
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Introduction
Cancer is considered as one of the deadliest human diseases today. Angiogenesis, the propagation of new blood vessels from pre-existing vasculature, is a critical step in the progression of cancer as it is essential in the growth and metastasis of tumors. Hence, suppression of angiogenesis is a promising approach in cancer therapy. Syringin, a phenylpropanoid glycoside with a molecular formula of C17H24O9, has been found to exhibit chemopreventive effects. However, its anti-angiogenic activity and the underlying mechanism of action are still unknown.
Methods
In this work, in ovo chorioallantoic membrane (CAM) assay has been conducted to evaluate the effect of syringin on neovascularization. Additionally, reverse molecular docking studies have been performed in order to identify the probable enzyme targets in the angiogenesis pathway.
Results
Treatment with syringin showed significant dose-dependent inhibition of blood vessel length and junctions in the CAM of duck eggs; the anti-angiogenic activity of syringin at 100 µM and 200 µM is comparable with 200 µM of the positive control celecoxib. The results of reverse docking studies indicate that syringin binds the strongest to dihydrofolate reductase (DHFR) and, to some extent, with transforming growth factor-beta receptor type 1 (TGF-βR1), vascular endothelial growth factor receptor 2 (VEGFR2), and matrix metalloproteinase-2 (MMP-2). Furthermore, ADMET models revealed that syringin potentially possesses excellent pharmacokinetic and toxicity profiles.
Conclusion
This study demonstrates the potential of syringin as an anti-angiogenic agent and elicits further investigations to establish its application in cancer suppression.
Introduction
Cancer ranks as the second leading cause of death worldwide, accounting to 9.6 million mortalities in 2018.Citation1 It is a complex disorder characterized by the abnormal proliferation and spread of malignant cells into surrounding tissues.Citation2 Progression of this condition relies on its capacity to promote angiogenesis,Citation3 which is the intricate process of initiating new blood vessels from pre-existing ones.Citation4 The new capillary vessels are fundamental as they supply oxygen and sustenance that are indispensable in the growth and survival of malignancy.Citation5 Moreover, these vessels are essential in the spread of cancer into other organs for they function as the transport channels of metastatic cells, implying that neovascularization is necessary in metastasis. The dependency of tumor progression on angiogenesis,Citation3 together with its vital function in the pathological development and metamorphosis of neoplastic cells, makes this cascade an attractive target in cancer therapy.Citation6
Impairment of signal transduction, due to genetic and epigenetic modifications, is a defining characteristic of cancer.Citation7 Previous accounts have shed light on the significance of molecular interactions and signaling pathways in carcinogenesis.Citation8 Neovascularization is affected by a number of molecular mediators such as growth factors,Citation9 transcription factors,Citation10 signaling pathways,Citation11 extracellular matrix molecules,Citation12 cytokines,Citation11 and many others. Due to the undesirable effects that manifest when chemotherapeutic agents attack normal cells, the use of targeted cancer therapy has become more appealing.Citation13 Through the targeted approach, drug molecules specifically act at the pathways and proteins that serve critical roles in cancer and its microenvironment.Citation14
Overexpression of numerous angiogenesis-related proteins is typical in many forms of cancer as they are crucial in the growth, spread, and progression of tumors.Citation3,Citation15 The complexity of the angiogenic system provides many targets for therapeutic intervention.Citation16 With the current advances in molecular angiogenesis, novel molecular targets that are overexpressed in cancer have been identified.Citation17 The vascular endothelial growth factor (VEGF/VEGFR2) pathway constitutes a series of signaling mechanisms that control the proliferation, migration, survival, as well as penetrability of vascular endothelial cells.Citation18 The phosphorylation of VEGFR2 is recognized as a critical trigger in tumor angiogenesis.Citation19 Transforming growth factor-beta (TGF-β) is vital for the stabilization of new vessels and is responsible for the production of extracellular matrix and the appropriate interaction between endothelial cells and mural cells.Citation20 Matrix metalloproteinase-2 (MMP-2) is an extracellular remodeling enzyme that aids in the degradation of the basal membrane required for the migration and infiltration of proliferative endothelial cells in the course of angiogenesis.Citation21 Dihydrofolate reductase (DHFR) is an enzyme that plays a fundamental role in the synthesis of nucleic acid precursors, which are essential for cell proliferation and growth.Citation22 Fibroblast growth factor receptors (FGFRs) regulate cell proliferation, differentiation, and angiogenesis.Citation23 Tie2 is vital in vascular network formation, and its receptors act as regulators of angiogenesis and vessel maturation.Citation24,Citation25 Insulin-like growth factor-1 receptor (IGF-1R) is commonly overexpressed in cancer.Citation26 Its signaling initiated by ligand binding mediates many crucial cell responses including angiogenesis, invasion, and metastasis of neoplasia.Citation27,Citation28 The complex and multifactorial nature of tumor angiogenesis, especially in advanced tumors, necessitates the use of therapeutic compounds that act broadly against cancer-specific targets and pathways in order to reduce challenges associated with developing resistance. Therefore, broadly acting natural drugs with very low toxicity have promising roles in preventing tumor angiogenesis.Citation16
The majority of the standard chemotherapeutic agents cause systemic toxicity which impairs several healthy organs/tissues.Citation29 The toxicities of these drugs on the processes of metabolism and excretion could continuously lead to serious injury to the kidneys, liver, and heart that could, later on, cause coagulopathy and peripheral neuronal toxicity.Citation30 Thus, there is an urgent need for less toxic drugs that can intercept and cure cancer.
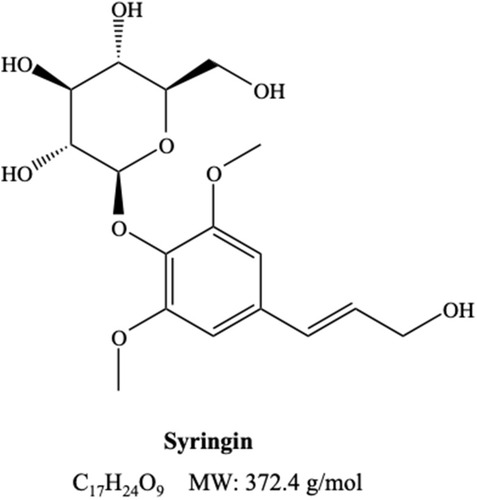
Syringin, also known as Eleutheroside B or in its formal name 4-[(1E)-3-Hydroxy-1-propen-1-yl]-2,6-dimethoxyphenyl D-glucopyranoside (), is a phenylpropanoid glycoside present in plants such as Eleutherococcus senticosus,Citation31 Syringa velutina,Citation32 Tinospora cordifolia,Citation33 and numerous others. This compound was known to possess cytotoxic, apoptotic, and antitumor functions in an array of human cancer cell lines.Citation34–Citation37 These studies suggest that syringin is a promising agent for cancer treatment. Nonetheless, the role of syringin in the inhibition of angiogenesis and its underlying mechanism remains unknown.
Drug discovery and development is a very costly and extensive procedure.Citation38 Fortunately, computer-aided drug design (CADD) provides efficiency in minimizing the time, labor, and cost of drug research by the use of computational methods that can speed-up the entire process.Citation39 These methods were instrumental in the discovery of new antineoplastic drugs that are used today. These include Gefitinib,Citation40 Erlotinib,Citation41 Sorafenib,Citation42 Lapatinib,Citation43 Abiraterone,Citation44 and Crizotinib,Citation45 which are all approved medications that are initially discovered using computational methods.Citation46
In this work, reverse molecular docking techniqueCitation47,Citation48 was conducted to understand the interaction of syringin with the molecular mediators involved in the multifaceted interplay in angiogenesis. The anti-angiogenic activity of syringin was evaluated through the chorioallantoic membrane (CAM) assay and the probable molecular targets of the title compound were identified using the reverse docking technique. Furthermore, in silico assessment of drug-likeness and the compound’s absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties were also performed to predict the safety and oral drug-likeness of syringin.
Figure 1 The structure of syringin.
Materials and Methods
Reagents and Materials
Syringin (≥98% purity) was acquired from Chemfaces (Wuhan, China), and the standard Celecoxib (≥98% purity) was purchased from Sigma Aldrich (St. Louis, MO, USA). Phosphate buffer saline (PBS) was bought from Gibco (Waltham, MA, USA), while dimethylsulfoxide (DMSO) was obtained from J.T. Baker (Center Valley, PA, USA). All the other chemicals and reagents used in this study were of analytical reagent grade. Additionally, fertilized duck eggs were purchased from a hatchery in Baliuag, Bulacan, Philippines.
Chick Chorioallantoic Membrane (CAM) Assay
The effect of syringin on angiogenesis was evaluated through chorioallantoic membrane (CAM) assay as per the protocol of Thanekar et alCitation49 with minor modifications on the method of analyzing vascular branching and choice of positive control. A total of six fertilized duck eggs were tested per group and were incubated using an Incubox automatic turning incubator maintained at 37°C with 85% relative humidity. Eggs between the 8th and 10th day of embryonic development (the peak of CAM neovascularizationCitation50) were utilized. Prior to the assay, the access portal was prepared by locating the embryo using a light source. Thereafter, the eggshells were marked and sanitized using 70% alcohol to create a 1-cm2 window. The entire experiment was performed under sterile conditions to avoid contaminants. The incubator was disinfected and the tools were autoclaved before use.
Celecoxib (200 µM) and syringin at various concentrations (12.5 µM, 25 µM, 50 µM, 100 µM, 200 µM) were dissolved in dimethylsulfoxide (DMSO, 0.1% v/v) with phosphate buffer saline (PBS). At day 8 of embryonic development, sterile filter paper discs (10 mm) loaded with 100 µL of syringin, celecoxib, and 0.1% DMSO were aseptically placed over the blood vessel of a growing CAM through the access portal. Later on, adhesive tape was used to seal the eggshell, and the eggs were returned to the humidified incubator. After 48 hours, the eggs were reopened and the CAMs were observed and photographed. Photographs of the chorioallantoic membranes (CAMs) with the highest quality were chosen. The chosen visual field represents the region around the filter paper disc with the tested compounds. Photographs were taken at the same distance from the egg window to the lens of the camera. Through Angioquant version 1.33 (MATLAB Inc., Tampere, Finland), a software program for the analysis of blood vessels in images, the digital figures of the CAMs were inspected and parameters such as vessel length and its junctions were quantified.Citation51 The anti-angiogenic effect was then calculated using the following formula:Citation52
Molecular Modeling
Computational Tools
Protein-Ligand docking was accomplished with the use of Autodock Vina 1.1.2Citation53 in Python Prescription (PyRx) software, installed in an iMac desktop computer, equipped with 3.1 GHz Intel Core i7 processor, 16GB RAM, and an NVIDIA GeForce 750M graphics card. Autodock Vina utilizes AMBER force field as the scoring parameter for molecular docking.
Preparation of 3D Structure of Ligands and Protein Targets
The crystal structures of dihydrofolate reductase (PDB ID: 1DRE), transforming growth factor-beta receptor type 1 (PDB ID: 1VJY), vascular endothelial growth factor receptor 2 (PDB ID: 3EWH), matrix metalloproteinase-2 (PDB ID: 1HOV), and other angiogenesis-related proteins were downloaded from Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank. Protein preparation was carried out using the UCSF Chimera 1.13 software and AutoDock tools of PyRx. Co-crystallized ligands, solvents, water molecules, and ions were removed, except for the ions that present essential roles in the ligand–target interaction. The location of the reference ligand in the protein structure was recorded and used to serve as the target site. Afterwards, the co-crystallized ligand was deleted to allow the interaction of test compounds with the binding site. The three-dimensional (3D) structure of ligands such as syringin (CID 5316860) (https://pubchem.ncbi.nlm.nih.gov/compound/Syringin), as well as the other inhibitors evaluated, were retrieved from the PubChem compound database. The ligands were minimized in the OpenBabel tool using Universal Force Field (UFF) and then converted into AutoDock ligand format (PDBQT), which is suitable for docking in PyRx.
Reverse Molecular Docking
The potential targets of syringin in the cascade of angiogenesis were virtually examined by reverse docking approach to determine the probable targets of this ligand. Commonly known molecular targets involved in angiogenesis were chosen, and the binding modes, binding affinity, as well as the residue interaction of syringin on the active site of these proteins were determined. A grid-based docking method using a rigid protein receptor and flexible ligand was utilized in the experiment.Citation53 The grid box was optimized to attain the best binding pose of the redocked native ligand which exhibits high similarity to the original co-crystallized ligand. The residues featured in the docking interaction, RMSD value, and generated pose were the basis in validating the docking parameters. The grid optimization protocol was executed using Autodock Vina, and the grid box encompasses the area containing the amino acid residues of the target binding site.
Listed in are the important residues enclosed in the grid box as well as the grid box parameters of selected target proteins. The featured residues, RMSD value, and pose were the basis in evaluating the validity of the docking procedure.
Table 1 Residues and Grid Box Parameters of Selected Protein Targets
The structures of the crystallized and redocked ligands were inspected using UCSF chimera 1.13 software (San Francisco, CA, USA).Citation54 The two structures were superimposed and each pair of atoms was manually selected to calculate the root main square deviation (RMSD) values of the overlapped structures. The RMSD is a measure of the accuracy of the docking protocol used,Citation55 an RMSD value of <2 Å being deemed successful.Citation56,Citation57 Moreover, the docking poses and ligand interactions were viewed using Biovia Discovery Studio Visualizer 2019 (Accelrys Inc., San Diego, CA, USA) to visually confirm if the essential residues and interactions observed in the crystal structure were maintained in the simulated structure.
ADMET and Toxicity Predictions
Aside from having therapeutic efficacy, it is also of prime importance that drug candidates possess excellent ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties.Citation58 In this study, syringin was analyzed using the ADMET and TOPKAT (Toxicity Prediction by Komputer Assisted Technology) modules of Biovia Discovery Studio 2.5 (Accelrys Inc., San Diego, CA, USA). The ADMET descriptors protocol includes the determination of human intestinal absorption, aqueous solubility, blood-brain barrier (BBB) penetration, plasma protein binding, CYP2D6 inhibition, and hepatotoxicity. A quantitative structure–activity relationship (QSAR) model was utilized by this module to examine various attributes of the test compound. Meanwhile, carcinogenicity, mutagenicity, skin irritant activity, and others were evaluated using the TOPKAT module. The toxicity prediction protocol performs the assessment on a series of toxicity endpoints frequently employed in drug development.Citation59 Furthermore, the conformance of syringin to Lipinski’s rule of fiveCitation60 was also assessed.
Statistical Analysis
Analysis of data was conducted using one-way analysis of variance (ANOVA), together with post hoc Tukey’s comparison test in GraphPad Prism version 6.01 (San Diego, CA, USA). Values were presented as mean ± SD, and findings were regarded as statistically significant when P < 0.05.
Results and Discussion
Anti-Angiogenic Activity
Assessment of the anti-angiogenic responses of the drug was accomplished using the chorioallantoic membrane (CAM) assay. This assay is considered as the most widely used method in the evaluation of agents that affect blood vessel formation.Citation61,Citation62 DMSO at 0.1% concentration was used as the vehicle of the compounds and served as the negative control. Celecoxib was used as the positive control due to its established inhibitory activity in angiogenesis.Citation63–Citation68
The results of the chorioallantoic membrane (CAM) assay are summarized in . Celecoxib at 200 µM inhibited the total number of junctions by 62.08 ± 5.60% (P < 0.0001). Meanwhile, at 12.5 µM, 25 µM, 50 µM, 100 µM, and 200 µM concentrations, syringin was able to decrease the total number of junctions by 22.83 ± 2.3%, 29.81 ± 3.4%, 39.61 ± 3.3%, 63.19 ± 2.34%, and 66.98 ± 3.62%, respectively. When it comes to vessel length, celecoxib at 200 µM was able to shorten it by 45.83±9.94% (P < 0.0001), whereas syringin at 12.5 µM, 25 µM, 50 µM, 100 µM, and 200 µM reduced vessel length by 10.38 ± 4.16%, 14.64 ± 3.15%, 25.42 ± 6.74%, 44.15 ± 8.20%, and 55.14 ± 8.94%, respectively. It is also noteworthy that at 100 µM and 200 µM, the activity of syringin is comparable with the percentage inhibition of 200 µM celecoxib. These results demonstrate that syringin exhibits a significant dose-dependent decrease in the total number of junctions from 12.5 µM to 200 µM, and mean vessel length from 50 µM to 200 µM in comparison with the negative control group (P < 0.0001). Representative images of treated membranes (), and the percentage inhibition of the total number of junctions and mean length of blood vessels, as functions of concentration, are illustrated in , respectively.
Figure 2 Representative images of chorioallantoic membranes (CAMs) upon exposure to various treatment groups.
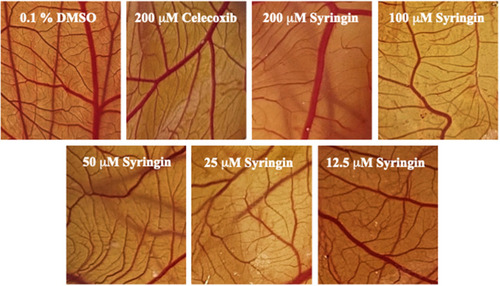
Figure 3 Chorioallantoic membranes (CAMs) treated with 0.1% DMSO, celecoxib, and syringin after 48 hours. (A) Graph demonstrating the percentage inhibition of the total number of junctions. (B) Graph denoting the percentage difference in mean length of blood vessels. Statistical analysis was calculated using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. There were statistically significant differences between the average total number of junctions of the negative control group (0.1% DMSO group) against all the concentrations of syringin and celecoxib. Meanwhile, syringin at 25 uM to 200 uM, along with 200 µM celecoxib, exhibited significant differences in the mean length of vessels parameter compared to the negative control group. Remarkably, syringin at 100 µM and 200 µM is statistically comparable with the 200 µM positive control. Data are presented as mean ± standard deviation, (n = 6), * P value < 0.0001 vs 0.1% DMSO (negative control group), ♦ P value < 0.05 vs 0.1% DMSO (negative control group), ns – non-significant as compared with the positive control (celecoxib).
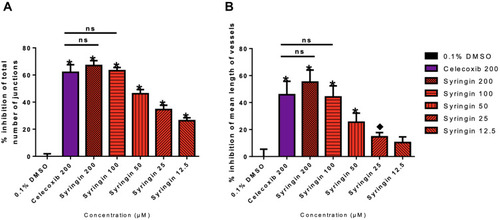
Table 2 Percentage Inhibition of Total Number of Junctions and Mean Length of Blood Vessels Treated with Syringin, Celecoxib, and 0.1% DMSO After 48 Hours of Treatment in CAM Assay
Reverse Molecular Docking Analysis
It has been established that molecular docking is considered as a top screening approach in drug discovery and development.Citation69 We demonstrated the utility of this technique in discovering new inhibitors of various druggable targets in M. tuberculosis.Citation70–Citation76 Reverse or inverse molecular docking helps in determining the probable protein targets of a ligand and shed some light on the possible mechanism of action of a drug.Citation47,Citation48 In this study, reverse docking was conducted to investigate the interaction between syringin and various angiogenesis-related proteins at the molecular level, particularly examining the hydrogen bonds, van der Waals and hydrophobic interactions, which are the principal driving force in maintaining a stable ligand-protein complex.Citation77
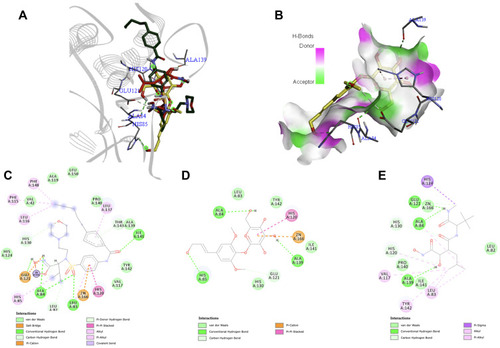
Syringin–DHFR Interaction
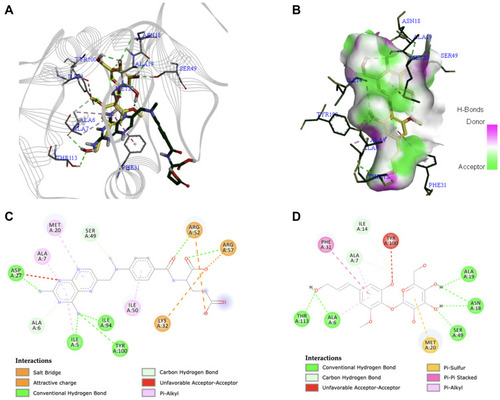
The folate pathway has been regarded as a crucial target in cancer chemotherapy. Dihydrofolate reductase (DHFR) is an enzyme that plays a fundamental role in the synthesis of nucleic acid precursors and is essential for cell proliferation and growth.Citation22 It catalyzes the reduction of dihydrofolate to tetrahydrofolate through NADPH, a pocket situated deep within the enzyme.Citation78 Meanwhile, methotrexate is an anti-folate agent available in the market for various types of cancer.Citation79 However, aside from its poor pharmacokinetic property, its side effects include myelosuppression as well as impairment of the gastrointestinal tract, kidneys, and liver.Citation80 Unfortunately, there is no available structure of DHFR from a human organism that is in complex with methotrexate, a standard antifolate drug. In contrast, ample literature shows that DHFR (eg, 1DRE) with methotrexate is common in the E. coli organization. A recent paper by Hobani et al also used the 1DRE structure to compare the effect of curcumin to methotrexate at the active site of DHFR protein.Citation81 Thus, the interaction of syringin with DHFR using the 1DRE crystal structure was investigated. The RMSD value of the redocked ligand was 0.844 Å, which is within the accepted range. Syringin formed a complex with DHFR through six conventional hydrogen bonds with Ala6, Asn18, Ala19, Ser49, and Thr113. The other residues that had an interaction with the ligand include Ala7, Ile14, Met20, Phe31, and Tyr100. Syringin had a binding affinity of −9.0 kcal/mol, whereas the redocked methotrexate ligand had −8.0 kcal/mol. These results indicate that the binding of syringin with DHFR is stronger than the known ligand, methotrexate. shows the docked structures of syringin together with the antifolate drug methotrexate on the active site of 1DRE, shows the 3D image of syringin on the hydrogen bond surface of the protein, while demonstrate the 2D interaction diagrams of methotrexate and syringin with 1DRE, respectively.
Figure 4 (A) Binding mode of syringin (yellow sticks) and methotrexate (green sticks) on the active site of dihydrofolate reductase (DHFR); (B) 3D docking snapshot with syringin on the surface of DHFR; (C) 2D interaction diagram of the native ligand (methotrexate) with DHFR; (D) 2D interaction diagram of syringin with DHFR.
Syringin–TGF-βR1 Interaction
The transforming growth factor-β (TGF-β) pathway has been recognized as a mediator of numerous cellular responses affecting cancer.Citation82 In the case of human transforming growth factor-beta receptor type 1 (PDB ID: 1VJY), the key residues of this protein are Lys232, Leu260, Ser280, His283, and Asp351.Citation83 As shown in , syringin had an interaction with all of the key residues, such as conventional hydrogen bond with Asp351 (), pi-cation with Lys232, pi-alkyl with Leu260, and van der Waals interaction with Ser280 and His283. Ser280 is considered a critical residue required for inhibitor selectivity, to which syringin had a van der Waals interaction. Compound 460 (), the redocked reference ligand, had a binding energy of −10.2 kcal/mol, while syringin had −8.7 kcal/mol. LY-580276 (), an established inhibitor, had a binding energy of −9.6 kcal/mol. The superimposed 3D interaction diagrams of syringin, compound 460, and LY-580276 at the active site of 1VJY are presented in . Although considered comparable, the binding affinity of syringin with TGF-βR1 protein target is slightly inferior compared to compound 460 and LY-580276.
Figure 5 (A) Binding mode of syringin (yellow sticks), compound 460 (green sticks), and LY-580276 (red sticks) on the active site of transforming growth factor-beta receptor type 1 (TGF-βR1); (B) 3D docking snapshot showing syringin on the surface of TGF-βR1; (C) 2D interaction diagram of the native ligand (compound 460) with TGF-βR1; (D) 2D interaction diagram of syringin with TGF-βR1; (E) 2D interaction diagram of the established inhibitor (LY-580276) with TGF-βR1.
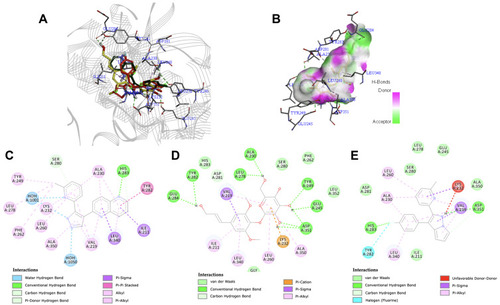
Syringin–VEGFR2 Interaction
The VEGF/VEGFR2 pathway constitutes a series of signaling mechanism that controls the proliferation, migration, survival, as well as penetrability of vascular endothelial cells.Citation18 For the VEGFR2 kinase domain (PDB ID: 3EWH), Glu885, Cys919, and Asp1046 are recognized as the key residues to which a compound will bind to for optimal interaction with the protein. Cys919 acts as a predominant residue responsible for the maintenance of inhibitor activity.Citation84 As shown in , syringin had a conventional hydrogen bond with Cys919 similar to the co-crystallized ligand, k11 (); while vandetanib (), an established inhibitor of VEGFR2, had an unfavorable interaction with the said residue. Furthermore, syringin formed van der Waals interaction with Glu885 and conventional hydrogen bond with Asp1046; vandetanib had a van der Waals interaction with both Glu885 and Asp1046; whereas k11 had an unfavorable interaction with Glu885 and Asp1046. The binding energy of syringin was −7.5 kcal/mol, while that of vandetanib and k11 were −8.8 kcal/mol and −12.2 kcal/mol, respectively. depicts the binding mode of syringin along with vandetanib and the native ligand at the active site of 3EWH. Despite the excellent binding affinity of k11, it has a molecular weight of 527.511 g/mol,Citation85 which makes it quite undesirable as an oral drug candidate according to the Lipinski’s rule.Citation60
Figure 6 (A) Binding mode of syringin (yellow sticks), compound K11 (green sticks), and vandetanib (red sticks) on the active site of vascular endothelial growth factor receptor 2 (VEGFR2); (B) 3D docking snapshot showing syringin on the surface of VEGFR2; (C) 2D interaction diagram of the native ligand (compound K11) with VEGFR2; (D) 2D interaction diagram of syringin with VEGFR2; (E) 2D interaction diagram of the established inhibitor (vandetanib) with VEGFR2.
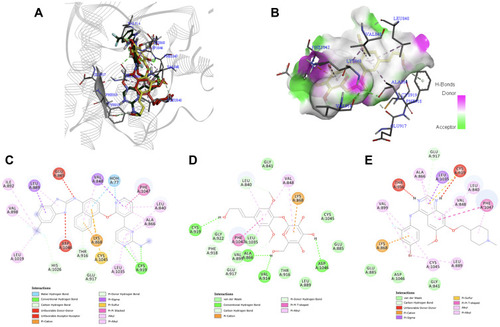
Syringin–MMP-2 Interaction
Matrix metalloproteinase-2 (MMP-2) is capable of degrading elastin, fibronectin, gelatin, as well as collagen types IV, V, VI, and X,Citation86 which are all recognized as part of the principal components of the extracellular matrix.Citation87 Numerous accounts supported the claim that a decrease in MMP-2 expression resulted in significant inhibition of angiogenesis in cancer.Citation88–Citation91 According to Agrawal et al, binding with zinc is usually regarded as a requirement for MMP inhibitors.Citation92 shows that when syringin was docked against MMP-2 (PDB ID: 1HOV), it exhibited pi–cation interaction with Zinc166. It also had a conventional hydrogen bond with Ala84 (), an extremely conserved residue in the target site of MMP. Moreover, His120 and His130 are some of the residues also present at the active site of the protein, and the compound of interest demonstrated pi-pi stacked and van der Waals interaction with those residues, respectively. Van der Waals interaction was also formed with Leu83, another backbone residue of 1HOV.Citation93,Citation94
The other interactions of syringin include conventional hydrogen bond with His85 and Ala139, carbon hydrogen bond with Glu121, and van der Waals interaction with Ile141 and Tyr142. The native ligand (i52) had a pi–cation interaction with Zinc166, conventional hydrogen bond with Ala84, pi–pi stacked interaction with His120, and carbon hydrogen bond with His130 (). Meanwhile, the known inhibitor marimastat () had the following interactions with the crucial amino acid residues at the active site: conventional hydrogen bond with Ala84, carbon hydrogen bond with His120 and His130, and van der Waals interaction with Zinc166. Syringin, marimastat, and i52 were observed to bind into the active site of the protein cavity with a good fit (). Syringin exhibited a binding energy of −6.9 kcal/mol, while marimastat and i52 had −6.6 kcal/mol and −8.0 kcal/mol, respectively. Although i52 exhibited better binding affinity than syringin, i52 and marimastat both contain a hydroxamic acid moiety, which is linked to toxicity as well as mutagenicity, and has been documented to manifest unfavorable pharmacokinetics.Citation93,Citation95 In this work, the ADMET and TOPKAT calculations (vide infra) indicate that syringin would most likely be nontoxic especially to the liver. Due to the unwanted effects exhibited by known inhibitors of MMP-2,Citation96 new and effective MMP-2 suppressive agents with a safer profile must be explored.
Figure 7 (A) Binding mode of syringin (yellow sticks), i52 (green sticks), and marimastat (red sticks) on the active site of matrix metalloproteinase-2 (MMP-2); (B) 3D docking snapshot showing syringin on the surface of MMP-2; (C) 2D interaction diagram of the native ligand (i52) with MMP-2; (D) 2D interaction diagram of syringin with MMP-2; (E) 2D interaction diagram of the established inhibitor (marimastat) with MMP-2.
As summarized in , syringin potentially binds to numerous angiogenesis-related proteins such as DHFR, TGF-βR1, VEGFR2, and MMP-2. Although its binding affinity with these proteins may not be as high as some of the co-crystallized ligands and known inhibitors, syringin virtually binds with a multitude of proteins involved in neovascularization indicating the possible mechanism behind its anti-angiogenic activity.
Table 3 Root-Mean-Square Deviation (RMSD) Values and Binding Energies for Syringin and Native Ligands in Complex with Various Angiogenesis Proteins
ADMET Profile
The utmost reasons for the failure of the majority of drug candidates in clinical trials are inadequate ADME (absorption, distribution, metabolism, excretion) and high toxicity.Citation97 Therefore, early assessment of the pharmacokinetics and toxicity profiles is vital in drug discovery. Syringin was reported to possess hepatoprotectiveCitation98 and cardioprotective properties,Citation99 which are desirable properties of an anti-cancer/anti-angiogenic agent. The majority of the standard chemotherapeutic medications cause systemic toxicity, which leads to serious injury to the liver and heart.Citation30 Hence, prevention of these toxicities is vital for cancer survival.Citation100 Thus, ADMET predictions were performed on syringin. indicates that syringin would exhibit outstanding qualities as a drug candidate. Since the brain is not the target location in this study, it is desirable that the experimental compound does not penetrate the blood-brain barrier (BBB) to diminish any possible disturbances to the brain homeostasis.Citation101 Delightfully, the ADMET results show that syringin has a very low permeation ability to the BBB. Furthermore, the title compound exhibits optimal solubility, a vital characteristic that affects the bioavailability of the drug.Citation102 On the other hand, the absorption level of syringin was shown to be moderate, a characteristic that can be easily addressed by formulating the compound into a dosage form suitable for drugs with moderate absorption. Syringin was also predicted to be nontoxic to the liver, not bound to plasma proteins, and is not an inhibitor of CYP 2D6, suggesting that it will not cause drug-induced hepatotoxicity, is highly efficient in traversing cell membranes, therefore greater possibility to get to the target site, and less likely to affect the plasma concentration of other medications. These information indicate that there is a low probability that syringin will elicit drug–drug interactions at the pharmacokinetic level.
Table 4 ADMET Profile of Syringin
Furthermore, the TOPKAT data () show that syringin is a non-carcinogenic, non-mutagenic, and non-skin irritant agent. Additionally, indicates that the computed lethal dose 50 (LD50) of syringin in rats is 10,000 mg/kg, which, according to Hodge and Sterner toxicity scale, is classified as practically non-toxic.Citation103 Notably, the predicted maximum tolerated dose of the compound in rat models is 861 mg/kg. These findings matched the in vivo study of Krishnan et al who conducted a toxicity investigation of syringin. Signs and symptoms of toxicity such as agitation, pulmonary distress, diarrhea, convulsions, and coma did not occur on Wistar rats administered with 10, 20, 30, 50, and 100 mg/kg body weight of the test compound.Citation104 These results imply that syringin is safe to use in in vivo analyses and possesses features worthy of further development into a clinically applicable drug.
Table 5 Toxicity Profile of Syringin
Table 6 Toxicity Prediction of Syringin in Rat Models
The drug-like properties of syringin were also evaluated based on the Lipinski’s rule of five. According to this rule, compounds with a partition coefficient (LogP) ≤5, molecular weight ≤500, quantity of hydrogen bond acceptors ≤10, quantity of hydrogen bond donors ≤5, and topological polar surface area (TPSA) <140ÅCitation2 are likely to exhibit good oral bioavailability, smooth membrane permeability as well as favorable gastrointestinal absorption.Citation60 reveals that syringin perfectly complies with the criteria of the Lipinski’s rule, suggesting that this compound can be further developed as an oral drug for cancer. Furthermore, since it is predicted that the drug has a high maximum tolerated dose index, moderate to high doses may be considered to attain optimal inhibition of neovascularization.
Table 7 Drug-Like Properties of Syringin
Conclusion
The ever increasing incidence of cancer is making it a very serious health concern worldwide and attracts great interest in the field of medicinal research. For many years, plants, herbs, and other natural products have been utilized as therapeutic agents. Syringin, a phenylpropanoid compound present in diverse plant families, has been reported to elicit cytotoxicity and apoptotic activity.Citation34–Citation37 In this work, the chorioallantoic membrane (CAM) assay was conducted to assess the effect of the test compound on neovascularization. The results showed that the anti-angiogenic activity of syringin at 100 µM and 200 µM is comparable with 200 µM of celecoxib, the positive control. Furthermore, the reverse molecular docking study unveiled the probable angiogenesis targets that were inhibited by syringin as manifested in the CAM assay. Specifically, the activity of syringin was likely mediated by the inhibition of dihydrofolate reductase (DHFR), transforming growth factor-beta receptor type 1 (TGF-βR1), vascular endothelial growth factor receptor 2 (VEGFR2), and matrix metalloproteinase-2 (MMP-2). These results will guide future enzyme-based assays that will further validate the anti-angiogenic action of syringin. In fact, further in vitro investigations are underway in our group. It is also noteworthy that syringin is a good candidate for drug development as it exhibited outstanding ADMET and drug-like properties. This is the first study to substantiate the angiogenesis inhibitory action of syringin and its probable mechanism of action, demonstrating that it may be a feasible multi-targeted agent for anti-angiogenic cancer therapy.
Disclosure
The authors report grants from the National Research Council of the Philippines during the conduct of the study. The authors report no other potential conflicts of interest in this work.
Acknowledgments
This study was funded by the Institutional Grant for Invigorating Basic Research on Health Sciences Phase II research program of the Department of Science and Technology-National Research Council of the Philippines (Project No. Q-006). The authors are also thankful to The Graduate School and the Research Center for the Natural and Applied Sciences, University of Santo Tomas, Manila, Philippines, as well as to the staff of the Emerging Interdisciplinary Research (EIDR) laboratory (OVPAA-EIDR 12-001-121102) of the University of the Philippines Manila for the training, assistance, and for allowing them to use the facilities.
References
- Cancer. Available from: https://www.who.int/health-topics/cancer#tab=tab_1. Accessed 115, 2020.
- What Is Cancer? Available from: https://training.seer.cancer.gov/disease/cancer/. Accessed 15, 2020.
- NishidaN, YanoH, NishidaT, KamuraT, KojiroM. Angiogenesis in Cancer. Vasc Health Risk Manag. 2006;2(3):213–219. doi:10.2147/vhrm.2006.2.3.21317326328
- AdairTH, MontaniJ-P. Angiogenesis. San Rafael, CA: Morgan & Claypool Life Sciences; 2011.
- FolkmanJ. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi:10.1146/annurev.med.57.121304.13130616409133
- TahergorabiZ, KhazaeiM. A review on angiogenesis and its assays. Iran J Basic Med Sci. 2012;15(6):1110.23653839
- SeverR, BruggeJS. Signal transduction in cancer. Cold Spring Harb Perspect Med. 2015;5(4):a006098. doi:10.1101/cshperspect.a00609825833940
- KessenbrockK, PlaksV, WerbZ. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. doi:10.1016/j.cell.2010.03.01520371345
- FerraraN, KerbelRS. Angiogenesis as a therapeutic target. Nature. 2005;438(7070):967–974.16355214
- MafuTS, SeptemberA, ShamleyD. The potential role of angiogenesis in the development of shoulder pain, shoulder dysfunction, and lymphedema after breast cancer treatment. Cancer Manag Res. 2018;10:81–90.29391829
- UcuzianAA, GassmanAA, EastAT, GreislerHP. Molecular mediators of angiogenesis. J Burn Care Res. 2010;31(1):158–175. doi:10.1097/BCR.0b013e3181c7ed8220061852
- NeveA, CantatoreFP, MaruottiN, CorradoA, RibattiD. Extracellular matrix modulates angiogenesis in physiological and pathological conditions. Biomed Res Int. 2014;2014:1–10. doi:10.1155/2014/756078
- BaudinoT. Targeted cancer therapy: the next generation of cancer treatment. Curr Drug Discov Technol. 2015;12(1):3–20. doi:10.2174/157016381266615060214431026033233
- PadmaVV. An overview of targeted cancer therapy. BioMedicine. 2015;5:4.25705584
- BarronGA, GouaM, WahleKW, BermanoG. Circulating levels of angiogenesis-related growth factors in breast cancer: A study to profile proteins responsible for tubule formation. Oncol Rep. 2017;38(3):1886–1894. doi:10.3892/or.2017.580328714000
- WangZ, DabrosinC, YinX, et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin Cancer Biol. 2015;35(Suppl):S224–s243. doi:10.1016/j.semcancer.2015.01.00125600295
- FallahA, SadeghiniaA, KahrobaH, et al. Therapeutic targeting of angiogenesis molecular pathways in angiogenesis-dependent diseases. Biomed Pharmacother. 2019;110:775–785. doi:10.1016/j.biopha.2018.12.02230554116
- JiL, WuM, LiZ. Rutacecarpine inhibits angiogenesis by targeting the VEGFR2 and VEGFR2-Mediated Akt/MTOR/p70s6k signaling pathway. Molecules. 2018;23(8):2047. doi:10.3390/molecules23082047
- LuanX, GaoY-G, Guan-Y-Y, et al. Platycodin D inhibits tumor growth by antiangiogenic activity via blocking VEGFR2-mediated signaling pathway. Toxicol Appl Pharmacol. 2014;281(1):118–124. doi:10.1016/j.taap.2014.09.00925250884
- RouwkemaJ, KhademhosseiniA. Vascularization and angiogenesis in tissue engineering: beyond creating static networks. Trends Biotechnol. 2016;34(9):733–745. doi:10.1016/j.tibtech.2016.03.00227032730
- García-VilasJA, QuesadaAR, MedinaMÁ. Hydroxytyrosol targets extracellular matrix remodeling by endothelial cells and inhibits both ex vivo and in vivo angiogenesis. Food Chem. 2017;221:1741–1746. doi:10.1016/j.foodchem.2016.10.11127979155
- KalogrisC, GarulliC, PietrellaL, et al. Sanguinarine suppresses basal-like breast cancer growth through dihydrofolate reductase inhibition. Biochem Pharmacol. 2014;90(3):226–234. doi:10.1016/j.bcp.2014.05.01424875448
- MohammadiM, OlsenSK, IbrahimiOA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16(2):107–137. doi:10.1016/j.cytogfr.2005.01.00815863029
- SatoTN, TozawaY, DeutschU, et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376(6535):70–74. doi:10.1038/376070a07596437
- TeichertM, MildeL, HolmA, et al. Pericyte-expressed Tie2 controls angiogenesis and vessel maturation. Nat Commun. 2017;8:1. doi:10.1038/ncomms1610628232747
- WernerH. Tumor suppressors govern insulin-like growth factor signaling pathways: implications in metabolism and cancer. Oncogene. 2011;31(22):2703–2714. doi:10.1038/onc.2011.44721963847
- GrantMB, MamesRN, FitzgeraldC, EllisEA, AboufriekhaM, GuyJ. Insulin-like growth factor I acts as an angiogenic agent in rabbit cornea and retina: comparative studies with basic fibroblast growth factor. Diabetologia. 1993;36(4):282–291.7682976
- SamaniAA, YakarS, LeroithD, BrodtP. The role of the igf system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28(1):20–47.16931767
- SchirrmacherV. From chemotherapy to biological therapy: a review of novel concepts to reduce the side effects of systemic cancer treatment. Int J Oncol. 2019;54(2):407–419.30570109
- MaedaH, KhatamiM. Analyses of repeated failures in cancer therapy for solid tumors: poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin Transl Med. 2018;7(1):11. doi:10.1186/s40169-018-0185-629541939
- HuangL, ZhaoH, HuangB, ZhengC, PengW, QinL. Acanthopanax senticosus: review of botany, chemistry and pharmacology. Die Pharmazie Int J Pharm Sci. 2011;66(2):83–97.
- ParkHJ, LeeMS, LeeKT, SohnIC, HanYN, MiyamotoKI. Studies on constituents with cytotoxic activity from the stem bark of Syringa velutina. Chem Pharm Bull (Tokyo). 1999;47(7):1029–1031. doi:10.1248/cpb.47.102910434406
- HerowatiR, WidodoGP. Molecular Docking studies of chemical constituents of Tinospora cordifolia on glycogen phosphorylase. Procedia Chem. 2014;13:63–68. doi:10.1016/j.proche.2014.12.007
- LeeC-H, HuangC-W, ChangP-C, et al. Reactive oxygen species mediate the chemopreventive effects of syringin in breast cancer cells. Phytomedicine. 2019;61:152844. doi:10.1016/j.phymed.2019.15284431029906
- XiaN. Syringin exhibits anticancer effects in HeLa human cervical cancer cells by inducing apoptosis, cell cycle arrest and inhibition of cell migration. Bangladesh J Pharmacol. 2016;11(4):838–843. doi:10.3329/bjp.v11i4.27755
- XuJP. Cancer Inhibitors from Chinese Natural Medicines. Taylor & Francis Group: Boca Raton: CRC Press; 2016.
- YangX, YuanJ, WanJ. Cytotoxic phenolic glycosides from Boschniakia himalaica. Chem Nat Compounds. 2012;48(4):555–558. doi:10.1007/s10600-012-0308-z
- MohsRC, GreigNH. Drug discovery and development: role of basic biological research. Alzheimer’s Dementia. 2017;3(4):651–657.
- LeelanandaSP, LindertS. Computational methods in drug discovery. Beilstein J Org Chem. 2016;12(1):2694–2718. doi:10.3762/bjoc.12.26728144341
- MuhsinM, GrahamJ, KirkpatrickP. Gefitinib. Nat Rev Drug Discov. 2003;2(7):515–516. doi:10.1038/nrd113612841190
- GrunwaldV, HidalgoM Development of the epidermal growth factor receptor inhibitor Tarceva (TM) (OSI-774). New Trends in Cancer for the 21st Century 2003, 235–246.
- WilhelmS, CarterC, LynchM, LowingerT, DumasJ, SmithRA. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–844. doi:10.1038/nrd213017016424
- WoodER, TruesdaleAT, McdonaldOB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;2004(64):6652–6659. doi:10.1158/0008-5472.CAN-04-1168
- JarmanM, BarrieSE, LleraJM. The 16,17-double bond is needed for irreversible inhibition of human cytochrome P450 (17 alpha) by abiraterone (17-(3-pyridyl)androsta-5,16-dien-3 beta-ol) and related steroidal inhibitors. J Med Chem. 1998;41:5375–5381. doi:10.1021/jm981017j9876107
- ButrynskiJE, D’adamoDR, HornickJL, Dal CinP, AntonescuCR, JhanwarSC. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 1727–1733;2010(363).
- CuiW, AouidateA, WangS, YuQ, LiY, YuanS. Discovering anti-cancer drugs via computational methods. Front Pharmacol. 2020;11.
- KharkarPS, WarrierS, GaudRS. Reverse docking: a powerful tool for drug repositioning and drug rescue. Future Med Chem. 2014;6(3):333–342.24575968
- BillonesJB. Reverse docking study unravels the potential Mycobacterium tuberculosis enzyme targets of agelasine F. Oriental J Chem. 2016;32(2):851–858. doi:10.13005/ojc/320210
- ThanekarD, DhodiJ, GawaliN, et al. Evaluation of antitumor and anti-angiogenic activity of bioactive compounds from Cinnamomum tamala: in vitro, in vivo and in silico approach. South Afr j Botany. 2016;104:6–14. doi:10.1016/j.sajb.2015.09.014
- DeryuginaEI, QuigleyJP. Chick embryo chorioallantoic membrane model systems to study and visualize human tumor cell metastasis. Histochem Cell Biol. 2008;130(6):1119–1130. doi:10.1007/s00418-008-0536-219005674
- NiemistoA, DunmireV, Yli-HarjaO, ZhangW, ShmulevichI. Robust quantification of in vitro angiogenesis through image analysis. IEEE Trans Med Imaging. 2005;24(4):549–553. doi:10.1109/TMI.2004.83733915822812
- ZamanK, RahimF, TahaM, et al. Synthesis, thymidine phosphorylase, angiogenic inhibition and molecular docking study of isoquinoline derivatives. Bioorg Chem. 2019;89:102999. doi:10.1016/j.bioorg.2019.10299931151055
- TrottO, OlsonAJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461.19499576
- PettersenEF, GoddardTD, HuangCC, et al. System for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi:10.1002/jcc.2008415264254
- KufarevaI, AbagyanR. Methods of protein structure comparison. Methods Mol Biol Homol Modeling. 2011;231–257.
- MorrisGM, Lim-WilbyM. Molecular Docking. Mol Modeling Proteins. 2008;443:365–382.
- Saleh-E-InMM, RoyA, Al-MansurMA, et al. Isolation and in silico prediction of potential drug-like compounds from anethum Sowa L. Root extracts targeted towards cancer therapy. Comput Biol Chem. 2019;78:242–259.30584950
- GuanL, YangH, CaiY, et al. ADMET-score–a comprehensive scoring function for evaluation of chemical drug-likeness. Medchemcomm. 2019;10(1):148–157.30774861
- WangJ, PengW, LiX, et al. Towards to potential 2-cyano-pyrimidines cathepsin-K inhibitors: an in silico design and screening research based on comprehensive application of quantitative structure–activity relationships, molecular docking and ADMET prediction. J Mol Struct. 2019;2019(1195):914–928. doi:10.1016/j.molstruc.2019.06.020
- LipinskiCA, LombardoF, DominyBW, FeeneyPJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;23(1–3):3–25. doi:10.1016/S0169-409X(96)00423-1
- VargasA, Zeisser-LabouèbeM, LangeN, GurnyR, DelieF. The chick embryo and its chorioallantoic membrane (CAM) for the in vivo evaluation of drug delivery systems. Adv Drug Deliv Rev. 2007;59(11):1162–1176. doi:10.1016/j.addr.2007.04.01917870202
- MousaSA, YalcinM, DavisPJ. Models for assessing anti-angiogenesis agents: appraisal of current techniques. Anti-Angiogenesis Strategies Cancer Ther. 2017;21–38.
- GungorH, IlhanN, EroksuzH. The effectiveness of cyclooxygenase-2 inhibitors and evaluation of angiogenesis in the model of experimental colorectal cancer. Biomed Pharmacother. 2018;102:221–229. doi:10.1016/j.biopha.2018.03.06629562216
- KangKB, WangTT, WoonCT, et al. Enhancement of glioblastoma radioresponse by a selective COX-2 inhibitor celecoxib: inhibition of tumor angiogenesis with extensive tumor necrosis. Int J Radiat Oncol Biol Phys. 2007;67(3):888–896.17293239
- MandracchiaD, TripodoG, TrapaniA, et al. Inulin based micelles loaded with curcumin or celecoxib with effective anti-angiogenic activity. Eur J Pharm Sci. 2016;93:141–146. doi:10.1016/j.ejps.2016.08.02727539141
- RautCP, NawrockiS, LashingerLM, et al. Celecoxib inhibits angiogenesis by inducing endothelial cell apoptosis in human pancreatic tumor xenografts. Cancer Biol Ther. 2004;3(12):1217–1224. doi:10.4161/cbt.3.12.122115477758
- RosasC, SinningM, FerreiraA, FuenzalidaM, LemusD. Celecoxib decreases growth and angiogenesis and promotes apoptosis in a tumor cell line resistant to chemotherapy. Biol Res. 2014;47(1):27. doi:10.1186/0717-6287-47-2725027008
- VaishV, SanyalSN. Role of Sulindac and Celecoxib in the regulation of angiogenesis during the early neoplasm of colon: exploring PI3-K/PTEN/Akt pathway to the canonical Wnt/β-catenin signaling. Biomed Pharmacother. 2012;66(5):354–367. doi:10.1016/j.biopha.2012.01.00422397759
- RoyS, KumarA, BaigMH, MasaříkM, ProvazníkI. Virtual screening, ADMET profiling, molecular docking and dynamics approaches to search for potent selective natural molecules based inhibitors against metallothionein-III to study Alzheimer’s disease. Methods. 2015;83:105–110. doi:10.1016/j.ymeth.2015.04.02125920949
- BillonesJB, CarrilloMCO, OrganoVG, et al. Toward antituberculosis drugs: in silico screening of synthetic compounds against Mycobacterium tuberculosis l,d-transpeptidase 2. Drug Des Devel Ther. 2016;10:1147–1157. doi:10.2147/DDDT.S97043
- BillonesJB, CarrilloMCO, OrganoVG, et al. In silico discovery and in vitro activity of inhibitors against Mycobacterium tuberculosis 7,8-diaminopelargonic acid synthase (MtbBioA). Drug Des Devel Ther. 2017;11:563–574. doi:10.2147/DDDT.S119930
- BillonesJB, CarrilloMCO, OrganoVG, MacalinoSJY, EmnacenIA, SyJBA. Virtual screening against Mycobacterium tuberculosis lipoate protein ligase B (MtbLipB) and in silico ADMET evaluation of top hits. Oriental J Chem. 2013;29(4):1457–1468. doi:10.13005/ojc/290423
- UyVCC, BillonesJB. Towards antituberculosis drugs: virtual screening for potential inhibitors of pantothenate synthetase of Mycobacterium tuberculosis. Philippine Sci Letters. 2012;5(2):122–130.
- YangCTM, BillonesJB. Towards antituberculosis drugs: molecular docking of curcumin and its analogues to pantothenate synthetase. Philippine J Sci. 2012;141(2):187–196.
- BillonesJB, ValleAMF. Structure-based design of inhibitors against maltosyltransferase GlgE. Oriental J Chem. 2014;30(3):1137–1145. doi:10.13005/ojc/300326
- SampacoIII, BillonesAB, BJ. Virtual screening of natural products, molecular docking and dynamics simulations on M. tuberculosis S-adenosyl-L-homocysteine hydrolase. Oriental J Chem. 2015;31(4):1859–1865.
- WengC, FuY, JiangH, ZhuangS, LiH. Binding interaction between a queen pheromone component HOB and pheromone binding protein ASP1 of apis cerana. Int J Biol Macromol. 2015;72:430–436. doi:10.1016/j.ijbiomac.2014.08.04625195542
- SinghA, DeshpandeN, PramanikN, JhunjhunwalaS, RangarajanA, AtreyaHS. Optimized peptide based inhibitors targeting the dihydrofolate reductase pathway in cancer. Sci Rep. 2018;8(1):1–8.29311619
- HagnerN, JoergerM. Cancer chemotherapy: targeting folic acid synthesis. Cancer Manag Res. 2010;2:293.21301589
- AvendañoC, MenéndezJC. Antimetabolites In: Medicinal Chemistry of Anticancer Drugs. Eds. Avendaño C, Menéndez JC, Amsterdam: Elsevier; 2008:9–52.
- HobaniY, JerahA, BidwaiA. A comparative molecular docking study of curcumin and methotrexate to dihydrofolate reductase. Bioinformation. 2017;13(3):63–66. doi:10.6026/9732063001306328584445
- GuerreroPA, MccartyJH. TGF-β activation and signaling in angiogenesis. Physiol Pathol Angiogenesis Signaling Mech Targeted Ther. 2017.
- GellibertF, WoolvenJ, FouchetM-H, et al. Identification of 1,5-Naphthyridine derivatives as a novel series of potent and selective TGF-β Type I receptor inhibitors. J Med Chem. 2004;47(18):4494–4506. doi:10.1021/jm040024715317461
- ZhangY, ChenY, ZhangD, WangL, LuT, JiaoY. Discovery of novel potent VEGFR-2 inhibitors exerting significant antiproliferative activity against cancer cell lines. J Med Chem. 2018;61(1):140–157. doi:10.1021/acs.jmedchem.7b0109129189002
- K11. 3-[(4,6-Dipyridin-4-yl-1,3,5-triazin-2-yl)amino]-4-methyl-N-[3-(trifluoromethyl)phenyl] benzamide. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/138857895. Accessed 110, 2020.
- Jabłońska-TrypućA, MatejczykM, RosochackiS. Matrix Metalloproteinases (MMPs), the Main Extracellular Matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J Enzyme Inhib Med Chem. 2016;31(sup1):177–183.27028474
- YueB. Biology of the extracellular matrix. J Glaucoma. 2014;23.22706338
- BerglinL, SarmanS, PloegIVD, et al. Reduced choroidal neovascular membrane formation in matrix metalloproteinase-2–deficient mice. Invest Ophthalmol Visual Sci. 2003;44(1):403. doi:10.1167/iovs.02-018012506102
- ItohY, ItoA, IwataK, TanzawaK, MoriY, NagaseNH. Plasma Membrane-Bound Tissue Inhibitor of Metalloproteinases (TIMP)-2 Specifically Inhibits Matrix Metalloproteinase 2 (Gelatinase A) Activated on the Cell Surface. J Biol Chem. 1998;273(38):24360–24367. doi:10.1074/jbc.273.38.243609733724
- Ohno-MatsuiK, UetamaT, YoshidaT, et al. Reduced retinal angiogenesis in MMP-2–deficient mice. Invest Ophthalmol Visual Sci. 2003;44(12):5370. doi:10.1167/iovs.03-024914638740
- PfeiferA, KesslerT, SillettiS, ChereshDA, VermaIM. Suppression of angiogenesis by lentiviral delivery of PEX, a noncatalytic fragment of matrix metalloproteinase 2. Proc Natl Acad Sci. 2000;97(22):12227–12232. doi:10.1073/pnas.22039959711035804
- AgrawalA, Romero-PerezD, JacobsenJA, VillarrealFJ, CohenSM. Zinc-binding groups modulate selective inhibition of MMPs. ChemMedChem. 2008;3(5):812–820. doi:10.1002/cmdc.20070029018181119
- AhmadA, SayedA, GinnebaughKR, et al. Molecular docking and inhibition of matrix metalloproteinase-2 by novel difluorinatedbenzylidene curcumin analog. Am J Transl Res. 2015;7(2):298.25901198
- MukherjeeA, AdhikariN, JhaT, Pentanoic AcidA. Derivative targeting Matrix Metalloproteinase-2 (MMP-2) induces apoptosis in a chronic myeloid leukemia cell line. Eur J Med Chem. 2017;141:37–50. doi:10.1016/j.ejmech.2017.09.05229028530
- SmithGF. Designing drugs to avoid toxicity. Prog Med Chem Progress Med Chem. 2011;50:1–47.
- FingletonB. MMPs as therapeutic targets—still a viable option?Semin Cell Dev Biol. 2008;19(1):61–68. doi:10.1016/j.semcdb.2007.06.00617693104
- GombarVK, SilverIS, ZhaoZ. Role of ADME characteristics in drug discovery and their in silico evaluation: in silico screening of chemicals for their metabolic stability. Curr Top Med Chem. 2003;3(11):1205–1225. doi:10.2174/156802603345201412769701
- GongX, ZhangL, JiangR, WangCD, YinXR, WanJY. Hepatoprotective effects of syringin on fulminant hepatic failure induced by D‐galactosamine and lipopolysaccharide in mice. J Appl Toxicol. 2014;34(3):265–271. doi:10.1002/jat.287623620140
- LiF, ZhangN, WuQ, et al. Syringin prevents cardiac hypertrophy induced by pressure overload through the attenuation of autophagy. Int J Mol Med. 2016;39(1):199–207. doi:10.3892/ijm.2016.282427959392
- KalamK, MarwickTH. Role of cardioprotective therapy for prevention of cardiotoxicity with chemotherapy: a systematic review and meta-analysis. Eur J Cancer. 2013;49(13):2900–2909. doi:10.1016/j.ejca.2013.04.03023706982
- El-GamalKM, El-MorsyAM, SaadAM, EissaIH, AlswahM. Synthesis, docking, QSAR, ADMET and antimicrobial evaluation of new quinoline-3-carbonitrile derivatives as potential DNA-gyrase inhibitors. J Mol Struct. 2018;1166:15–33. doi:10.1016/j.molstruc.2018.04.010
- ChungTD, TerryDB, SmithLH. In vitro and in vivo assessment of ADME and PK properties during lead selection and lead optimization–guidelines, benchmarks and rules of thumb In: Assay Guidance Manual. Eds. Markossian S, Sittampalam GS, Grossman A, et al. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2015.
- Hodge, HC, Sterner, JH. Tabulation of Toxicity Classes. American Industrial Hygiene Association Quarterly1949;10(4):93–96. doi:10.1080/00968204909344159
- KrishnanSS, SubramanianIP, SubramanianSP. Isolation, characterization of syringin, phenylpropanoid glycoside from Musa paradisiaca tepal extract and evaluation of its antidiabetic effect in streptozotocin-induced diabetic rats. Biomed Prev Nutri. 2014;4(2):105–111. doi:10.1016/j.bionut.2013.12.009