRApproval summary for erlotinib for treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of at least one prior chemotherapy regimenClin Cancer Res2005116414642116166415
- MokTSWuYLThongprasertSGefitinib or carboplatin-paclitaxel in pulmonary adenocarcinomaN Engl J Med200936194795719692680
- FukuokaMWuYLThongprasertSBiomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS)J Clin Oncol2011292866287421670455
- MitsudomiTMoritaSYatabeYGefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trialLancet Oncol20101112112820022809
- MaemondoMInoueAKobayashiKGefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFRN Engl J Med20103622380238820573926
- ZhouCWuY-LChenGErlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small cell lung cancer (OPTIMAL, CTONG-0802): a multi-centre, open-label, randomized phase 3 studyLancet Oncol20111273574221783417
- RosellRGervaisRVergnenegreAErlotinib versus chemotherapy (CT) in advanced non-small cell lung cancer (NSCLC) patients (p) with epidermal growth factor receptor (EGFR) mutations: Interim results of the European Erlotinib Versus Chemotherapy (EURTAC) phase III randomized trialJ Clin Oncol.201129Suppl Abstr 7503
- TohCKGaoFLimWTNever-smokers with lung cancer: epidemiologic evidence of a distinct disease entityJ Clin Oncol2006242245225116710022
- MorrisSWKirsteinMNValentineMBFusion of a kinase gene ALK, to a nuclear gene, NPM, in non-Hodgkin’s lymphomaScience1994263128112848122112
- ChiarieRVoenaCAmbrogioCThe anaplastic lymphoma kinase in the pathogenesis of cancerNat Rev Cancer20088112318097461
- SodaMChoiYLEnomotoMIdentification of the transforming EML4-ALK fusion gene in non-small-cell lung cancerNature200744856156617625570
- RikovaKGuoAZengQGlobal survey of phosphotyrosine signaling identifies oncogenic kinase in lung cancerCell20071311190120318083107
- HornLPaoWEML4-ALK: Honing in on a new target in non-small-cell lung cancerJ Clin Oncol2009264232423519667260
- SasakiTRodigSJChirieacLRThe biology and treatment of EML4-ALK non-small cell lung cancerEur J Cancer2010461773178020418096
- SodaMTakadaSTakeuchKA mouse model for EML4-ALK positive lung cancerProc Natl Acad Sci U S A2008105198931989719064915
- ShawATSolomonBTargeting anaplastic lymphoma kinase pathway in lung cancerClin Cancer Res2011172081208621288922
- CuiJJTran-DubeMShenHStructure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK)J Med Chem2011546342636321812414
- ChristensenJGZouHYArangoMECytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphomaMol Cancer Ther200763314332218089725
- KwakELCamidgeDRClarkJClinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066J Clin Oncol.20092715 Suppl Abstr 3509
- TanWWilnerKDBangYPharmacokinetics (PK) of PF-02341066, a dual ALK/MET inhibitor after multiple oral doses to advanced cancer patientsJ Clin Oncol201028Suppl 15 Abstr 2596
- CostaDBKobayashiSPandyaSSCSF concentration of the anaplastic lymphoma kinase inhibitor crizotinibJ Clin Oncol.201129e443e44521422405
- KwakELBangY-JCamidgeDRAnaplastic lymphoma kinase inhibition in non-small-cell lung cancerN Engl J Med20103631693170320979469
- CamidgeDRBangYKwakELProgression-free survival (PFS) from a phase I study of crizotinib (PF-02341066) in patients with ALK-positive non-small cell lung cancer (NSCLC)J Clin Oncol.201129Suppl Abstr 2501
- OuS-HIBazhenovaLCamidgeDRRapid and dramatic radiographic and clinical response to an ALK inhibitor (crizotinib, PF02341066) in an ALK translocation-positive patient with non-small cell lung cancerJ Thorac Oncol201052044204621102269
- CrinòLKimDRielyGJInitial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): PROFILE 1005J Clin Oncol201129Suppl Abstr 7514
- Xalkori [crizotinib, package insert]New York, NYPfizer Inc2011
- NickensDTanWWilnerKPharmacokinetic/pharmacodynamic evaluation of the concentration-QTc relationship of crizotinib (PF-02341066), an anaplastic lymphoma kinase and c-MET/hepatocyte growth factor receptor dual inhibitor administered orally to patients with advanced cancerPoster presented at the 101st annual meeting of the American Association for Cancer ResearchWashington, DCApril 17–21, 2010 Abstr 1673
- OuS-HIAzadaMDyJAsymptomatic profound sinus brady-cardia (heart rate <45) in non-small cell lung cancer patients treated with crizotinibJ Thorac Oncol201162135213722088989
- OuSISalgiaRClarkJWComparison of crizotinib (PF-02341066) pharmacokinetics between Asian and non-Asian patients with advanced malignanciesJ Thorac Oncol20105Suppl 5S382
- CamidgeDRKonoSAFlaccoAOptimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatmentClin Cancer Res2010165581559021062932
- TakeuchiKChoiYLSodaMMultiplex reverse transcription-PCR screening for EML4-ALK fusion transcriptsClin Cancer Res2008146618662418927303
- Mino-KenudsonMChirieacLRLawKA novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistryClin Cancer Res2010161561157120179225
- YiESBolandJMMaleszewskiJJCorrelation of IHC and FISH for ALK gene rearrangement in non-small cell lung carcinoma. IHC score algorithm for FISHJ Thorac Oncol2011645946521278610
- PaikJHChoeGKimHScreening of anaplastic lymphoma kinase rearrangement by immunohistochemistry in non-small cell lung cancer. Correlation with fluorescence in situ hybridizationJ Thorac Oncol2011646647221258247
- CataldoKAJalalSMLawMEDetection of t(2;5) in anaplastic large cell lymphoma: comparison of immunohistochemical studies, FISH, and RT-PCR in paraffin-embedded tissueAm J Surg Pathol1999231386139210555007
- ButrynskiJED’AdamoDRHornickJLCrizotinib in ALK-rearranged inflammatory myofibroblastic tumorN Engl J Med20103631727173320979472
- Gambacorti-PasseriniCMessaCCrizotinib in anaplastic large-cell lymphomaN Engl J Med201136477577621345110
- OuS-HIKwakELSiwak-TappCActivity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplificationJ Thorac Oncol2011694294621623265
- LennerzJKEwakELMichaelMIdentification of small and lethal subgroup of esophagogastric adenocarcinoma with evidence of response to crizotinib by MET amplificationJ Clin Oncol.201129Suppl Abstr 4130
- ChiASKwakELClarkJWClinical improvement and rapid radiographic regression induced by a MET inhibitor in a patient with MET-amplified glioblastomaJ Clin Oncol.201129Suppl Abstr 2072
- RobinsonDRWuY-MLinS-FThe protein tyrosine receptor kinase family of the human genomeOncogene2000195548555711114734
- BergethonKShawATOuS-HIROS1 rearrangements define a unique class of lung cancersJ Clin Oncol2012 In press
- PaoWChmieleckiJRational, biologically based treatment of EGFR-mutant non-small-cell lung cancerNat Rev Cancer20101076077420966921
- ChoiYLSodaMYamashitaYEML4-ALK mutations in lung cancer that confer resistance to ALK inhibitorsN Engl J Med20103631734173920979473
- KatayamaRKhanTMBenesCTherapeutic strategies to over-come crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALKProc Natl Acad Sci U S A20111087535754021502504
- SasakiTOkudaKZhangWThe neuroblastoma associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK translocated cancersCancer Res201070100381004321030459
- SakamotoHTsukaguchiTHiroshimaSCH542802, a selective ALK inhibitor capable of blocking the resistance gatekeeper mutantCancer Cell20111967969021575866
- SasakiTKoivunenJOginoAA novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitorsCancer Res2011716051606021791641
- HeuckmannJMHolzelMSosMLALK mutations conferring differential resistance to structurally diverse ALK inhibitorsClin Cancer Res September 262011 [Epub ahead of print.]
- TrapelJMollapourMGiacconeGTargeting the dynamic HSP90 complex in cancerNat Rev Cancer20101053754920651736
- NormantEPaezGWestKAThe Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC modelsOncogene2011302581258621258415
- SequistLVGettingerSNeilNNActivity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancerJ Clin Oncol2010284953496020940188
- WongKKoczywasMGoldmanJWAn open-label phase II study of the Hsp90 inhibitor ganetespib (STA-9090) as monotherapy in patients with advanced non-small cell lung cancer (NSCLC)J Clin Oncol201129Suppl Abstr 7500
- CohenMHWilliamsGASridharaRUnited States Food and Drug Administration drug approval summary: gefitinib (ZD1839; Iressa) tabletsClin Cancer Res2004101212121814977817
- ThatcherNChangAParikhPGefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer)Lancet20053661527153716257339
- ChangAParikhPThongprasertSGefitinib (IRESSA) in patients of Asian origin with refractory advanced non-small cell lung cancer: subset analysis from the ISEL studyJ Thorac Oncol2006184785517409969
- CamidgeDRKonoSALuXAnaplastic lymphoma kinase gene rearrangements in non-small cell lung cancer are associated with prolonged progression-free survival on pemetrexedJ Thorac Oncol2011677478021336183
- LeeJ-OKimTMLeeS-HAnaplastic lymphoma kinase translocation. A predictive biomarker of pemetrexed in patients with non-small cell lung cancerJ Thorac Oncol201161474148021642865
- Paz-AresLGDe MarinisFDediuMPARAMOUNT: Phase III study of maintenance pemetrexed (pem) plus best supportive care (BSC) versus placebo plus BSC immediately following induction treatment with pem plus cisplatin for advanced nonsquamous non-small cell lung cancer (NSCLC)J Clin Oncol201129Suppl Abstr CRA7510
- PaikPKArcilaMEFaraMClinical characteristics of patients with lung adenocarcinomas harboring BRAF mutationsJ Clin Oncol2011292046205021483012
- MarchettiAFelicioniLMalatestaSClinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutationsJ Clin Oncol2011293574257921825258