
Abstract
Objective
Angiotensin II (Ang II) is known to contribute to the pathogenesis of heart failure by eliciting cardiac remodeling and dysfunction. The glucagon-like peptide-1 (GLP-1) has been shown to exert cardioprotective effects in animals and patients. This study investigates whether GLP-1 receptor agonist liraglutide inhibits abdominal aortic constriction (AAC)-induced cardiac fibrosis and dysfunction through blocking Ang II type 1 receptor (AT1R) signaling.
Methods
Sprague-Dawley rats were subjected to sham operation and abdominal aortic banding procedure for 16 weeks. In treated rats, liraglutide (0.3 mg/kg) was subcutaneously injected twice daily or telmisartan (10 mg/kg/day), the AT1R blocker, was administered by gastric gavage.
Results
Relative to the animals with AAC, liraglutide reduced protein level of the AT1R and upregulated the AT2R, as evidenced by reduced ratio of AT1R/AT2R (0.59±0.04 vs. 0.91±0.06, p<0.05). Furthermore, the expression of angiotensin converting enzyme 2 was upregulated, tissue levels of malondialdehyde and B-type natriuretic peptide were reduced, and superoxide dismutase activity was increased. Along with a reduction in HW/BW ratio, cardiomyocyte hypertrophy was inhibited. In coincidence with these changes, liraglutide significantly decreased the populations of macrophages and myofibroblasts in the myocardium, which were accompanied by reduced protein levels of transforming growth factor beta1, Smad2/3/4, and upregulated smad7. The synthesis of collagen I and III was inhibited and collagen-rich fibrosis was attenuated. Consistent with these findings, cardiac systolic function was preserved, as shown by increased left ventricular systolic pressure (110±5 vs. 99±2 mmHg, p<0.05), ejection fraction (83%±2% vs. 69%±4%, p<0.05) and fraction shortening (49%±2% vs. 35%±3%, p<0.05). Treatment with telmisartan provided a comparable level of protection as compared with liraglutide in all the parameters measured.
Conclusion
Taken together, liraglutide ameliorates cardiac fibrosis and dysfunction, potentially via suppressing the AT1R-mediated events. These data indicate that liraglutide might be selected as an add-on drug to prevent the progression of heart failure.
Introduction
Maladaptive cardiac remodeling referring to molecular, cellular and interstitial alterations in the myocardium, is characterized by histopathological changes in the structure, shape, and function of the heart, and often leads to the development of heart failure.Citation1–Citation3 Animal studies and clinical observations have shown that cardiac remodeling exhibits progressive worsening of cardiac dysfunction with a higher risk of cardiovascular morbidity and mortality.Citation4–Citation6 Tissue fibrosis as a key component of cardiac remodeling occurs inevitably in the chronic phases of the different pathological conditions, including hypertension, myocardial infarction, inflammation, and hypertrophic cardiomyopathy.Citation6,Citation7
Angiotensin II (Ang II), the major bioactive peptide product of the renin-angiotensin-aldosterone system, plays an important pathophysiological role in the cardiovascular system, including blood pressure regulation, systemic inflammatory response, interstitial collagen deposition, and tissue fibrotic formation through its interaction with the type 1 receptor (AT1R).Citation8,Citation9 The inhibition of Ang II formation or the blockade of the AT1R activation with Ang II-converting enzyme inhibitors (ACEi) or AT1 receptor blocker (ARBs) has emerged as an appealing therapeutic approach to prevent cardiac dysfunction and heart failure by attenuating progression of cardiac remodeling.Citation10,Citation11 Our laboratory has had a longstanding interest in the areas of Ang II-induced cardiomyocyte hypertrophy, tissue fibrosis, and cardiac dysfunction. We have previously reported that pharmacological inhibition of the AT1R significantly reduces cardiac fibrosis and improves cardiac function.Citation12,Citation13 Although ACEi or ARBs have achieved good clinical results, according to the reports of recent years, there might be substantial side effects such as cough, angioedema, and hypotension.Citation14,Citation15 Therefore, adjunctive therapies to reduce cardiac remodeling and promote cardiac recovery through modulating the Ang II system still merits further investigation.
Glucagon-like peptide-1 (GLP-1) is an incretin peptide hormone produced in the intestinal endocrine L-cells, and plays an important role in glucose homeostasis to control food intake. Plasma half-life of endogenous GLP-1 is short, and can be rapidly degraded by the enzyme dipeptidyl peptidase-4.Citation16 Liraglutide is a long-acting GLP-1 receptor agonist, and has been widely used in the treatment of type 2 diabetes mellitus.Citation17 Recently, extensive research studies have shown that liraglutide not only possesses the glucose-dependent hypoglycemic effects, but also exerts the protection in variety of cardiovascular diseases independent of the mechanism of blood sugar regulation, such as preservation in vascular endothelial cell function, regulation in blood pressure, attenuation in cardiomyocyte injury, and improvement in heart function.Citation18–Citation20 Our laboratory has previously reported that liraglutide protects the heart against Ang II infusion-induced cardiac fibrosis via inhibiting the AT1R-activated signaling.Citation12,Citation13,Citation21 However, it is unknown whether liraglutide attenuates the pressure overload-induced heart failure. In the present study, we selected a rat model of transverse abdominal aortic constriction (AAC) to demonstrate the potential beneficial effects of liraglutide on cardiac fibrosis and heart function underlying mechanisms through modulating adverse actions of Ang II, including expression of the AT1R, activation of oxidative stress, migration of macrophages, proliferation of myofibroblasts, and deposition of collagens. To further support whether liraglutide exerted effects are mediated by blocking activation of the AT1R signaling, telmisartan, the AT1R antagonist, was selected for comparison. Cardiomyocyte hypertrophy and myocardial interstitial fibrosis were defined by morphological staining, and heart contractile function was assessed using two-dimensional echocardiography.
Materials and methods
Animals and AAC model
Male Sprague-Dawley rats weighting 200–250 g from 6 to 8 weeks were obtained from the Animal Laboratory Center of Shanxi Medical University, Taiyuan, People’s Republic of China. The procedures were in compliance with “The Guide for the Care of Use of Laboratory Animals” published by the US National Institute of Health (8th edition, revised, 2011)”. The experimental protocol was approved by the Experimental Animal Management Committee of Shanxi Medical University. Rats were housed under normal conditions (a 12 hrs light/dark cycle, temperature at 20–25°C, and relative humidity at 50–60%).
The rats were placed in a supine position on a surgery platform and an incision was made along the manubrium and midline of the abdomen. After the abdominal cavity was opened, the abdominal aorta was ligated using a 4–0 silk suture with a 7-gauge needle that is placed parallel to the aortic arch at a point of 1 cm above the left renal artery. The degree of AAC was controlled by withdrawing the needle under the suture. After the operation was completed, the penicillin was injected and animals were allowed to recover for 1 week before signing the experimental groups.
Experimental group and study protocol
The rats were randomly divided into four groups (n=6 in each group) after surgery: 1) Sham control: rats underwent the same surgical procedure without banding the aorta; 2) AAC: rats were subjected to AAC for 16 weeks; 3) liraglutide treatment: rats were received a subcutaneous injection of liraglutide (Novo Nordisk Pharma Ltd., Gladsaxe, Denmark) at a dose of 0.3 mg/kg twice daily after AAC; 4) telmisartan treatment: rats were administered telmisartan (Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA) via gastric gavage at a dose of 10 mg/kg/day after AAC. The doses selected for liraglutide and telmisartan were based on previous experiments.Citation12,Citation21
Measurement of the body weight and blood glucose
Rat body weight and blood glucose value were measured every 2 weeks. The rats were fasted overnight (14–17 hrs) before the blood glucose was measured. The blood was collected from the tail vein of the rats on the next day, and the level of blood glucose was measured with blood glucose test paper (Glucometer, Blood sugar paper, Johnson, USA).
Measurement of the lipid peroxidation level, antioxidant enzyme activity, N terminal pro B type natriuretic peptide
The hearts were homogenized in phosphate buffer. The level of malonaldehyde (MDA) in the left ventricle was measured as the index of lipid peroxidation with an MDA detection kit (Jiancheng Bioengineering Institute, Nanjing, People’s Republic of China). Superoxide dismutase (SOD) activity to represent the ability of trapping oxygen radicals was determined with a SOD detection kit (Jiancheng Bioengineering Institute). N-terminal pro-B-type natriuretic peptide (NT-pro-BNP) level in response to pressure overload were determined using a detection kit (Xitang Bioengineering Institute, Shanghai, China)
Determination of heart/body weight (HW/BW) ratio and myocyte sectional area (MSA)
At the end of the experiment, the rat heart was rapidly removed and cleaned in the ice saline. After drying, the heart/body weight (HW/BW) index was calculated as heart weight divided by body weight (mg/g tissue). The left ventricle was then divided into two parts: one part was frozen immediately in the liquid nitrogen and the other part was paraffin-embedded. The MSA of cardiomyocytes was measured after conventional HE staining. Six areas in a high-powered field were randomly selected from each tissue section with a digital camera through 20X objective lens under light microscopy. Thirty cardiomyocytes as having a visible nucleus and intact cellular membrane were selected for the measurement and analysis using image analysis software (Image J; NIH, Rockville, MD, USA).
Expression of AT1R, AT2R, TGF-β1, Smads, and collagens by Western blot assay
The protein contents of the AT1R and AT2R, TGF-β1, Smads, and collagens were determined by Western blot assay.Citation12,Citation21 In brief, the heart tissue samples from different groups were homogenized in a lysis buffer and the protein concentration was measured by the BCA Protein Assay reagent kit (Boster Biotech, Wuhan, People’s Republic of China). The protein was then boiled and loaded onto SDS-PAGE to perform a electrophoresis. The protein was then transferred from the gel to the nitrocellulose membrane. Membranes were subsequently exposed to one of the following antibodies: the rabbit anti-AT1 and AT2 receptor polyclonal antibodies (Santa Cruz Biotechnology Inc., Dallas, TX, USA), a mouse anti-TGF-β1 monoclonal antibody (Abcam, Inc. MA, USA), the rabbit anti-p-Smad2, p-Smad3, anti-Smad2, Smad3, Smad4 and Smad7 monoclonal antibodies (Cell Signaling Technology, MA, USA), the mouse anti-collagen type I and III monoclonal antibodies (Abcam). Appropriate secondary antibodies were used, and the antibody-antigen complexes in all membranes were detected by the ECL PLUS kit (Boster Biotech). Subsequently, the protein bands exposed by the UVP gel imaging system (UVP, MA, USA) were analyzed by gray value using image J software, and the protein gray value of β-actin was used as an internal parameter for protein loading control.
Immunohistochemical staining for ACE2, macrophages, myofibroblasts, and TGF-β1
The heart samples were cut into 4 μm thickness and immunohistochemical staining was performed to identify the expression of the ACE2, macrophages, myofibroblasts, and TGF-β1 in the myocardium as we reported previously.Citation12,Citation21 In brief, the tissue sections were deparaffinized in xylene, dehydrated in graded ethanol and stained using a rabbit polyclonal antibody against ACE2 (Abcam), a rabbit monoclonal antibody against macrophages (CD68; EMD Millipore, Billerica, MA, USA), a monoclonal antibody against alpha-smooth muscle actin (α-SMA, Abcam), and a mouse anti-TGF-β1 monoclonal antibody (Abcam), respectively. Bound antibodies were detected by horseradish peroxidase-conjugated corresponding IgG, and diaminobenzidine tetrahydrochloride was used to observe the presence and location of ACE2, macrophages, myofibroblasts, and TGF-β1. Five sections from the different animals in each group were selected for an image acquisition using Aperio Digital pathological scanning system (Vista, CA, USA). The mean optical density (MOD) in the expression of ACE2, TGF-β1, and the number of macrophages and myofibroblasts per high-powered field were determined using cytoplasmic v2 software (Vista).
Detection of tissue fibrosis by Masson’s trichrome staining
Masson’s trichrome staining was used to evaluate the interstitial collagen deposition in the myocardium and quantitatively analyzed by morphometry as previously reported.Citation12,Citation21 In brief, the tissue samples were sectioned to a thickness of 6 μm using a Microtome (Leica RM2135, Meyer Instruments, TX, USA). Six randomly selected high-powered fields per section were selected to determine the areas of positively stained collagen (ImageJ, NIH, MA, USA). The staining pattern by Masson’s trichrome turns collagen blue, nuclei black, and viable muscle fiber as red.
Evaluation of global cardiac function by echocardiography and hemodynamic analysis
At the end of the observation period, the rats were anesthetized with inhaled 1.5% isoflurane, and transthoracic echocardiography was performed using a 15s MHz linear transducer. Percent fraction shortening, ejection fraction (EF), and left ventricular internal dimension (LVIDd) were calculated using a two-dimensional guided M-mode ultrasound system (Vivid 7, GE Healthcare, Waukesha, WI, USA).Citation13 Over three consecutive cardiac cycles were averaged for measurements. For hemodynamic analysis, both the carotid arteries were cannulated with a polyethylene catheter connected to a Statham transducer to measure the mean carotid pressure via a BL-410 biological signal acquisition and processing system (Techman Software Co., Ltd., Chengdu, China). The polyethylene catheter within the right carotid artery was then inserted into the left ventricle to determine the cardiac performance, including heart rate, left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), and maximum positive (+dp/dtmax)/negative (-dp/dtmax) values of the first derivative of left ventricular pressure.Citation13
Statistical analysis
All data were showed as the mean±standard error using Prism v 7.01 analysis system (GraphPad Software Inc, La Jolla,CA, USA). A one-way ANOVA followed by Student–Newman–Keul’s post hoc test was used to analyze group differences in the band intensity of AT1R, AT2R, TGF-β1, Smads, the MOD in the expression of ACE2, TGF-β1, collagens, the number of macrophages and myofibroblasts accumulated in the myocardium. The value of p<0.05 was accepted as a statistical significance.
Results
Effects of liraglutide and telmisartan on blood sugar, oxidative stress, antioxidant enzyme, cardiac hypertrophy, and HW/BW ratio after AAC
The body weight and blood sugar were monitored every 2 weeks starting from the second week of the observation. As shown in , no statistically significant difference in these two parameters was found between the Sham and AAC groups. Treatment with liraglutide and telmisartan also did not alter the body weight and blood sugar during the course of the experiment.
Figure 1 Effects of liraglutide and telmisartan on body weight, blood sugar, MDA, SOD, NT-pro-BNP, and cardiac hypertrophy after abdominal aortic constriction (AAC). (A) body weight and blood glucose were measured among different groups. (B and C) Levels of MDA, SOD, and NT-pro-BNP activity in myocardial tissue were determined using Elisa kits. (D) MSA in series was measured morphometrically at a HPF (original magnification: 200; scale bars: 100 μm). (E) Heart/body weight ratio (HW/BW, mg/g) was calculated by weight. Values are mean±SEM (n=6/group). *p<0.05 AAC vs Sham; #p<0.05 liraglutide (Lira) or telmisartan (Telmi) vs AAC.
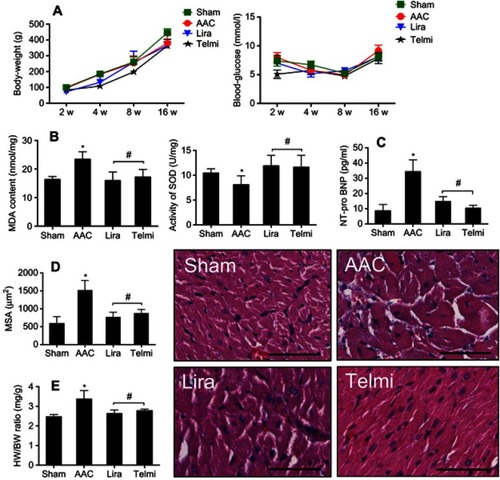
The content of MDA and the activity of SOD in the heart were used to estimate the levels of lipid peroxidation and tissue antioxidant ability. As shown in , AAC significantly increased MDA (23.4±1.3 vs 16.3±0.6 nmol/mg, p<0.05) and reduced SOD (8.1±0.8 vs 10.4±0.4 U/mg, p<0.05) compared with the Sham group, suggesting that chronic pressure overload increases cardiac oxidative status. Relative to the AAC group, administration of liraglutide or telmisartan reduced MDA level to 15.9±1.6 and 17.2±1.4 nmol/mg (all p<0.05 vs AAC group), respectively, and increased SOD enzyme activity by 11.9±1.2 and 11.6±1.1 U/mg, all p<0.05 vs AAC group, respectively, implying improved antioxidant ability of the heart.
The measurement of NT-pro-BNP levels were used to estimate the degree of heart failure and the calculation of MSA and HW/BW ratio was selected to evaluate cardiac hypertrophy. As shown in , AAC significantly increased NT-pro-BNP level at 16 weeks of observation relative to the Sham animals, which were reduced by liraglutide (14.8±1.3 pg/mL) and telmisartan (10.3±0.8 pg/mL) compared with AAC group (48.4±8.6 pg/mL), respectively, all p<0.05. Consistent with this change, the MSA in the AAC group increased significantly relative to the Sham group (1512±99 vs 586±98 µm2, p<0.05). Treatment with liraglutide or telmisartan comparatively reduced the MSA to 766±61 µm2 or 871±51 µm2 (all p<0.05 vs AAC group, ). The inhibitory effect of liraglutide or telmisartan on cardiac hypertrophy was further confirmed by the HW/BW ratio (). Relative to the Sham group, AAC group significantly increased the HW/BW ratio relative to the Sham group (3.38±0.17 vs 2.46±0.05, p<0.05), which was significantly reduced by liraglutide or telmisartan (2.63±0.09 and 2.77±0.05 vs AAC group, all p<0.05).
Effects of liraglutide and telmisartan on expression of AT1R, AT2R, AT1R/AT2R ratio, and ACE2 after AAC
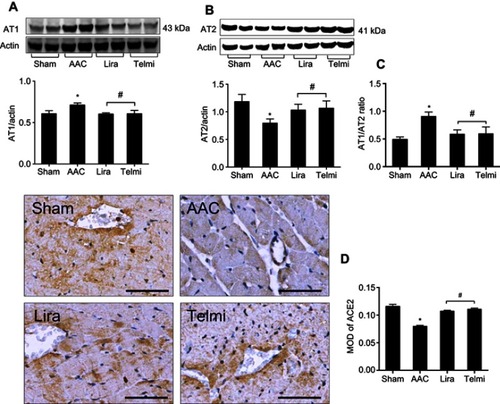
The expression of Ang II receptors was detected using Western blot assay. At the end of 16 weeks of AAC, relative to the Sham control, the protein level of the AT1R was significantly increased by 10.55±2.5% (), and the protein expression of the AT2R was decreased by 36.43±5.6% (), all p<0.05, whereas the expression patterns of AT1R and AT2R in the liraglutide and the telmisartan groups were reversed, as evidenced by a reduced AT1R/AT2R ratio (). Immunohistochemical staining showed that the expression of ACE2 in the perivascular and interstitial myocardium is significantly attenuated in the AAC group relative to the Sham group (). Relative to the AAC group, administration of liraglutide or telmisartan preserved the expression of ACE2 at week 16.
Figure 2 Effects of liraglutide and telmisartan on the expression of AT1R, AT2R, AT1R/AT2R ratio, and ACE2 after AAC. (A and B) Protein levels of AT1R and AT2R were detected by Western blot assay. (C) AT1R/AT2R ratio was calculated from the intensity of each individual bands. (D) ACE2 expression in the intermyocardium and intracardiac vessels was determined using immunohistochemical staining and calculated as mean optical density (MOD) in the tissue section (magnification: 200; scale bars: 100 μm). Values are mean±SEM (n=6/group). *p<0.05 AAC vs Sham; #p<0.05 Lira or Telmi vs AAC.
Effects of liraglutide and telmisartan on the macrophage infiltration and TGF-β1 expression
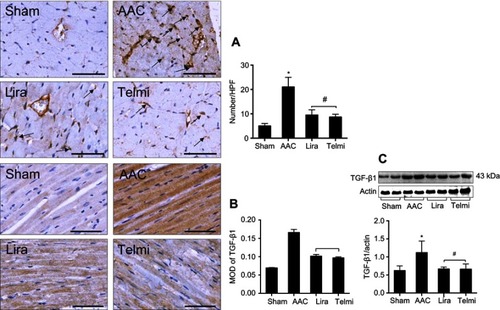
The detection in the accumulation of CD68 positive cells in the myocardium by immunohistochemistry was represented as an indicator of macrophage infiltration. The results showed that in the AAC group, the large number of macrophages is recruited in the peri-vascular region and interstitial myocardium at week 16 (), which was attenuated by treatment with liraglutide or telmisartan. Consistent with enhanced macrophage infiltration in the AAC group, the expression of TGF-β1 in the myocardium was also significantly increased (). The measurement of protein level of TGF-β1 by Western blot assay further confirmed the change in the TGF-β1 expression (). Administration of liraglutide or telmisartan during AAC comparatively reduced the infiltration of macrophages and expression of TGF-β1 at week 16 compared with the AAC group ().
Figure 3 Effects of liraglutide and telmisartan on the accumulation of macrophages and the expression TGF-β1. (A) The numbers of macrophages were determined by positively stained cells (arrows) using immunohistochemistry. (B) TGF-β1 expression in the intermyocardium was determined using immunohistochemistry and calculated as MOD in the tissue section (magnification: 200; scale bars: 100 μm). (C) Protein level of TGF-β1 was detected by Western blot assay. Values are mean±SEM (n=6/group). *p<0.05 AAC vs Sham; #p<0.05 Lira or Telmi vs AAC.
Effects of liraglutide and telmisartan on the myofibroblast proliferation and Smad expression
The detection in the number of α-SMA positive cells in the myocardium was indicated as a parameter to reflect the fibroblast proliferation. As shown in , relative to the Sham control, the abundant α-SMA positive myofibroblasts were identified by immunohistochemical staining following 16 weeks of AAC, predominantly located in the outside of the blood vessels in the myocardium. However, the appearance of α-SMA positive myofibroblasts at week 16 was inhibited by administration of liraglutide or telmisartan compared with the AAC group.
Figure 4 Effects of liraglutide and telmisartan on the proliferation of myofibroblasts and the expression of Smads. (A) The numbers of myofibroblasts were determined by α-SMA positive cells (arrows) using immunohistochemistry. (B) Phosphorylated and total protein levels of Smad2 and Smad3 were detected by Western blot assay. (C and D) Protein levels of Smad4 and Smad7 were detected by Western blot assay. Values are mean±SEM (n=6/group). *p<0.05 AAC vs Sham; #p<0.05 Lira or Telmi vs AAC.
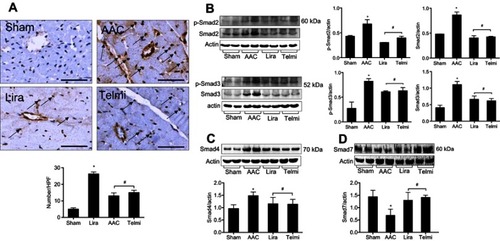
The disruption in the protein expression of Smad family members following TGF-β1 signaling is associated with myofibroblast activation and collagen deposition. Smad2 and Smad3 were barely phosphorylated in the Sham control. However, at 16 weeks of AAC as analyzed by Western blot assay, the phosphorylation of Smad2/3 was markedly enhanced, consistent with increased total protein levels of Smad2/3 (). Furthermore, the total protein level of Smad4 was significantly upregulated (), and total protein expression of Smad7 was downregulated () relative to the Sham group. However, all these changes in the protein expression of Smad family members induced by AAC were significantly reversed by administration of liraglutide or telmisartan (–).
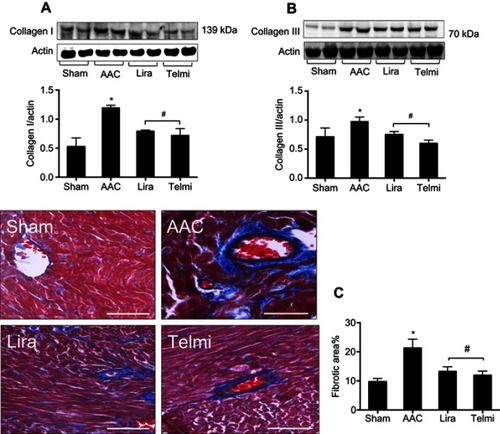
Effects of liraglutide and telmisartan on collagen synthesis and tissue fibrosis after AAC
Phosphorylation of Smads is responsible for induction of collagens. Along with enhanced phosphorylation of Smad2/3, the protein levels of collagens I and III examined by Western blot assay were significantly increased at 16 weeks of AAC compared with the Sham control (). Administration of liraglutide or telmisartan was equally effective in reducing the production of collagen I and III after the rats were treated with these drugs. Collagen deposition was evaluated using Masson’s trichrome staining. Consistent with upregulated expression of collagens I and III at 16 weeks of AAC, the region of deposited collagens as defined by percent collagen-rich areas was significantly expanded in the perivascular and interstitial myocardium. No newly synthesized collagens were detected in the Sham control throughout the experiment. The hearts treated with liraglutide or telmisartan showed a significant reduction in collagen deposition in both perivascular region and intermyocardium as defined by reduced collagen-rich area ().
Figure 5 Effects of liraglutide and telmisartan on the synthesis of collagens and tissue fibrosis after AAC. (A and B) Protein levels of collagens I and III were analyzed by Western blot assay. All bands were normalized by actin as illustrated in the bar graphs. (C) Interstitial and perivascular fibrosis in the myocardium was identified as collagen deposition shown as positive areas of blue staining using Masson’s trichrome method. All images were viewed under magnification 200 (scale bars: 100 μm). Values are mean±SEM (n=6/group). *p<0.05 AAC vs Sham; #p<0.05 Lira or Telmi vs AAC.
Effects of liraglutide and telmisartan on cardiac performance after AAC
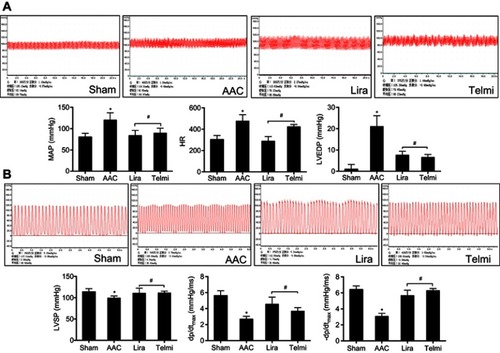
Two-dimensional ultrasound system was used to detect the cardiac function after AAC and interventions. As shown in , AAC caused a significant increase in the LVIDd relative to the Sham group at 16 weeks of the experiment (), suggesting left ventricular enlargement (dilation and hypertrophy), consistent with increased cardiomyocyte hypertrophy (). Furthermore, cardiac systolic function as assessed by LVEF () and LVFS () was reduced by AAC relative to the Sham group. Treatment with liraglutide or telmisartan enhanced cardiac performance compared with results in AAC group (). To confirm the echocardiography results, cardiac function was further analyzed by a BL-410 biological signal acquisition and processing system. At week 16, AAC significantly increased MAP, HR and LVEDP (), and reduced LVSP () relative to the Sham control. Furthermore, the maximal rate in increase of LV pressure (+dp/dtmax) and decrease of LV pressure (−dp/dtmax) were significantly reduced (). Treatment with liraglutide and telmisartan for 16 weeks effectively prevented the progression of cardiac dysfunction (), consistent with the inhibition of cardiac hypertrophy and fibrosis.
Figure 6 Effects of liraglutide and telmisartan on cardiac performance after AAC. (A) Cardiac global function was quantitatively assessed from two-dimensional echocardiographic short axis images among the different groups. (B) Left ventricular internal diameter end diastole (LVIDd) was measured at end-diastole. (C and D) Cardiac systolic function was determined by measuring ejection fraction (EF) and fraction shortening (FS). Values are mean±SEM (n=6/group). *p<0.05 AAC vs Sham; #p<0.05 Lira or Telmi vs AAC.
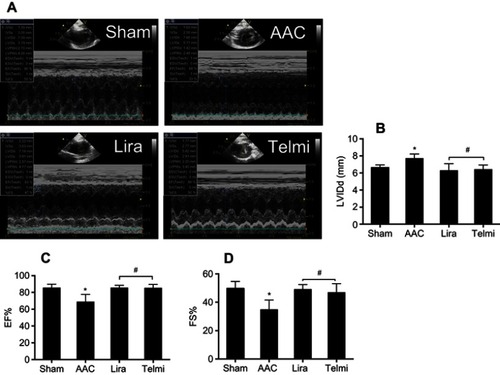
Figure 7 Effects of liraglutide and telmisartan on cardiac hemodynamics after AAC. (A) Representative tracing of the invasive cardiac hemodynamics recording among the different groups. MAP, HR, and left ventricular end-diastolic pressure (LVEDP) were obtained using a pressure transducer inserted into the left ventricle through the right carotid artery. (B) Left ventricular systolic pressure (LVSP) and ±dp/dtmax values of the first derivative of left ventricular pressure were obtained using a pressure transducer inserted into the left ventricle. Values are mean±SEM (n=6/group). *p<0.05 AAC vs Sham; #p<0.05 Lira or Telmi vs AAC.
Discussion
We have previously demonstrated that Ang II acts via its AT1R to increase blood pressure and cardiac fibrosis.Citation12,Citation13 In the present study, we evaluated the effect of exogenous administration of liraglutide on AAC-induced cardiac fibrosis and dysfunction, primarily focusing on the modulation on Ang II AT1R-mediated signaling. We found that treatment with liraglutide rebalances the pro- and anti-oxidative stress, including MDA, SOD, and NT-pro-BNP. Following the downregulation in AT1R expression and upregulation in the AT2R/ACE2 expression, the migration of macrophages and proliferation of myofibroblasts were inhibited, and TGF-β1/Smads-mediated collagen deposition was attenuated. In association with an inhibition of myocardial hypertrophy and HW/BW ratio, cardiac performance was improved, suggesting that stimulation in GLP-1 receptor is effective to reduce Ang II-mediated deleterious effects on the heart. Comparative protection with telmisartan confirmed that the cardioprotection by liraglutide is achieved through blocking the AT1R.Citation12
Published data have suggested that the balance in the protein expression between AT1R and AT2R plays a fundamental role in the development of cardiac fibrosis and progression of cardiac dysfunction.Citation22–Citation25 Pharmacological blockade of the AT1R is associated with the improvement of cardiac function and the attenuation of hypertension.Citation26,Citation27 Based on the results shown in , we found that there is a reciprocal relationship in the protein expression between the AT1R and the AT2R after AAC. Stimulation of the AT1R signaling perturbs the AT2R expression. We have previously shown that blockade of the AT1R after Ang II infusion is accompanied with an upregulation of the AT2R, suggesting that the AT1R interacts with the AT2R.Citation12,Citation13 The data presented in the present study further confirmed this reciprocal expression relationship between the AT1R and AT2R. Furthermore, activation of Ang II AT1R by AAC resulted in excessive production of reactive oxygen species, manifested by a significant elevation of MDA content and reduction of SOD activity. Comparative modulation in MDA and SOD between liraglutide and telmisartan suggested that inhibition of oxidant production with liraglutide is potentially achieved by blocking the AT1R. Followed by the AT2R internalization along with AT1R antagonism by liraglutide, macrophage migration, myofibroblast proliferation, and collagen deposition were inhibited. However, it is warranted to further check whether direct blockade with the AT2R-specific inhibitor or using an AT2R knockout model could interfere the pressure overload-induced cardiac remodeling and dysfunction.
ACE2 is an exopeptidase that catalyzes the conversion of Ang II to Ang (1–7). An increase in ACE2 activity is associated with a decrease in tissue level of Ang II.Citation28–Citation30 In contrast, loss of ACE2 leads to an increase in blood pressure, progressive ventricular dilatation, and poor systolic performance, which all are accompanied with an elevation in Ang II level in the heart.Citation31 It is known from the literature that at protein level, the expression of the AT1R and ACE can be modified by the AT1R blockade.Citation32 In this regard, we have previously reported that Ang II infusion-induced upregulation in AT1 receptor and reduction in ACE2 expression can be blocked by liraglutide and telmisartan.Citation12,Citation21 In the present study, AAC caused significant reduction in the expression of ACE2 that is also blocked by the AT1R blocker, further confirming an involvement of the AT1R in interfering ACE2 expression. Being consistent with downregulation of the AT1R with liraglutide,Citation12 ACE2 expression was enhanced, suggesting that this action may further enhance the inhibitory effects of the AT1R antagonism on AAC-induced cardiac remodeling and dysfunction. We speculate that an alteration in the balance of AT1R and ACE2 by liraglutide might protect the heart against Ang II mediated adverse effects through the beneficial modulation in response to Ang (1–7) generation. However, in future studies, it is interesting to further evaluate the efficacy of cardioprotection between liraglutide and agonist of ACE2–Ang-(1–7) axis.
Ventricular remodeling often occurs as a consequence of chronic pressure overload, resulting in progressive alterations in size, mass, and structure of the heart. The process is associated with poor prognosis due to ventricular dysfunction.Citation33 Animal studies and clinical implications of cardiac remodeling have provided strong evidence showing that stimulation of Ang II, production of reactive oxygen species, release of inflammatory mediators, and deposition of collagen are mostly implicated in the development of cardiac remodeling.Citation34 In the present study, cardiac remodeling is chartered by cardiac myocyte hypertrophy and tissue fibrosis. At 16 weeks of AAC, cardiomyocyte cross-sectional analysis by HE staining revealed a significant increase in cardiomyocyte size and Masson’s trichrome staining confirmed an abundant perivascular and interstitial collagen deposition. In the cellular level, AAC caused a significant increase in oxidative stress, migration of macrophages, and proliferation of myofibroblasts. In the protein level, expression of TGF-β1/Smads was upregulated and deposition of collagens was enhanced. However, these changes induced by AAC were significantly inhibited with 16 weeks of liraglutide treatment. Comparative protection between liraglutide and telmisartan confirmed the protection is mediated by modulating the AT1R. These results were consistent with our previous report showing that inhibition of TGFβ1 expression with an anti-oxidant compound suppresses fibroblast proliferation and interrupts phosphorylation of Smad2/3 to form a heterotrimeric complex with Smad4 whereas Smad7, an inhibitor of phosphorylation of Smad2/3, is upregulated.Citation13
Increased fibrosis of the ventricle can result in a failure to relax appropriately which impairs cardiac filling to lead to heart failure with reduced EF.Citation35–Citation37 We selected 16 weeks of experimental period for abdominal aortic stenosis because it can reflect the natural progression of cardiac dysfunction. We may miss a time window to reflect compensatory response, ie, concentric hypertrophy with normal EF in the early stage of AAC, but did find a reduction in EF following 16 weeks of period of chronic hemodynamic overload. Data clearly showed a significant ventricular dilatation and systolic dysfunction at this time point, as evidenced by increased HW/BW ratio, LVID, and MSA in series, suggesting the development of the eccentric hypertrophy. At the same time, LVEDP was increased, and LVSP, dp/dtmax, and EF were reduced, indicating that there is an increase in ventricular compliance and elevated filling pressure, often attributing to cardiac systolic dysfunction. Treatment with liraglutide for 16 weeks significantly reduced collagen deposition/fibrosis with less chamber dilatation and enhanced cardiac systolic function, suggesting that mechanisms of action underlying liraglutide protection under a condition of chronic pressure-overload are associated with an inhibition of tissue fibrosis.Citation38–Citation40 Although we cannot conclude whether liraglutide has a direct inotropic effect on the heart, the comparative inhibition in lipid peroxidation, hypertrophy, and fibrosis demonstrated between liraglutide and telmisartan in the present study suggest that these beneficial effects are primarily mediated by blockade of the AT1R.
Conclusion
In summary, we demonstrate that liraglutide protects the heart against the pressure overload with AAC induced cardiac injury in rat. The signaling pathways underlying inhibition of myocardial fibrosis and prevention of cardiac systolic dysfunction is primarily mediated by a downregulation of Ang II AT1R. Blockade of the AT1R with liraglutide, and also by application of telmisartan come along with upregulation of the AT2R and ACE2 as well as inhibition of the downstream fibrotic mediators (ie, TGFβ1 and Smad2/3). In line with these findings, cardiac hypertrophy and fibrosis were attenuated. Since cardiac fibrosis has been confirmed to be the final pathway leading to ventricular dysfunction and heart failure, liraglutide in addition to its effect on the blood glucose control might be considered as a second-line add-on therapy to protect the heart against fibrosis-induced heart failure in patients.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgment
This study was supported in part by grants from the Mercer University School of Medicine, the Medcen Community Health Foundation, Georgia, and the National Natural Science Foundation of China (81470436).
References
- Tham YK, Bernardo BC, Ooi JY, et al. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. 2015;89(9):1401–1438. doi:10.1007/s00204-015-1477-x25708889
- Jackson JD, Cotton SE, Bruce Wirta S, et al. Burden of heart failure on patients from China: results from a cross-sectional survey. Drug Des Devel Ther. 2018;12:1659–1668. doi:10.2147/DDDT.S148949
- Liehn EA, Postea O, Curaj A, Marx N. Repair after myocardial infarction between fantasy and reality. J Am Coll Cardiol. 2011;58(23):2357–2362. doi:10.1016/j.jacc.2011.08.03422115639
- Cohn JN, Ferrari R, Sharpe N; Behalf of an International Forum on Cardiac Remodeling. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol. 2000;35(3):569–582. doi:10.1016/s0735-1097(99)00630-010716457
- Ammar KA, Jacobsen SJ, Mahoney DW, et al. Prevalence and prognostic significance of heart failure stages: application of the American College of Cardiology/American Heart Association heart failure staging criteria in the community. Circulation. 2007;115(12):1563–1570. doi:10.1161/CIRCULATIONAHA.106.66681817353436
- Wu QQ, Xiao Y, Yuan Y, et al. Mechanisms contributing to cardiac remodeling. Clin Sci (Lond). 2017;131(18):2319–2345. doi:10.1042/CS2017116728842527
- Suthahar N, Meijers WC, Silljé HHW, de Boer RA. From inflammation to fibrosis-molecular and cellular mechanisms of myocardial tissue remodeling and perspectives on differential treatment opportunities. Curr Heart Fail Rep. 2017;14(4):235–250. doi:10.1007/s11897-017-0343-y28707261
- Bacmeister L, Schwarzl M, Warnke S, et al. Inflammation and fibrosis in murine models of heart failure. Basic Res Cardiol. 2019;114(3):19–54. doi:10.1007/s00395-019-0722-530887214
- Ames MK, Atkins CE, Pitt B. The renin-angiotensin-aldosterone system and its suppression. J Vet Intern Med. 2019;33(2):363–382. doi:10.1111/jvim.1545430806496
- Côté N, Mahmut A, Fournier D, et al. Angiotensin receptor blockers are associated with reduced fibrosis and interleukin-6 expression in calcific aortic valve disease. Pathobiology. 2014;81(1):15–24. doi:10.1159/00035089623969418
- Müller P, Kazakov A, Semenov A, et al. Ramipril and telmisartan exhibit differential effects in cardiac pressure overload-induced hypertrophy without an additional benefit of the combination of both drugs. J Cardiovasc Pharmacol Ther. 2013;18(1):87–93. doi:10.1177/107424841143477322368266
- Zhang LH, Pang XF, Bai F, et al. Preservation of glucagon-like peptide-1 level attenuates angiotensin II-induced tissue fibrosis by altering AT1/AT2 receptor expression and angiotensin-converting enzyme 2 activity in rat heart. Cardiovasc Drugs Ther. 2015;29(3):243–255. doi:10.1007/s10557-015-6592-725994830
- Zhang WW, Bai F, Wang J, et al. Edaravone inhibits pressure overload-induced cardiac fibrosis and dysfunction by reducing expression of angiotensin II AT1 receptor. Drug Des Devel Ther. 2017;11:3019–3033. doi:10.2147/DDDT.S144807
- Crowley MJ, Powers BJ, Myers ER, McBroom AJ, Sanders G. Angiotensin converting enzyme inhibitors and angiotensin II receptor blockers for treatment of ischemic heart disease: future research needs prioritization. Am Heart J. 2012;163:777–782. doi:10.1016/j.ahj.2012.02.01622607854
- Ram CVS. Angiotensin receptor blockers: current status and future prospects. Am J Med. 2008;121:656–663. doi:10.1016/j.amjmed.2008.02.03818691475
- Pabreja K, Mohd MA, Kode C, Wootten D, Furness SGB. Molecular mechanisms underlying physiological and receptor pleiotropic effects mediated by GLP-1R activation. Br J Pharmacol. 2014;171:1114–1128. doi:10.1111/bph.1231323889512
- Liu J, Liu Y, Chen L, et al. Glucagon-like peptide-1 analog liraglutide protects against diabetic cardiomyopathy by the inhibition of the endoplasmic reticulum stress pathway. J Diabetes Res. 2013;2013(5):1–8.
- Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311–322. doi:10.1056/NEJMoa160382727295427
- Abdul-Ghani M, DeFronzo RA, Del Prato S, et al. Cardiovascular disease and type 2 diabetes: has the dawn of a new era arrived? Diabetes Care. 2017;40(7):813–820. doi:10.2337/dc16-273628637886
- Svanström H, Ueda P, Melbye M, et al. Use of liraglutide and risk of major cardiovascular events: a register-based cohort study in Denmark and Sweden. Lancet Diabetes Endocrinol. 2019;7(2):106–114. doi:10.1016/S2213-8587(18)30320-630527909
- Bai F, Pang XF, Zhang LH, et al. Angiotensin II AT1 receptor alters ACE2 activity, eNOS expression and CD44–hyaluronan interaction in rats with hypertension and myocardial fibrosis. Life Sci. 2016;153:141–152. doi:10.1016/j.lfs.2016.04.01327085217
- Meyers TA, Heitzman JA, Krebsbach AM, et al. Acute AT1R blockade prevents isoproterenol-induced injury in mdx hearts. J Mol Cell Cardiol. 2019;128:51–61. doi:10.1016/j.yjmcc.2019.01.01330664850
- Jones ES, Black MJ, Widdop RE. Angiotensin AT2 receptor contributes to cardiovascular remodelling of aged rats during chronic AT1 receptor blockade. J Mol Cell Cardiol. 2004;37(5):1023–1030. doi:10.1016/j.yjmcc.2004.08.00415522279
- Namsolleck P, Recarti C, Foulquier S, Steckelings UM, Unger T. AT2 receptor and tissue injury: therapeutic implications. Curr Hypertens Res. 2014;16:416–426. doi:10.1007/s11906-013-0416-6
- Oishi Y, Ozono R, Yoshizumi M, Akishita M, Horiuchi M, Oshima T. AT2 receptor mediates the cardioprotective effects of AT1 receptor antagonist in post-myocardial infarction remodeling. Life Sci. 2006;80(1):82–88. doi:10.1016/j.lfs.2006.08.03317023005
- Perret-Guillaume C, Joly L, Jankowski P, Benetos A. Benefits of the RAS blockade: clinical evidence before the ONTARGET study. J Hypertens Suppl. 2009;27(2):S3–S7. doi:10.1097/01.hjh.0000354511.14086.f1
- Minas JN, Thorwald MA, Conte D, Vázquez-Medina JP, Nishiyama A, Ortiz RM. Angiotensin and mineralocorticoid receptor antagonism attenuates cardiac oxidative stress in angiotensin II-infused rats. Clin Exp Pharmacol Physiol. 2015;42(11):1178–1188. doi:10.1111/1440-1681.1247326234762
- Keidar S, Kaplan M, Gamliel-Lazarovich A. ACE2 of the heart: from angiotensin I to angiotensin (1–7). Cardiovasc Res. 2007;73(3):463–469. doi:10.1016/j.cardiores.2006.09.00617049503
- Wang W, McKinnie SM, Farhan M, et al. Angiotensin-converting enzyme 2 metabolizes and partially inactivates Pyr-Apelin-13 and Apelin-17: physiological effects in the cardiovascular system. Hypertension. 2016;68(2):365–377. doi:10.1161/HYPERTENSIONAHA.115.0689227217402
- Zhang W, Miao J, Wang S, Zhang Y. The protective effects of beta-casomorphin-7 against glucose -induced renal oxidative stress in vivo and vitro. PLoS One. 2013;8(5):e63472. doi:10.1371/journal.pone.006347223658831
- Crackower MA, Sarao R, Oudit GY, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–828. doi:10.1038/nature0078612075344
- Wang X, Ye Y, Gong H, et al. The effects of different angiotensin II type 1 receptor blockers on the regulation of the ACE-AngII-AT1 and ACE2-Ang(1–7)-Mas axes in pressure overload-induced cardiac remodeling in male mice. J Mol Cell Cardiol. 2016;97:180–190. doi:10.1016/j.yjmcc.2016.05.01227210827
- Fabiani I, Pugliese NR, La Carrubba S, et al. Incremental prognostic value of a complex left ventricular remodeling classification in asymptomatic for heart failure hypertensive patients. J Am Soc Hypertens. 2017;11(7):412–419. doi:10.1016/j.jash.2017.05.00528619598
- Forrester SJ, Booz GW, Sigmund CD, et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. 2018;98(3):1627–1738. doi:10.1152/physrev.00038.201729873596
- Triposkiadis F, Giamouzis G, Boudoulas KD, et al. Left ventricular geometry as a major determinant of left ventricular ejection fraction: physiological considerations and clinical implications. Eur J Heart Fail. 2018;20(3):436–444. doi:10.1002/ejhf.105529105899
- Fabijanovic D, Milicic D, Cikes M. Left ventricular size and ejection fraction: are they still relevant? Heart Fail Clin. 2019;15(2):147–158. doi:10.1016/j.hfc.2018.12.01230832807
- Tomoaia R, Beyer RS, Simu G, Serban AM, Pop D. Understanding the role of echocardiography in remodeling after acute myocardial infarction and development of heart failure with preserved ejection fraction. Med Ultrason. 2019;21(1):69–76. doi:10.11152/mu-176830779834
- Esposito G, Cappetta D, Russo R, et al. Sitagliptin reduces inflammation, fibrosis and preserves diastolic function in a rat model of heart failure with preserved ejection fraction. Br J Pharmacol. 2017;174(22):4070–4086. doi:10.1111/bph.1368627922176
- Nauck MA, Meier JJ, Cavender MA, Abd El Aziz M, Drucker DJ. Cardiovascular actions and clinical outcomes with glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Circulation. 2017;136(9):849–870. doi:10.1161/CIRCULATIONAHA.117.02813628847797
- Gaspari T, Brdar M, Lee HW, et al. Molecular and cellular mechanisms of glucagon-like peptide-1 receptor agonist-mediated attenuation of cardiac fibrosis. Diab Vasc Dis Res. 2016;13(1):56–68. doi:10.1177/147916411560500026408644