Abstract
Cystic fibrosis (CF) is a life-limiting, multisystem disease characterized by thick viscous secretions leading to recurrent lung infections, bronchiectasis, and progressive deterioration in lung function. CF is caused by loss or dysfunction of the CF transmembrane conductance regulator (CFTR) protein which is responsible for transepithelial chloride and water transport. Improved understanding of CFTR protein dysfunction has allowed the development of mutation-specific small-molecule compounds which directly target the underlying CFTR defect. Ivacaftor is the first licensed small-molecule compound for CF patients which targets the CFTR gating mutation Gly551Asp (previously termed G551D) and has the potential to be truly disease-modifying. Ivacaftor is an oral medication given twice daily and has shown benefit in terms of an increase in lung function, decreased sweat chloride, weight gain, improvement in patient-reported quality of life, and reduction in number of respiratory exacerbations in clinical trials. Although ivacaftor is currently only licensed for use in approximately 5% of the CF population (those who have at least one Gly551Asp mutation), the developmental pathway established by ivacaftor paves the way for other CFTR modulators that may benefit many more patients. In particular, a CFTR modulator for those with the Phe508del deletion (previously ∆F508) would allow 90% of the CF population to benefit from disease-modifying treatment.
Background
Cystic fibrosis (CF) is the most common autosomal recessive disorder in Caucasians and affects approximately 28,000 patients in the United States and approximately 36,000 patients in Europe.Citation1 It is a multisystem disease affecting primarily the lungs, causing thick viscous secretions and recurrent respiratory tract infections, but also affects the pancreas, hepatobiliary system, reproductive tract, gastrointestinal tract, and bones. Death is usually due to pulmonary damage from infections and inflammation ().
Figure 1 Model for cystic fibrosis lung disease.
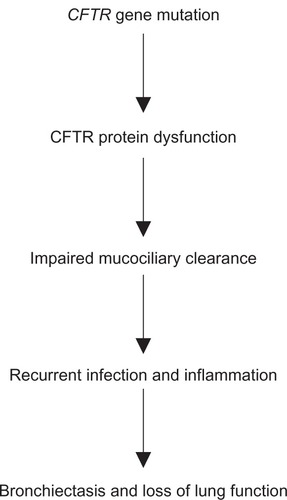
Although the first clear description of CF was written in 1938 by Dr Dorothy Andersen, a pathologist in New York, the gene that encodes the CF transmembrane conductance regulator (CFTR) protein on the long (q) arm of chromosome 7 was only identified in 1989.Citation2–Citation4 More than 1900 CFTR gene mutations with the potential to cause disease have been described to date.Citation5,Citation6
The life expectancy of patients with CF has risen steadily over the last 25 years, from a median predicted age of survival of 25 years in 1985 to 37 years in 2011.Citation7 Current treatment strategies in CF, including aggressive and early treatment of respiratory infections, mucociliary clearance, nutritional supplementation, and treatment of airway inflammation, are associated with improved pulmonary function and reduced exacerbations.Citation8 Until now, all treatments have targeted the downstream consequences of CFTR dysfunction rather than addressing the underlying CFTR defect. Ivacaftor, licensed in 2012, is the first available treatment to address directly the underlying genetic defect.
Structure of CFTR protein
The CFTR protein is a relatively large glycoprotein containing 1480 amino acids,Citation4 with a molecular mass of 170,000.Citation9 It is a member of the adenosine triphosphate (ATP) binding cassette family of proteins that are primarily concerned with transmembrane transport functions.Citation4 The protein consists of two transmembrane regions, each linked to a nucleotide binding domain. ATP binds to both nucleotide binding domains to control gating of the channel. Spanning the region between nucleotide binding domain 1 and the second transmembrane region is a cytoplasmic regulatory domain (R-domain) which is comprised of many charged amino acids. The R-domain is a unique feature of CFTR.Citation10 It contains several potential sites for phosphorylation by protein kinases. The activity of CFTR as an ion channel depends upon both phosphorylation of the R-domain and binding of ATP to the nucleotide binding domains.
Many of the mutations identified in CF, including the most common mutation, Phe508del (previously ∆F508), occur in the first nucleotide binding domain 1, while very few occur in the second. Around 75% of CF alleles contain the Phe508del mutation, a three base pair deletion resulting in loss of a phenylalanine residue at position 508 in the protein.
Function of CFTR protein
CFTR functions as a regulated chloride ion channel and localizes to the apical cell membrane of secretory and absorptive epithelial cells within the airway, pancreas, liver, intestine, sweat gland, and vas deferens. ATP-driven conformational changes in CFTR open and close a gate to allow transmembrane flow of anions down their electrochemical gradient.Citation11 There is also some evidence that CF lung disease relates to loss of regulation of the epithelial sodium channel transporter (ENaC) by CFTR, rather than being due just to chloride transport abnormalities.Citation12,Citation13
In CF, dysfunctional CFTR protein leads to absent or decreased chloride secretion, increased sodium and water absorption, and liquid depletion on the surface of the airway. This causes thick viscous secretions and impairs mucociliary clearance, resulting in the classical manifestations of CF, ie, infection, inflammation, and eventual bronchiectasis with loss of lung function. Sweat chloride levels are usually elevated (<60 mmol/L) in patients with CF, whereas people without CF have sweat chloride levels <40 mmol/L.Citation14
Classification of CFTR mutations
There are currently over 1900 known mutations affecting CFTR, many of which give rise to a disease phenotype.Citation5 Six different classes are described. A brief description of the CFTR classification is described here and in .
Table 1 CFTR protein defects and their potential therapies
Class I CFTR mutations affect biosynthesis of CFTR. They either prevent synthesis of a stable protein due to a nonsense mutation or result in production of a truncated protein due to creation of a premature stop codon. These truncated proteins are unstable and are rapidly degraded by proteins in the endoplasmic reticulum. Class 1 mutations include the most severe phenotypes because no functional protein is synthesized. This type of mutation is found in 5%–10% of CF chromosomes worldwide, but account for around 60% of mutations in Jewish people with CF.Citation15 The designation for nonsense mutation ends with an X and examples are Trp1282X (previously W1282X) and Gly542X.
Class II CFTR mutations affect CFTR maturation. They result in misprocessing of CFTR and give rise to misfolded CFTR protein, producing a lack of functional protein at the cell membrane. This altered protein fails to be trafficked to the correct location in the apical cell membrane, resulting in premature degradation by the proteasome. The small amount that does reach the correct location functions poorly. The Phe508del deletion accounts for the majority of mutations in this group. Around 90% of individuals with CF are heterozygous and 40%–50% are homozygous for the Phe508del.Citation6,Citation7
Class III CFTR mutations affect chloride channel regulation/gating. CFTR protein production and trafficking are normal, but when the protein arrives at the cell surface it does not respond to cAMP stimulation. This prevents binding of ATP and/or phosphorylation. The CFTR protein fails to open in response to intracellular signals at the cell surface, and remains closed for the majority of the time. The missense mutation Gly551Asp (previously termed G551D) is the third most common CFTR mutation and causes a glycine-to-aspartate substitution at residue 551.Citation7,Citation16 Approximately 4%–5% of all patients with CF have the Gly551Asp mutation on at least one allele,Citation6,Citation7 although this mutation is more frequent in those of Celtic (Scottish, Irish, English) origin.Citation17
Class IV CFTR mutations affect chloride conductance in the pore region of the channel. The CFTR protein reaches the cell surface, but the abnormal conformation of the pore leads to poor conductance of chloride ions because of reduced chloride permeation and open channel probability. Arg117His (previously termed R117H) is the most common class IV mutation, with a frequency of 2%–4%.Citation6,Citation7
Class V CFTR mutations affect mRNA stability. The splicing machinery is altered, resulting in generation of both aberrantly and correctly spliced mRNA. This leads to a reduced number of functioning CFTR proteins in the plasma membrane, as was demonstrated for the 3849+10 kb C<T mutation.Citation18 Less than 1% of patients with CF have class V mutations. The level of functioning CFTR can vary between different patients and even between different organs of the same patient and often results in a milder clinical phenotype.
Class VI CFTR mutations affect CFTR stability. Truncation of the C terminus of CFTR produces a functional protein that is unstable at the apical membrane surface, eg, Gln1412X (previously termed Q1412X).
Expression of the clinical phenotype
The CF phenotype is highly heterogenous among individual patients, and even between siblings carrying identical CFTR mutations. Genotype-phenotype relationships are complex, and are affected by many factors, including alternative genomic loci containing modifier genes as well as environmental factors such as pollution, smoking, bacterial infection, malnutrition, and certain therapeutic agents.Citation19 Of the various clinical symptoms of CF, only pancreatic function has been shown to correlate well with CFTR genotype.Citation20 In particular, substantial phenotypic variability is seen for lung disease, and there is often poor genotype-phenotype correlation for individual patients.
CFTR homozygous or compound heterozygous class I–III CFTR mutations are associated with an absence of or severely reduced production of functional CFTR. In general, this correlates well with a more severe phenotype involving pancreatic insufficiency (<95% of cases), liver disease (3%–5% of cases), young age at diagnosis (usually less than one year), high sweat chloride levels (<80 mmol/L), and meconium ileus (around 20% of cases).Citation21–Citation24 Class IV, V, and VI CFTR mutations are usually associated with some residual CFTR function and a more variable and sometimes milder disease with pancreatic sufficiency (70%–80% of cases), lower sweat chloride levels, no meconium ileus, and milder pulmonary disease.Citation24 The class IV, V, and VI mutations usually dominate in patients who are compound heterozygotes.
The CFTR classification system has a number of limitations. Many of the mutations identified remain unclassified.Citation5 In particular, the functional consequences of many rare mutations (particularly missense mutations) are unknown and cannot be predicted. Furthermore, a number of mutations give rise to more than one functional defect, eg, Phe508del, which may explain some of the clinical variability observed between subjects.Citation25 The CFTR2 study is an international initiative that seeks to address this by providing expert-reviewed functional and clinical information on CFTR mutations.Citation5,Citation26
Development of mutation-specific gene therapy
After the CFTR gene was discovered in 1989, there was considerable hope that gene therapy could be rapidly developed. However, this has proven to be extremely challenging. There have been over 25 gene therapy trials using either viral or cationic lipid-based vector systems which have shown limited success.Citation27 Even after successful uptake of the CFTR gene, the cells lose expression after a few weeks. Gene therapy, should it prove to be therapeutically effective, could be used to treat all patients with CF regardless of CFTR mutation. Currently, a gene therapy study is actively enrolling patients in the United Kingdom using a cationic lipid-based vector (GL67A/pGM169).Citation28
An alternative to gene therapy, which shows more promise, is the development of small-molecule compounds that target specific CFTR mutations. These exciting new developments are based on improved understanding of CFTR structure and dysfunction in CF. Small-molecule compounds are divided into three groups, ie, CFTR correctors, CFTR potentiators, and CFTR suppressors. CFTR correctors facilitate the trafficking of more CFTR molecules to the airway epithelial surface. CFTR potentiators activate appropriately located mutant CFTR by increasing the time that activated CFTR channels at the cell surface remain open. CFTR suppressors of premature termination codons promote ribosomal read through of nonsense mutations. Each of these agents has therapeutic potential alone or in combination for a specific patient population. Small molecules that improve chloride ion transport in human bronchial CF cells are identified through high throughput screening which can simultaneously look at over 100,000 potential drugs.Citation29,Citation30 Molecules that look “active” on screening are then investigated further.
Ivacaftor
Ivacaftor (VX-770) is an oral, bioavailable potentiator that is designed to increase the time that activated CFTR channels at the cell surface remain open. It was licensed in 2012 both in the United States and Europe for use in patients with CF aged six years and over who carry at least one copy of the class III mutation, Gly551Asp. Ivacaftor is the first licensed CF medication actually addressing the primary consequences of CFTR dysfunction, rather than the downstream consequences of the disease, and is therefore potentially truly disease-modifying.
Development
Ivacaftor was identified by screening over 228,000 small-molecule compounds using high throughput screening with a cell-based fluorescence membrane potential assay designed to identify CFTR potentiators.Citation31 Initial in vitro studies of the effect of ivacaftor on CFTR-mediated chloride secretion have been performed on both recombinant cell lines and primary cultures of human bronchial epithelial cells. These studies have demonstrated that ivacaftor increases chloride transport by increasing the probability of the CFTR channel being open, and increases apical fluid height and ciliary beat frequency.Citation31 The exact mechanism of action of ivacaftor at the CFTR channel is not completely understood. Further in vitro studies have elaborated on the mechanism further and have shown that ivacaftor opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner.Citation32
The efficacy and safety of ivacaftor in patients with CF with at least one Gly551Asp mutation in CFTR has been evaluated in two large, multicenter, randomized, double-blind, placebo-controlled trials. The subjects in these trials were clinically stable inpatients aged <12 years (n = 161)Citation33 and children aged 6–12 years (n = 52).Citation34 The primary efficacy endpoint in both studies was the mean absolute change from baseline in forced expiratory volume in one second (FEV1) percent predicted at 24 weeks from baseline. Patients in both trials were randomized in a 1:1 ratio to receive either 150 mg of ivacaftor or placebo orally every 12 hours for 48 weeks, in addition to their usual CF therapies prescribed prestudy.Citation33,Citation34 In the adult and adolescent group, 70% of trial participants were on regular dornase alfa and 50% were on nebulized tobramycin.Citation33 Use of inhaled hypertonic saline was not permitted because it does not have regulatory approval in the United States as a therapy for CF.Citation33 Ivacaftor was given with foods containing fat because this increases the exposure by 2–4-fold.Citation34 Patients from both studies were enrolled in a further open-label extension study for which interim results were available after a further 48 weeks for adults and adolescents and 24 weeks for children.Citation35
The first of these studies recruited adults and adolescents (mean age 26 years) with CF and a mean FEV1 of 63.6% at entry to the study. The treatment effect of ivacaftor was an increase in FEV1 of 10.6%. This was seen within two weeks of treatment and was sustained to week 48 and then to week 96 in the extended open-label study.Citation33,Citation34 Those treated with ivacaftor also had a 55% decrease in respiratory exacerbations, a reduction in sweat chloride values (a measure of CFTR function) in the order of 50–60 mmol/L, and a weight gain of 2.7 kg more than in the placebo group.Citation33 Improvements in quality of life as measured by the Cystic Fibrosis Questionnaire-Revised were also seen in those treated with ivacaftor. After the initial 24-week trial period, those who had been treated with placebo (n = 63) were started on treatment with ivacaftor. After 48 weeks on treatment, similar improvements in FEV1 of 9.4% were seen.Citation34
The second study evaluated ivacaftor in 52 children (mean age nine years) who had a higher FEV1 (mean 84%) than the previous group at entry to the study.Citation34 Only four children had an FEV1 < 70% at baseline. An overall ivacaftor treatment effect of 9.3% predicted FEV1 was shown. A smaller difference was shown between ivacaftor (n = 11) and placebo (n = 10) in the subgroup with a baseline FEV1 < 90% (6.9% improvement). Improvements were sustained to week 72 in the open-label extension study.Citation34 Although there were similar increases in weight and sweat chloride as in the adult study, no improvement was seen in the CF Quality of Life Questionnaire-Revised at either 24 or 48 weeks.Citation34 Again, those children who were switched from placebo to ivacaftor (n = 22) in the follow-on open-label study showed a mean improvement in FEV1 of 8.1% after 24 weeks.Citation34,Citation35
The ivacaftor trials have highlighted the need for new clinical endpoints, particularly in children or older patients with mild lung disease.Citation36 Although reductions in decline of FEV1 are often used as a measure of success in clinical trials, the sensitivity of this measure is limited in those with mild lung disease, such as the majority of the study group aged 6–12 years.Citation37,Citation38 This has provoked interest in the search for establishing reliable endpoints including sweat chloride, nasal potential difference, and intestinal current, all of which are surrogate markers of CFTR function.Citation36
Ivacaftor is the first agent to show a reduction in the sweat chloride level to values below the diagnostic threshold for CF (60 mmol/L).Citation33 Interestingly, in the trials, decreases in sweat chloride did not correlate with improvement in FEV1 in those treated with ivacaftor.Citation39 Further, there was not a threshold level for change in sweat chloride above which an improvement in FEV1 was apparent.Citation39 The lack of correlation of sweat chloride with improvement in FEV1 underlines the many physiological, environmental, and genetic factors that are likely to modulate CF disease severity and the difficulty in finding robust endpoints for clinical trials.
Safety
There are no identified adverse effects when ivacaftor is compared with placebo. However, although safety data are available for 60 weeks of treatment in the trials, the long-term safety profile of ivacaftor over a lifetime or its safety in young children is unknown. Most recently, an animal study showed that juvenile rats treated with ivacaftor from postnatal days 7–35 at dose levels of 10 mg/kg/day and higher (approximately one tenth the maximum recommended human dose) developed cataracts.Citation40 There is uncertainty about the relevance of this risk to children, given that there are differences in eye development between humans and rats.Citation40 A further safety study is being conducted in children younger than 11 years of age (when eye development is usually complete) who will be followed up for at least two years with assessment at six-monthly intervals for both visual acuity and cataracts. The safety of ivacaftor in pregnancy and breast feeding has also not been established. As women remain healthier, with increased life expectancy and increased expectations from life, the safety of CFTR modulators in pregnancy needs to be considered.
Clinical utility
Current data suggest that ivacaftor could potentially prevent progressive lung damage and loss of lung function in patients with CF. Although it is unlikely that established structural lung damage will be reversed, correction of the chloride transport defect may allow aggressive symptomatic therapy to become more effective. Infants identified with the Gly551Asp mutation at newborn screening are most likely to benefit over their lifetime, potentially with a reduction in the need for other long-term treatments. The safety profile of ivacaftor in infants has not yet been established, but a trial of ivacaftor in children under six years of age with the Gly551Asp mutation is planned. However, starting ivacaftor therapy at birth will not prevent damage that is already present (such as congenital bilateral absence of the vas deferens and meconium ileus).
The financial viability of long-term use of ivacaftor is a concern, given that the cost per patient year in the United States is in the region of US $294,000.Citation41 Although ivacaftor is currently licensed only for the Gly551Asp mutation, which affects less than 5% of the CF population, it represents a tremendous financial burden over a lifetime. It is also important to put in perspective that the increase in FEV1 and reduction in pulmonary exacerbations are similar to those seen in clinical trials for dornase alpha and azithromycin.Citation42,Citation43
Other class III CFTR mutations
In vitro data suggest that ivacaftor has a similar effect on other CFTR forms with gating defects (class III mutations) and support investigation of the potential clinical benefit of ivacaftor in CF patients who have CFTR gating mutations beyond Gly551Asp.Citation44 Extending the ivacaftor license further to other CFTR class III mutations (gating defects) would allow 10%– 15% of patients with CF to benefit from treatment.Citation45 However, clinical studies in patients with individual class III mutations are difficult because of the low frequency at which many of these mutations occur. Expanding the pool of patients who are eligible for treatment with ivacaftor might be accomplished by allowing in vitro data or in vivo “n-of-1” studies (with, for example, reduction in sweat chloride concentration as an outcome measure) to validate the use of the drug.Citation45
Phe508del mutations
A specific small-molecule compound or combination of compounds for Phe508del, the most common CFTR proteins mutation, would allow over 90% of the CF population to be treated with a disease-modifying therapy.Citation6,Citation7 Phe508del CFTR proteins are largely destroyed in the endoplasmic reticulum, but channels that reach the cell surface do not open normally and are retrieved much faster than normal CFTR. In vitro studies have also shown that ivacaftor increases the activity of Phe508del CFTR channels, provided that they reach the cell surface.Citation25
Ivacaftor monotherapy has been trialed in patients with Phe508del CFTR with much less success than in those with Gly551Asp. Efficacy results from an early clinical study in which patients homozygous for the Phe508del mutation (n = 140) were randomized (4:1 ratio) to receive 150 mg of ivacaftor every 12 hours or placebo for 16 weeks in addition to their prescribed CF therapies were disappointing.Citation46 Treatment with ivacaftor showed no improvement in the primary outcome measure, ie, an increase in predicted FEV1 after 16 weeks, or in any secondary outcome measures, eg, weight gain or improvement in quality of life.Citation46 There was no clinically meaningful change in sweat chloride levels, although there was a slight decrease (2.9 mmol/L) from baseline in the ivacaftor group.Citation46
In combination with other small-molecule compounds
Another small-molecule compound, VX-809, a CFTR corrector, was developed after screening 164,000 small molecules for compounds that increased Phe508del CFTR-mediated chloride transport.Citation47 VX-809 showed some efficacy in vitro by enhancing Phe508del CFTR processing and chloride transport in cultured human bronchial epithelial cells.Citation47 However, the effect was much less than that seen with ivacaftor, and only 14% of epithelial cells showed improvement in chloride processing and secretion.Citation47 Although the exact level of CFTR correction required to prevent or slow disease progression is unknown, it has been suggested that CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to CF airway epithelium.Citation48 Early human studies with VX-809 monotherapy indicated a small dose-dependent effect on sweat chloride (<10 mmol/L) but no effect on lung function or patient-reported outcomes over the 28-day study period.Citation49
However, it was hypothesized, based on in vitro data in cultured epithelial cells, that the combination of a CFTR corrector (VX-809) and a potentiator (ivacaftor) may be more effective than either agent alone. A VX-809-ivacaftor combination approach has progressed to Phase II investigation. Interim analysis of results from this ongoing Phase II trial in those homozygous for the Phe508del CFTR mutation are encouraging, demonstrating an 8.5% mean improvement in predicted FEV1 when compared with placebo.Citation50 However, the decrease in sweat chloride was again small at 10 mmol/L.Citation50 Secondly, there was a much smaller treatment effect on FEV1 predicted in those heterozygous for the Phe508del mutation.Citation50 Larger clinical trials are awaited.
An ongoing multicenter, double-blind, placebo-controlled Phase II trial is examining the therapeutic efficacy and safety of VX-661, another CFTR corrector, both alone and in combination with ivacaftor in adults homozygous for the Phe508del mutation.Citation51 Initial reports have observed dose-dependent mean relative improvements of up to 9% in FEV1 predicted only in patients receiving the combination (VX-661 and ivacaftor) treatment at high doses versus placebo at day 28, which returned to baseline during a 28-day post-washout treatment.Citation52 There are plans to conduct further studies of VX-661 to evaluate further its potential for late-stage development, pending regulatory discussions.
Small-molecule “CFTR suppressor” compounds
Another small molecule compound, ataluren (PTC124), a CFTR suppressor, has been designed to overcome the adverse effects of a nonsense mutation by inducing selective ribosomal read through of mRNA containing a premature stop codon and allowing translation of a full length protein.Citation53 Nonsense mutations are particularly common in Jewish communities, as previously stated. Ataluren is an orally bioavailable drug currently being investigated both in patients with CF and in those with Duchenne muscular dystrophy.
A small early clinical study in 23 patients from Israel with nonsense mutations involved taking ataluren three times daily for two weeks (4 mg/kg morning, 4 mg/kg midday, 8 mg/kg evening) followed by a washout period and a further two weeks at a higher dose (10 mg/kg, 10 mg/kg, 20 mg/kg).Citation54 Ataluren improved chloride ion transport as assessed by nasal potential difference (measuring the voltage across the nasal epithelium).Citation54 In contrast, a previous trial with ataluren in 11 patients from the United States did not show electrophysiological improvement.Citation55 Results from a larger, multinational, randomized, double-blind, placebo-controlled trial including both adults and children (n = 238) with CF who received ataluren 10 mg/kg (morning and midday) or 20 mg/kg (evening) or placebo has not shown a significant change in FEV1 or in respiratory exacerbations.Citation56,Citation57
Ataluren has also been trialed in boys with Duchenne muscular dystrophy, a genetic disease caused by a premature stop mutation in 5%–13% of cases.Citation58 In early studies involving 38 boys with the disease who took ataluren for about a month, there were indications that muscle destruction had lessened, and dystrophin production increased in at least some trial participants. However, a larger randomized, multicenter, double-blind, placebo-controlled trial which followed 174 boys with Duchenne muscular dystrophy showed the primary endpoint of the six-minute walk test did not reach statistical significance.Citation58
Other potential treatments in clinical trials
The knowledge base surrounding the structure and function of CFTR that has accumulated in the last 20 years continues to grow. Translational research feeding from this is now yielding compounds that provide real prospects of pharmacotherapy for this disease.
A major function of CFTR in healthy airways is to maintain an adequate liquid layer on the airway surface. In CF, in addition to chloride channel dysfunction, defective regulation of epithelial sodium channels (ENaC) leads to transport of salt and water out of the airways, exacerbating the reduction in airway surface liquid. Therefore, strategies designed to inhibit ENaC function may result in clinical benefit. A potential for treatment of CF is to use agents that inhibit ENaC either alone or as adjuncts to CFTR correctors and/or potentiators.
The possibility of using RNA interference to reduce expression of ENaC in the mouse lung has been investigated as a strategy to reduce sodium hyperabsorption in the CF lung. Potent ENaC alpha-targeted siRNA molecules capable of efficient knockdown of the ENaC alpha, beta, and gamma subunits have been demonstrated to inhibit the expression of ENaC alpha mRNA in murine and human cells grown in vitro and following in vivo delivery to the mouse lung.Citation59 In addition, ENaC alpha-targeted shRNA can be used to knock down both human and mouse ENaC alpha mRNA.Citation60 Further work is needed to assess the functional consequences of inhibiting ENaC in the lung.
Cell-based assays have shown improvements in trafficking and function of CFTR at the cell membrane with application of partial siRNA knockdown of the heat shock protein 90 cochaperone ATPase regulator Aha1.Citation61 Histone deacetylase inhibitors and siHDAC7 may also be used to direct a global tissue response to Phe508del dysfunction that may ultimately improve surface hydration and reduce the inflammatory responses characteristic of the disease.Citation62 A further way in which a pharmacological approach to CF can be considered is to recruit alternative chloride channels, such as calcium-activated chloride channel, to act as surrogates for CFTR. A number of P2Y2 receptor agonists have been investigated and found to operate by increasing [Ca2+]i which, in turn, activates calcium-activated chloride channels. Some of these compounds are currently undergoing clinical trials.
Conclusion
In the last two decades, significant progress has been made in the field of CFTR discovery, with improved understanding and the development of potentially disease-modifying treatments. Three new agents, ie, ivacaftor, VX-809, and ataluren, target the basic defects in production of CFTR. At present, ivacaftor has shown the greatest therapeutic promise and trials have shown beneficial effects on lung function, number of respiratory exacerbations, surrogate markers of CFTR function, nutrition, and quality of life. However, ivacaftor does have significant limitations in terms of cost and the current licensing agreement is only for those patients with a Gly551Asp mutation, which represents only around 5% of the CF population. Despite these limitations, ivacaftor is an exciting development which may pave the way for other CFTR-modulating treatments which may benefit more patients. At present, efforts to treat the Phe508del CFTR mutation with ivacaftor or other small-molecule compounds have not shown adequate therapeutic efficacy. Further trials are currently underway and their results are awaited. Importantly, as future therapies aim to prevent rather than improve existing organ damage, FEV1 is unlikely to be a sensitive enough endpoint for future trials. The reliability of other endpoints, even established measurements such as sweat chloride, need to be examined further as clinical endpoints.
Disclosure
The authors report no conflicts of interest in this work.
References
- Farrell PM The prevalence of cystic fibrosis in the European Union J Cyst Fibros 2008 7 450 453 18442953
- Kerem B Rommens JM Buchanan JA Identification of the cystic fibrosis gene: genetic analysis Science 1989 245 1073 1080 2570460
- Rommens JM Iannuzzi MC Kerem B Identification of the cystic fibrosis gene: chromosome walking and jumping Science 1989 245 1059 1065 2772657
- Riordan JR Rommens JM Kerem B Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA Science 1989 245 1066 1073 2475911
- Cystic Fibrosis Mutation Database CFTR1 2013 Available from: http://www.genet.sickkids.on.ca/cftr/StatisticsPage.html Accessed May 16, 2013
- Cystic Fibrosis Registry Report Trust Annual Data Report 2010 12 2011 Available from: http://www.cftrust.org.uk/aboutcf/publications/cfregistryreports/UK_CF_Registry_-_Annual_Data_Report_2010.pdf Accessed May 16, 2013
- Patient Registry Annual Data Report Bethesda, MD Cystic Fibrosis Foundation 2011Available from: http://www.cff.org/LivingWithCF/QualityImprovement/PatientRegistryReport/Accessed May 16, 2013
- Cohen-Cymberknoh M Shoseyov D Kerem E Managing cystic fibrosis: strategies that increase life expectancy and improve quality of life Am J Respir Crit Care Med 2011 183 1463 1471 21330455
- Oxford University GeneMedicine CFTR Protein Structure Available from: http://www.genemedresearch.ox.ac.uk Accessed April 20, 2013
- Bear CE Li CH Kartner N Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR) Cell 1992 68 809 818 1371239
- Gadsby DC Vergani P Csanady L The ABC protein turned chloride channel whose failure causes cystic fibrosis Nature 2006 440 477 483 16554808
- Knowles M Gatzy J Boucher R Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis N Engl J Med 1981 305 1489 1495 7300874
- Mall M Grubb BR Harkema JR O’Neal WK Boucher RC Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice Nat Med 2004 10 487 493 15077107
- Farrell PM Rosenstein BJ White TB Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report J Pediatr 2008 153 S4 S14 18639722
- Kerem B Chiba-Falek O Kerem E Cystic fibrosis in Jews: frequency and mutation distribution Genet Test 1997 1 35 39 10464623
- McKone EF Emerson SS Edwards KL Aitken ML Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study Lancet 2003 361 1671 1676 12767731
- Hamosh A King TM Rosenstein BJ Cystic fibrosis patients bearing both the common missense mutation Gly-Asp at codon 551 and the delta F508 mutation are clinically indistinguishable from delta F508 homozygotes, except for decreased risk of meconium ileus Am J Hum Genet 1992 51 245 250 1379413
- Highsmith WE Burch LH Zhou Z A novel mutation in the cystic fibrosis gene in patients with pulmonary disease but normal sweat chloride concentrations N Engl J Med 1994 331 974 980 7521937
- Kulczycki LL Kostuch M Bellanti JA A clinical perspective of cystic fibrosis and new genetic findings: relationship of CFTR mutations to genotype-phenotype manifestations Am J Med Genet A 2003 116A 262 267 12503104
- Davis PB Schluchter MD Konstan MW Relation of sweat chloride concentration to severity of lung disease in cystic fibrosis Pediatr Pulmonol 2004 38 204 209 15274098
- McKone EF Goss CH Aitken ML CFTR genotype as a predictor of prognosis in cystic fibrosis Chest 2006 130 1441 1447 17099022
- de Gracia J Mata F Alvarez A Genotype-phenotype correlation for pulmonary function in cystic fibrosis Thorax 2005 60 558 563 15994263
- Shoshani T Augarten A Gazit E Association of a nonsense mutation (W1282X), the most common mutation in the Ashkenazi Jewish cystic fibrosis patients in Israel, with presentation of severe disease Am J Hum Genet 1992 50 222 228 1370365
- Rowntree RK Harris A The phenotypic consequences of CFTR mutations Ann Hum Genet 2003 67 471 485 12940920
- Van Goor F Straley KS Cao D Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules Am J Physiol Lung Cell Mol Physiol 2006 290 L1117 L1130 16443646
- Clinical and Functional Translation of CFTR CFTR2 Available from: http://www.cftr2.com Accessed January 19, 2013
- Oakland M Sinn PL McCray PBJr Advances in cell and gene-based therapies for cystic fibrosis lung disease Mol Ther 2012 20 1108 1115 22371844
- The UK Cystic Fibrosis Gene Therapy Consortium Gene therapy multi-dose clinical trial Available from: http://www.cfgenetherapy.org.uk Accessed April 20, 2013
- Carlile GW Robert R Zhang D Correctors of protein trafficking defects identified by a novel high-throughput screening assay Chembiochem 2007 8 1012 1020 17497613
- Galietta LV Jayaraman S Verkman AS Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists Am J Physiol Cell Physiol 2001 281 C1734 C1742 11600438
- Van Goor F Hadida S Grootenhuis PD Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770 Proc Natl Acad Sci U S A 2009 106 18825 18830 19846789
- Eckford PD Li C Ramjeesingh M Bear CE Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner J Biol Chem 2012 287 36639 36649 22942289
- Ramsey BW Davies J McElvaney NG A CFTR potentiator in patients with cystic fibrosis and the G551D mutation N Engl J Med 2011 365 1663 1672 22047557
- Vertex Pharmaceuticals Kalydeco© Available from: http://www.ema.europa.eu/docs/en_GB/document_library Acessed January 20, 2013
- [No authors listed] Ivacaftor (Kalydeco) for cystic fibrosis Med Lett Drugs Ther 2012 54 29 30 22499233
- De Boeck K Kent L Davies J CFTR biomarkers: time for promotion to surrogate end-point Eur Respir J 2013 41 203 216 22878883
- Konstan MW Wagener JS Yegin A Millar SJ Pasta DJ Vandevanter DR Design and powering of cystic fibrosis clinical trials using rate of FEV(1) decline as an efficacy endpoint J Cyst Fibros 2010 9 332 338 20646968
- Que C Cullinan P Geddes D Improving rate of decline of FEV1 in young adults with cystic fibrosis Thorax 2006 61 155 157 16384880
- Durmowicz AG Witzmann KA Rosebraugh CJ Chowdhury BA Change in sweat chloride as a clinical end point in cystic fibrosis clinical trials: the ivacaftor experience Chest 2013 143 14 18 23276841
- US Food and Drug Administration Notification to the Cystic Fibrosis Community on Kalydeco (ivacaftor) Available from: http://www.fda.gov/Drugs/DrugSafety Accessed April 20, 2013
- Bush A Simmonds NJ Hot off the breath: ‘I’ve a cost for’ – the 64 million dollar question Thorax 2012 67 382 384 22407889
- Fuchs HJ Borowitz DS Christiansen DH Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group N Engl J Med 1994 331 637 642 7503821
- Equi A Balfour-Lynn IM Bush A Rosenthal M Long term azithromycin in children with cystic fibrosis: a randomised, placebo-controlled crossover trial Lancet 2002 360 978 984 12383667
- Yu H Burton B Huang CJ Ivacaftor potentiation of multiple CFTR channels with gating mutations J Cyst Fibros 2012 11 237 245 22293084
- Davis PB Yasothan U Kirkpatrick P Ivacaftor Nat Rev Drug Discov 2012 11 349 350 22543461
- Flume PA Liou TG Borowitz DS Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation Chest 2012 142 718 724 22383668
- Van Goor F Hadida S Grootenhuis PD Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809 Proc Natl Acad Sci U S A 2011 108 18843 18848 21976485
- Zhang L Button B Gabriel SE CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium PLoS Biol 2009 7 e1000155 19621064
- Clancy JP Rowe SM Accurso FJ Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation Thorax 2012 67 12 18 21825083
- Boyle M The investigational CFTR corrector, VX-809, co-administered with the oral potentiator ivacaftor improved CFTR and lung function in F508del homozygous patients: Phase 2 study results Poster 260 presented at the 26th North American Cystic Fibrosis Conference Orlando, FL October 11–13, 2012
- ClinicalTrials.gov Study of VX-661 alone and in combination with VX-770 in subjects homozygous to the F508del-CFTR mutation Available from: http://clinicaltrials.gov/show/NCT01531673 Accessed April 20, 2013
- Vertex Treatment with VX-661 and ivacaftor in a Phase 2 study resulted in statistically significant improvements in lung function in people with cystic fibrosis who have two copies of the F508del mutation Available from: http://investors.vrtx.com/releasedetail.cfm?releaseid=757597 Accessed April 20, 2013
- Welch EM Barton ER Zhuo J PTC124 targets genetic disorders caused by nonsense mutations Nature 2007 447 87 91 17450125
- Kerem E Hirawat S Armoni S Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial Lancet 2008 372 719 727 18722008
- Clancy JP Rowe SM Bebok Z No detectable improvements in cystic fibrosis transmembrane conductance regulator by nasal aminoglycosides in patients with cystic fibrosis with stop mutations Am J Respir Cell Mol Biol 2007 37 57 66 17347447
- Kerem E Wilschanski M De Boeck K Phase 3 study of ataluren (PTC124®) in nonsense mutation cystic fibrosis (nmCF): baseline data J Cyst Fibros 2011 10 S17
- PTC Therapeutics Inc Top-line data from phase 3 trial of ataluren in patients with nonsense mutation cystic fibrosis show promising results Available from: http://ptct.Client.Shareholder.Com/releasedetail Accessed January 26, 2013
- Mendell JR Rodino-Klapac L Sahenk Z Gene therapy for muscular dystrophy: lessons learned and path forward Neurosci Lett 2012 527 90 99 22609847
- Hyde SC Painter H Gill DR EnaC knockdown in the mouse lung using RNAI Pediatr Pulmonol 2007 42 306
- Harding-Smith RE Gill DR Hyde SC Knockdown of ENaCá, as a treatment for cystic fibrosis lung disease Mol Ther 2011 19 1360 1398
- Wang X Venable J LaPointe P Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis Cell 2006 127 803 815 17110338
- Hutt DM Herman D Rodrigues AP Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis Nat Chem Biol 2010 6 25 33 19966789