Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized clinically by memory and cognitive dysfunction. Unfortunately, there is no effective therapeutic method for AD treatment or ways to halt disease progression. Many mechanisms are involved in the disease, including genes mutation and protein dysfunction. RNA interference (RNAi) technology may potentially be able to control AD. It can inhibit the protein expression of specific genes by activating a sequence-specific RNA degradation process. This is a powerful tool with which to study gene function, investigate the mechanism of the disease, and validate drug targets. In this review, we highlight the advances in RNAi technology in the investigation and treatment of AD.
Introduction
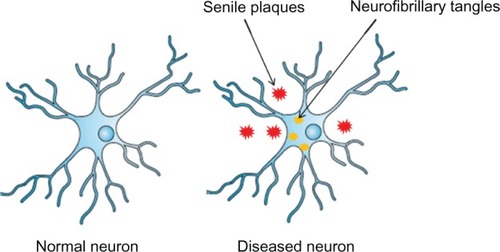
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized clinically by memory and cognitive dysfunction. It was first reported in 1907 by the German neurologist Alois Alzheimer.Citation1 The disease is generally classified into two types: sporadic AD (SAD) and familial AD (FAD). The former accounts for more than 90% of all cases and occurs in patients aged 65 years or older. The latter is rare and the age of onset is earlier than that of SAD, with symptoms appearing when patients are in their 40s or 50s.Citation2 Three genes lead to FAD – amyloid precursor protein (APP), presenilin 1 (PS1), and presenilin (PS2) – while the å4 allele of the apolipoprotein E gene has been identified as the major risk factor for SAD.Citation3 The neuropathology of AD is characterized by two types of lesions, extracellular senile plaques and intracellular neurofibrillary tangles (NFTs), which are composed of, respectively, â-amyloid (Aâ), a cleavage product of APP,Citation4 and aberrantly phosphorylated tau, a microtubule-associated protein ().Citation5,Citation6
Figure 1 A normal neuron (left) and an Alzheimer’s disease-affected neuron (right).
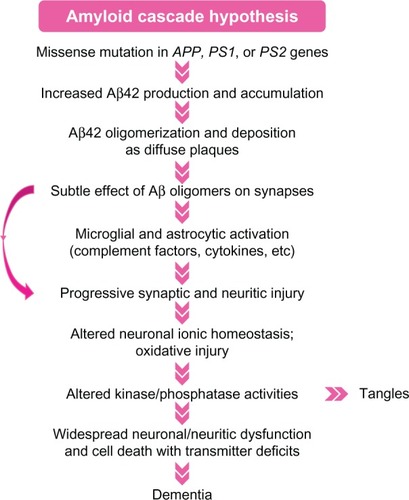
The detailed mechanism of the disease is still not clear and there are several hypotheses accounting for it, the most influential of which is the amyloid cascade hypothesis.Citation7–Citation9 According to this hypothesis, the accumulation of Aâ aggregates in the brain triggers a complex neurodegenerative cascade, which results in progressive cognitive impairment and dementia ().Citation7–Citation9 A variety of factors may induce AD and it is difficult to clarify what the functions of these factors are and how they work together to result in AD; however, RNA interference (RNAi) may make it easy to determine these.
Figure 2 Hypothetical pathogenetic steps of the amyloid cascade hypothesis.
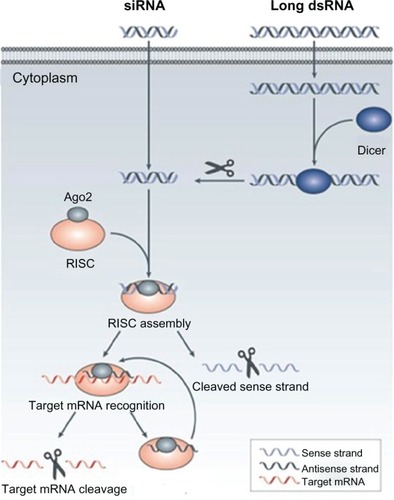
RNAi was discovered in 1998 by Andrew Fire and Craig Mello, who were awarded the Nobel Prize in Physiology and Medicine for their discovery 8 years later. RNAi regulates the expression of genes by controlling the synthesis of protein with a posttranscriptional gene-silencing mechanism. RNAi is triggered by the presence of long pieces of double-strand RNA, which cleave into the fragment known as small interfering RNA (siRNA) (21–23 nucleotides long) by the Dicer enzyme in the cytoplasm. Thereafter, siRNA is incorporated into a protein complex called the “RNA-induced silencing complex” and the sense strand of the siRNA is cleaved. The antisense strand guides the RNA-induced silencing complex to bind with messenger RNA (mRNA), which is complementary to the antisense strand and degrades it ().Citation10,Citation11
Figure 3 The mechanism of RNA interference.
RNAi is well suited to probe the biological function of individual genes, genes in pathways, genes known to be associated with diseases (including inherited genetic diseases) and viral pathogens. Further, RNAi may be used to discover novel genes critical to pathogenic processes.Citation12 Therefore, RNAi has major implications for basic and biomedical research that may lead to a number of clinical applications.Citation13–Citation15 Compared with other therapeutics, the principal advantage of RNAi is that all targets – including those that are usually unable to be targeted with drugs – are able to be drugged using RNAi. This is because, in theory, any transcript that encodes a protein that causes or contributes to a disease can be targeted by RNAi (see ).Citation16 To date, RNAi has been widely used in basic bioscience – including in the study of AD, which may be caused by different genes and proteins – and such usage may lead to novel therapies in the future.
Table 1 Comparison of RNAi with traditional pharmaceutical drugs
Here, we review the recent advances in the use of RNAi in AD research, investigation, and treatment.
RNAi and Aâ
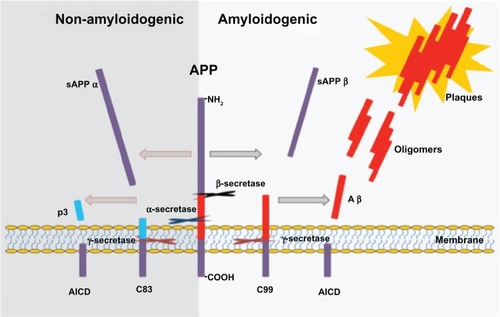
Aâ is a peptide of 40–42 amino acids that is processed from APP, which is a transmembrane protein expressed in many tissues and concentrated in neuron synapses. The primary function of APP is unknown, although it has been suggested that it is a regulator of synapse formation,Citation17 neural plasticity,Citation18 and iron export.Citation19 The metabolism of APP is well characterized, mediated by a series of enzymes termed “secretases” (á, ß, and ã).Citation20 Cleavage of APP by á-secretase occurs in the middle of the peptide, generating non-amyloidogenic APP fragments. In contrast, the sequential cleavage of APP by ß-secretase followed by ã-secretase generates a variety of potentially amyloidogenic Aß; the common isoforms are fragments of 40 or 42 amino acids called Aß40 and Aß42, respectively ().Citation20,Citation21 The Aß40 form is the more common of the two, but Aß42 is the more fibrillogenic and is considered to be primarily responsible for neuronal damage.Citation7 Aggregated Aâ forms insoluble filaments and these are deposited both intra- and extraneuronally.Citation22 These filaments constitute one of the main components of senile plaque, the primary hallmark of AD. In many cases of AD, the senile plaques are also found deposited in the walls of cerebral blood vessels.Citation4 Aâ is produced in a variety of subcellular locations, including the endoplasmic reticulum/intermediate compartment,Citation23 the trans-Golgi network (TGN),Citation24 and the endosomal/ lysosomal system.Citation25 The intracellular trafficking of Aâ production remains uncertain. The â-site APP-cleaving enzyme (BACE) is the main â-secretase synthesized as an immature and inactive form in the endoplasmic reticulum (ER)Citation26 and this undergoes maturation during its transport to the cell surface along the secretory pathway.Citation27–Citation31 The mature BACE is internalized from cell surface to endosomes,Citation27,Citation32 followed by sorting to the TGN for recycling or to lysosome for degradation.Citation30,Citation31 Using an RNAi approach, it has been found that the majority of APP is processed in the TGN to produce Aâ40 and that the retromer is key in mediating APP recycling from early endosomes to the TGN. This results in the accumulation of endocytosed APP in early endosomes and a reduction in APP processing.Citation33
Figure 4 Proteolytic processing of APP by á-, ß-, and ã-secretase.
Abbreviation: APP, amyloid precursor protein.
In addition, increasing levels of sortilin in post-mortem brain tissue of AD patients leads to increased BACE1-mediated cleavage of APP in cultured cells. RNAi suppression of sortilin results in decreased BACE1-mediated cleavage of APP. Further, sortilin expression redistributes BACE1 from the TGN to the endosomes and substantially reduces the retrograde trafficking of BACE1. It has been suggested that sortilin may be a modulator of BACE1 retrograde trafficking and subsequent generation of Aâ.Citation34
Receptor tyrosine kinases (RTKs) comprise a large family of cell surface receptors, which transduce various extracellular signals to the interior of cells.Citation35 BACE and subsequent Aâ production can be enhanced after stimulation of RTKs in cultured cells as well as in mouse hippocampus.Citation36 Stimulation of RTKs also induces BACE internalization into endosomes and Golgi apparatus. The enhancement of BACE activity and Aâ production on RTK activation could be specifically inhibited by Scr family kinase inhibitors and by depletion of endogenous c-Scr with RNAi.Citation36 GGA3, an adaptor protein which is recruited to the TGN by the Arf1-GTAase, is involved in BACE trafficking.Citation37–Citation39 If the levels of GGA3 are reduced, BACE levels are increased. RNAi silencing of GGA3 can elevate BACE and Aâ levels.Citation40
“Autophagy” is a conserved membrane trafficking pathway that mediates the delivery of cytoplasmic substrates to the lysosomes for degradation.Citation41 Impaired autophagic function is implicated in the pathology of various neurodegenerative diseases.Citation42 It has been found that Aß expression causes autophagosome accumulation.Citation43,Citation44 Moreover, decreased insulin-receptor signaling promotes the maturation of autophagosomes into degradative autolysosomes, whereas Aß impairs this process. “RNAi-mediated knockdown of lysosomal components results in enhanced Aß-toxicity and autophagosome accumulation. So, insulin-receptor signaling promotes the autophagic degradation of Aß.”Citation45 Amyloid precursor-like protein (apl-1) is a member of APP family, and loss of apl-1 leads to a severe molting defect and early larva lethalityCitation46 RNAi knockdown of apl-1 followed by drug testing on the acetylcholinesterase inhibitor aldicarb showed that loss of apl-1 leads to aldicarb hypersensitivity, indicating a defect in synaptic function.Citation47 Rab5, a small GTPase, regulates endosomal trafficking of vesicles from the plasma membrane to the early endosome.Citation46 Knockdown of the small GTPase rab-5 also leads to a dramatic decrease in the amount of apl-1 expression in neurons, suggesting that trafficking from the plasma membrane to the early endosome is important for apl-1 function.Citation47
It is assumed that á- and ß-secretase compete for APP as a substrate, but have opposite effects on Aß generation, as mentioned earlier. Increasing the APP á-secretase cleavage is considered a therapeutic approach for AD, as it is assumed to reduce Aß generation.Citation48 Alpha-secretase activities have been identified all belonging to a metalloprotease of the A disintegrin and metalloproteinase (ADAM) familyCitation49 RNAi-mediated knockdown of ADAM10 completely suppressed APP a-secretase cleavage in different cell lines and in primary murine neurons. In different cell lines, the reduction of a-secretase cleavage is not paralleled by a corresponding increase in the Aß generating ß-secretase cleavage, revealing that both alpha-secretase and beta-secretase do not always compete for APP as a substrate.Citation50
Thimet oligopeptidase (THOP1) is a thiol-sensitive metalloprotease with Zn2+, Mn2+, and Co2+ as cofactors, which cleaves peptide substrates of less than 18 amino acids.5152 It is primarily present in tissues rich in neuropeptides and hormones, suggesting a role in the processing of bioactive peptides.5354 The enzyme has been associated with the processing of APP at a ß-secretase site and with degradation of Aß peptide.Citation55,Citation56 Its overexpression is neuroprotective against Aß toxicity, while RNAi knockdown made neurons more vulnerable to amyloid peptide, which suggests that THOP1 operates against the toxic effects of Aß in the early stages of AD pathology and an increase in THOP1 expression might be part of a compensatory defense mechanism of the brain against an increased Aâ load.Citation57
Some other enzymes may also be involved with Aâ, such as acyl-coenzyme A cholesterol acyltransferase (ACAT-1), an enzyme that controls cellular equilibrium between free cholesterol and cholesteryl ester and modulates the proteolytic processing of APP in cell-based and animal models of AD.Citation58,Citation59 ACAT-1 RNAi, through a single transfection of ACAT-1 siRNA oligonucleotides, reduces cellular ACAT-1 protein by ~50%, cholesteryl ester levels by 22%, and causes a slight increase in the free cholesterol content of ER membranes, which correlates with reduced proteolytic processing of APP and a 40% decrease in Aâ secretion.Citation60 In addition, endogenous ferritin knockdown by RNAi can decrease iron in mammal cell cultures and promote the formation of well-ordered aggregates of Aâ while decreasing its toxicity.Citation61
RNAi and tau
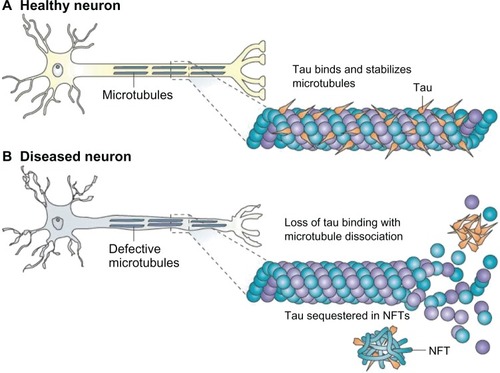
Tau proteins interact with tubulin, which makes up microtubules, to stabilize microtubules and promote the assembly of tubulin into microtubules. They are the product of alternative splicing from a single gene that in humans is designated “MAPT” (microtubule-associated protein tau). These proteins are mostly found in neurons rather than non-neuronal cells. When tau proteins are defective, they no longer stabilize microtubules properly.Citation62 They are capable of aggregating and fibrillating to form NFTs, another pathological hallmark of ADCitation5,Citation6 ().
Figure 5 (A) Tau facilitates microtubule stabilization within cells and is particularly abundant in neurons. (B) it is thought that tau function is compromised in Alzheimer’s disease and other tauopathies.
Notes: This probably results from both tau hyperphosphorylation, which reduces the binding of tau to microtubules, and the sequestration of hyperphosphorylated tau into neurofibrillary tangles (NFTs), which reduces the amount of tau that is available to bind microtubules. The loss of tau function leads to microtubule instability and reduced axonal transport, which could contribute to neuropathology. Copyright © 2009. Nature Publishing Group. Reproduced with permission from Brunden KR, Trojanowski JQ, Lee VM. Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat Rev Drug Discov. 2009;8(10):783–793.Citation97
Tau proteins play a critical role in AD pathogenesis; however, their molecular basis is unknown. Hyperphosphorylation of tau proteins can result in the self-assembly of paired helical filament and straight filament tangles, which are involved in the pathogenesis of AD.Citation63 SUT-2 is a gene required for tau neurotoxicity in a transgenic Caenorhabditis elegans model of tauopathy.Citation64 RNAi knockdown of MSUT2 (mammalian SUT-2) in cultured human cells overexpressing tau causes a marked decrease in tau aggregation.Citation65 Cyclin-dependent kinase 5 (CDK5) is a relevant kinase that has been hypothesized to contribute to the tau pathology.Citation66,Citation67 Silencing of CDK5 reduces the phosphorylation of tau in primary neuronal cultures and in the brain of wild-type C57BL/6 mice. Further, in one study, the knockdown of CDK5 strongly decreased the number of NFTs in the hippocampus of triple transgenic mice (3xTg-AD mice).Citation68
A recent concept that has emerged suggests that, rather than the amyloid plaques, small Aâ non-fibrillar oligomers could be the earliest mediators of neuronal dysfunction.Citation69,Citation70 Isolated Aâ dimers from the cortices of typical AD subjects first induce hyperphosphorylation of tau at AD-relevant epitopes in hippocampal neurons then disrupt the microtubule cytoskeleton and cause neuritic degeneration, all in the absence of amyloid fibrils. In one study, the researchers found that knocking down endogenous tau fully prevented the neurotic changes, whereas overexpressing human tau accelerated them. The results suggest natural Aâ dimers can induce AD-type tau phosphorylation then neuritic dystrophy.Citation71
RNAi and other investigation
AD is a neurodegenerative disorder characterized by deregulation of the neuronal cell cycle and differentiation control, eventually resulting in cell death.Citation72 During brain development, neuronal differentiation is regulated by Smad proteins, which are elements of the canonical transforming growth factor â (TGF-â) signaling pathway, linking receptor activation to gene expression.Citation73 In the normal adult brain, Smad proteins are constitutively phosphorylated and predominantly localized in neuronal nuclei.Citation74 Using RNAi in cell cultures to mimic the neuronal deficiency of Smad proteins observed in AD results in elevation of cell cycle-dependent kinase 4 and retardation of neurite outgrowth, giving rise to neuronal dedifferentiation and cell death.Citation75 Polo-like kinase 1 (Plk1) is an established regulator of many cell cycle-related events. Inhibition of Plk1 kinase activity or depletion of Plk1 by RNAi reduces Aâ-induced neuronal cell death.Citation76
While, as previously noted, three gene mutations lead to the development of FAD, SAD has only one clear genetic modifer: the å4 allele of the apolipoprotein E gene.Citation3 RNAi knockdown of apl-1, lipoprotein receptor-related protein 1, and cholesterol starvation leads to aldicarb hypersensitivity, indicating a defect in synaptic function.Citation77 Mutations associated with FAD change the ratio of Aâ42 to Aâ40 by increasing the production of Aâ42, which is linked to amyloid plaque formation.Citation78,Citation79 To determine whether the two types of AD share a common underlying molecular cause, RNAi has been used to systematically silence 24 genes linked to SAD. The results show that SAD genes do not specifically alter the Aâ42 to Aâ40 ratio, thus suggest that these genes probably contribute to AD through distinct mechanisms.Citation80
Mitochondrial function – which relies heavily on its morphology and distribution – and alterations in mitochondria morphology and distribution have been increasingly implicated in neurodegenerative diseases such as AD.Citation81 Dynamin-like protein 1 (DLP1), a regulator of mitochondrial fission and distribution,Citation82 has been found at significantly lower levels in SAD fibroblasts.Citation83 Expression of DLP1 by miR RNAi in human fibroblasts from normal subjects significantly increased mitochondrial abnormalities, which suggests that DLP1 reduction causes mitochondrial abnormalities in SAD fibroblasts.Citation84 Experiments have also demonstrated that elevated oxidative stress and increased Aâ production are likely to be the pathogenic factors that cause DLP1 reduction and abnormal mitochondrial distribution in AD cells.Citation84
RNAi and AD therapy
Over the past decade, at least 21 siRNA therapeutics have been developed for more than a dozen diseases, including various cancers, viruses, and genetic disorders.Citation84,Citation85 Many endeavors have also been made to improve AD treatment. The most advanced of these has focused on Aâ peptide production and clearance.Citation3 The therapeutic potential of RNAi in AD has been demonstrated through allele-specific gene silencing by short-hairpin RNA (shRNA) to selectively suppress mutant APP.Citation86 An anti-APPsw shRNA was delivered by the recombinant adeno-associated virus to the hippocampus of AD transgenic mice (APP/PS1). No neuronal toxicity was detected in short-and long-term transduction experiments. Over the long-term, bilateral hippocampal expression of anti-APPsw shRNA mitigated abnormal behaviors in this mouse model of AD. The difference in phenotype progression was associated with reduced levels of soluble Aâ but not with a reduced number of amyloid plaques.Citation87 Intravenously injected rabies viral glycoprotein-targeted exosome delivered siRNA specifically to neurons in mouse brain, resulting in strong mRNA (60%) and protein (62%) knockdown of BACE1 without the corresponding level of immune stimulation.Citation88 In addition, CBP-1 has been inhibited by RNAi to assess the age-dependent acceleration of the mortality rate of 30 drugs that reliably protected mammalian neurons.Citation89 Further, when a high-throughput RNAi approach is used to screen 572 kinases in the human genome for effects on tau hyperphosphorylation, EIF2AK2 effects may result from effects on tau protein expression, whereas DYRK1A and AKAP13 are likely to be more specifically involved in tau phosphorylation pathways.Citation90
However, several obstacles remain in the clinical development of RNAi-based therapeutics, the greatest of which is delivery. Suitable delivery methods are needed to transfer siRNA to the specified targets and control of potential off-target effects also needs to be considered.Citation85,Citation86 AD is genetically heterogeneous and multifactorial.Citation91 It may not be treated with a single siRNA. Thus, how to formulate the different RNAi components and deliver them into the target is a significant problem. Further, there have been few ideal vectors for clinical application.Citation92
Conclusion
AD has the greatest unmet medical needs in neurology.Citation93 Although a century has elapsed since the identification of AD, there are still no ideal therapeutic approaches for treating the disease. Current drugs improve symptoms but do not have profound disease-modifying effects.Citation94 A much deeper insight into this disease is required. RNAi has become a valuable research tool to help us understand this disease and to provide an efficient method for clinical applications.Citation95,Citation96 Due to its advantages over other current therapeutics, we strongly believe that RNAi technology will be helpful in addressing unresolved questions concerning AD via in vitro and in vivo approaches. Given the pace of new findings and discovery of applications, RNAi is likely to remain a major new therapy for the foreseeable future.
Acknowledgment
This research was supported in part by the Projects of National Science Foundation of China (No.81071025 and 81171203), and Projects of the Shanghai Committee of Science and Technology, China (No.11nm0503300, 11410708900 and 12XD1403800). The authors thank the Analytical Center of Shanghai JiaoTong University for technical support.
Disclosure
The authors declare no conflicts of interest in this work.
References
- Alzheimer A Über eine eigenartige Erkrankung der Hirnrinde [Concerning a novel disease of the cortex] Allgemeine Zeitschrift fur Psychiatrie und Psychisch-gerichtliche Medizin 1907 64 146 148 German
- Blennow K de Leon MJ Zetterberg H Alzheimer’s disease Lancet 2006 368 9533 387 403 16876668
- Citron M Alzheimer’s disease: strategies for disease modification Nat Rev Drug Discov 2010 9 5 387 398 20431570
- Glenner GG Murphy MA Amyloidosis of the nervous system J Neurol Sci 1989 94 1–3 1 28 2559163
- Goedert M Wischik CM Crowther RA Walker JE Klug A Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau Proc Natl Acad Sci U S A 1988 85 11 4051 4055 3131773
- Goedert M Spillantini MG Jakes R Rutherford D Crowther RA Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease Neuron 1989 3 4 519 526 2484340
- Hardy JA Selkoe DJ The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics Science 2002 297 5580 353 356 12130773
- Hardy J Alzheimer’s disease: the amyloid cascade hypothesis: an update and reappraisal J Alzheimers Dis 2006 9 Suppl 3 151 153 16914853
- Golde TE Petrucelli L Lewis J Targeting Abeta and tau in Alzheimer’s disease, an early interim report Exp Neurol 2010 223 2 252 266 19716367
- Fire A Xu S Montgomery MK Kostas ASA Driver SE Mello CC Potent and specifIc genetic interference by double-stranded RNA in Caenorhabditis elegans Nature 1998 391 6669 806 811 9486653
- Daneholt B Advanced information: RNA interference. Nobelprize.org [website on Internet]. 2012 Available from: http://www.nobelprize.org/nobel_prizes/medicine/laureates/2006/advanced.html AccessedNovember 8, 2012
- Seyhan AA Rya TE RNAi screening for the discovery of novel modulators of human disease Curr Pharm Biotechnol 2010 11 7 735 756 20420566
- Davidson BL Boudreau RL RNA interference: a tool for querying nervous system function and an emerging therapy Neuron 2007 53 6 781 788 17359914
- Gonzalez-Alegre P Paulson HL Technology insight: therapeutic RNA interference – how far from the neurology clinic? Nat Clin Pract Neurol 2007 3 7 394 404 17611488
- Lingor P Bähr M Targeting neurological disease with RNAi Mol Biosyst 2007 3 11 773 780 17940660
- Perrimon N Ni JQ Perkins L In vivo RNAi: today and tomorrow Cold Spring Harb Perspect Biol 2010 2 8 a003640 20534712
- Priller C Bauer T Mitteregger G Krebs B Kretzschmar HA Herms J Synapse formation and function is modulated by the amyloid precursor protein J Neurosci 2006 26 27 7212 7221 16822978
- Turner PR O’Connor K Tate WP Abraham WC Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory Prog Neurobiol 2003 70 1 32 12927332
- Duce JA Tsatsanis A Cater MA Iron-export ferroxidase activity of â-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease Cell 2010 142 6 857 867 20817278
- Zheng H Koo EH The amyloid precursor protein: beyond amyloid Mol Neurodegener 2006 1 5 16930452
- Forman MS Trojanowski JQ Lee VM Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs Nat Med 2004 10 10 1055 1063 15459709
- LaFerla FM Green KN Oddo S Intracellular amyloid-beta in Alzheimer’s disease Nat Rev Neurosci 2007 8 7 499 509 17551515
- Chyung AS Greenberg BD Cook DG Doms RW Lee VM Novel beta-secretase cleavage of beta-amyloid precursor protein in the endoplasmic reticulum/intermediate compartment of NT2N cells J Cell Biol 1997 138 3 671 680 9245794
- Xu H Sweeney D Wang R Generation of Alzheimer beta-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation Proc Natl Acad Sci U S A 1997 94 8 3748 3752 9108049
- Koo EH Squazzo SL Evidence that production and release of amyloid beta-protein involves the endocytic pathway J Biol Chem 1994 269 26 17386 17389 8021238
- Capell A Steiner H Willem M Maturation and pro-peptide cleavage of beta-secretase J Biol Chem 2000 275 40 30849 30854 10801872
- Huse JT Pijak DS Leslie GJ Lee VM Doms RW Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease beta-secretase J Biol Chem 2000 275 43 33729 33737 10924510
- Bennett BD Denis P Haniu M A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer’s beta-secretase J Biol Chem 2000 275 48 33712 33717 10922360
- Creemers JW Ines Dominguez D Plets E Processing of beta-secretase by furin and other members of the proprotein convertase family J Biol Chem 2001 276 6 4211 4217 11071887
- Koh YH von Arnim CA Hyman BT Tanzi RE Tesco G BACE is degraded via the lysosomal pathway J Biol Chem 2005 280 37 32499 32504 16033761
- Walter J Fluhrer R Hartung B Phosphorylation regulates intracellular trafficking of beta-secretase J Biol Chem 2001 276 18 14634 14641 11278841
- Vassar R Bennett BD Babu-Khan S Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE Science 1999 286 5440 735 741 10531052
- Choy RW Cheng Z Schekman R Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid â (Aâ) production in the trans-Golgi network Proc Natl Acad Sci U S A 2012 109 30 E2077 E2082 22711829
- Finan GM Okada H Kim TW BACE1 retrograde trafficking is uniquely regulated by the cytoplasmic domain of sortilin J Biol Chem 2011 286 14 12602 12616 21245145
- Hoyer S Glucose metabolism and insulin receptor signal transduction in Alzheimer disease Eur J Pharmacol 2004 490 1–3 115 125 15094078
- Zou L Wang Z Shen L Receptor tyrosine kinases positively regulate BACE activity and Amyloid-beta production through enhancing BACE internalization Cell Res 2007 17 5 389 401 17325690
- Bonifacino JS The GGA proteins: adaptors on the move Nat Rev Mol Cell Biol 2004 5 1 33 32 14708008
- Wahle T Prager K Raffler N Haass C Famulok M Walter J GGA proteins regulate retrograde transport of BACE1 from endosomes to the trans-Golgi network Mol Cell Neurosci 2005 29 3 453 461 15886016
- He X Li F Chang WP Tang J GGA proteins mediate the recycling pathway of memapsin 2 (BACE) J Biol Chem 2005 280 12 11696 11703 15615712
- Tesco G Koh YH Kang EL Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity Neuron 2007 54 5 721 737 17553422
- Cuervo AM Autophagy: in sickness and in health Trends Cell Biol 2004 14 2 70 77 15102438
- Chu CT Autophagic stress in neuronal injury and disease J Neuropathol Exp Neurol 2006 65 5 423 432 16772866
- Yu WH Kumar A Peterhoff C Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer’s disease Int J Biochem Cell Biol 2004 36 12 2531 2540 15325590
- Yu WH Cuervo AM Kumar A Macroautophagy – a novel Betaamyloid peptide-generating pathway activated in Alzheimer’s disease J Cell Biol 2005 171 1 87 98 16203860
- Florez-McClure ML Hohsfield LA Fonte G Bealor MT Link CD Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans Autophagy 2007 3 6 569 580 17675890
- Hornsten A Lieberthal J Fadia S APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability Proc Natl Acad Sci U S A 2007 104 6 1971 1976 17267616
- Wiese M Antebi A Zheng H Intracellular trafficking and synaptic function of APL-1 in Caenorhabditis elegans PLoS One 2010 5 9 pii:e12790
- Fahrenholz F Alpha-secretase as a therapeutic target Curr Alzheimer Res 2007 4 4 412 417 17908044
- Allinson TM Parkin ET Turner AJ Hooper NM ADAMs family members as amyloid precursor protein alpha-secretases J Neurosci Res 2003 74 3 342 352 14598310
- Kuhn PH Wang H Dislich B ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons Embo J 2010 29 17 3020 3032 20676056
- Dando PM Brown MA Barrett AJ Human thimet oligopeptidase Biochem J 1993 294 Pt 2 451 457 8373360
- Knight CG Dando PM Barrett AJ Thimet oligopeptidase specificity: evidence of preferential cleavage near the C-terminus and product inhibition from kinetic analysis of peptide hydrolysis Biochem J 1995 308 Pt 1 145 150 7755557
- Shrimpton CN Smith AI Soluble neutral metallopeptidases: physiological regulators of peptide action J Pept Sci 2000 6 6 251 263 10912905
- Shrimpton CN Smith AI Lew RA Soluble metalloendopeptidases and neuroendocrine signaling Endocr Rev 2002 23 5 647 664 12372844
- Koike H Seki H Kouchi Z Thimet oligopeptidase cleaves the full-length Alzheimer amyloid precursor protein at a beta-secretase cleavage site in COS cells J Biochem 1999 126 1 235 242 10393344
- Yamin R Malgeri EG Sloane JA McGraw WT Abraham CR Metalloendopeptidase EC 3.4.24.15 is necessary for Alzheimer’s amyloidbeta peptide degradation J Biol Chem 1999 274 26 18777 18784 10373494
- Pollio G Hoozemans JJ Andersen CA Increased expression of the oligopeptidase THOP1 is a neuroprotective response to Abeta toxicity Neurobiol Dis 2008 31 1 145 158 18571100
- Puglielli L Konopka G Pack-Chung E Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide Nat Cell Biol 2001 3 10 905 912 11584272
- Hutter-Paier B Huttunen HJ Puglielli The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease Neuron 2004 44 2 227 238 15473963
- Huttunen HJ Greco G Kovacs DM Knockdown of ACAT-1 reduces amyloidogenic processing of APP FEBS Lett 2007 581 8 1688 1692 17412327
- Liu B Moloney A Meehan S Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation J Biol Chem 2011 286 6 4248 4256 21147772
- Weingarten MD Lockwood AH Hwo SY Kirschner MW A protein factor essential for microtubule assembly Proc Natl Acad Sci U S A 1975 72 5 1858 1862 1057175
- Alonso A Zaidi T Novak M Grundke-Iqbal I Iqbal K Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments Proc Natl Acad Sci U S A 2001 98 12 6923 6928 11381127
- Guthrie CR Schellenberg GD Kraemer BC SUT-2 potentiates tauinduced neurotoxicity in Caenorhabditis elegans Hum Mol Genet 2009 18 10 1825 1838 19273536
- Guthrie CR Greenup L Leverenz JB Kraemer BC MSUT2 is a determinant of susceptibility to tau neurotoxicity Hum Mol Genet 2011 20 10 1989 1999 21355046
- Baumann K Mandelkow EM Biernat J Piwnica-Worms H Mandelkow E Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5 FEBS Lett 1993 336 3 417 424 8282104
- Paudel HK Lew J Ali Z Wang JH Brain proline-directed protein kinase phosphorylates tau on sites that are abnormally phosphorylated in tau associated with Alzheimer’s paired helical filaments J Biol Chem 1993 268 31 23512 23518 8226879
- Piedrahita D Hernández I López-Tobón A Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice J Neurosci 2010 30 42 13966 13976 20962218
- Haass C Selkoe DJ Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide Nat Rev Mol Cell Biol 2007 8 2 101 112 17245412
- Shankar GM Li S Mehta TH Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory Nat Med 2008 14 8 837 842 18568035
- Jin M Shepardson N Yang T Chen G Walsh D Selkoe DJ Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration Proc Natl Acad Sci U S A 2011 108 14 5819 5824 21421841
- Schmetsdorf S Arnold E Holzer M Arendt T Gärtner U A putative role for cell cycle-related proteins in microtubule-based neuroplasticity Eur J Neurosci 2009 29 6 1096 1107 19302146
- Heldin CH Miyazono K ten Dijke P TGF-beta signalling from cell membrane to nucleus through SMAD proteins Nature 1997 390 6659 465 471 9393997
- Ueberham U Ueberham E Gruschka H Arendt T Altered subcellular location of phosphorylated Smads in Alzheimer’s disease Eur J Neurosci 2006 24 8 2327 2334 17074053
- Ueberham U Hilbrich I Ueberham E Transcriptional control of cell cycle-dependent kinase 4 by Smad proteins – implications for Alzheimer’s disease Neurobiol Aging 2012 33 12 2827 2840 22418736
- Song B Davis K Liu XS Lee HG Smith M Liu X Inhibition of Polo-like kinase 1 reduces beta-amyloid-induced neuronal cell death in Alzheimer’s disease Aging (Albany N Y) 2011 3 9 846 851
- Wiese M Antebi A Zheng H Regulation of neuronal APL-1 expression by cholesterol starvation PLoS One 2012 7 2 e32038 22363792
- Borchelt DR Thinakaran G Eckman CB Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo Neuron 1996 17 5 1005 1013 8938131
- Duff K Eckman C Zehr C Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1 Nature 1996 383 6602 710 713 8878479
- Bali J Gheinani AH Zurbriggen S Rajendran L Role of genes linked to sporadic Alzheimer’s disease risk in the production of â-amyloid peptides Proc Natl Acad Sci U S A 2012 109 38 15307 15311 22949636
- Trimmer PA Swerdlow RH Parks JK Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines Exp Neurol 2000 162 1 37 50 10716887
- Shin HW Shinotsuka C Torii S Murakami K Nakayama K Identification and subcellular localization of a novel mammalian dynamin-related protein homologous to yeast Vps1p and Dnm1p J Biochem 1997 122 3 525 530 9348079
- Wang X Su B Fujioka H Zhu X Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients Am J Pathol 2008 173 2 470 482 18599615
- Burnett JC Rossi JJ Tiemann K Current progress of siRNA/shRNA therapeutics in clinical trials Biotechnol J 2011 6 9 1130 1146 21744502
- Burnett JC Rossi JJ RNA-based therapeutics: current progress and future prospects Chem Biol 2012 19 1 60 71 22284355
- Rodriguez-Lebron E Paulson HL Allele-specific RNA interference for neurological disease Gene Ther 2006 13 6 576 581 16355113
- Rodríguez-Lebrón E Gouvion CM Moore SA Davidson BL Paulson HL Allele-specific RNAi mitigates phenotypic progression in a transgenic model of Alzheimer’s disease Mol Ther 2009 17 9 1563 1573 19532137
- Alvarez-Erviti L Seow Y Yin H Betts C Lakhal S Wood MJ Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes Nat Biotechnol 2011 29 4 341 345 21423189
- Lublin A Isoda F Patel H FDA-approved drugs that protect mammalian neurons from glucose toxicity slow aging dependent on cbp and protect against proteotoxicity PLoS One 2011 6 11 e27762 22114686
- Azorsa DO Robeson RH Frost D High-content siRNA screening of the kinome identifies kinases involved in Alzheimer’s disease-related tau hyperphosphorylation BMC Genomics 2010 11 25 20067632
- Lambert JC Amouyel P Genetic heterogeneity of Alzheimer’s disease: complexity and advances Psychoneuroendocrinology 2007 32 Suppl 1 S62 S70 17659844
- Whitehead KA Langer R Anderson DG Knocking down barriers: advances in siRNA delivery Nat Rev Drug Discov 2009 8 2 129 138 19180106
- Alzheimer’s Association Alzheimer’s disease facts and figures Alzheimers Dement 2012 8 2 131 168 22404854
- Gomase VS Tagore S RNAi – a tool for target finding in new drug development Curr Drug Metab 2008 9 3 241 244 18336228
- Seyhan AA RNAi: a potential new class of therapeutic for human genetic disease Hum Genet 2011 130 5 583 605 21537948
- Bumcrot D Manoharan M Koteliansky V Sah DW RNAi therapeutics: a potential new class of pharmaceutical drugs Nat Chem Biol 2006 2 12 711 719 17108989
- Brunden KR Trojanowski JQ Lee VM Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies Nat Rev Drug Discov 2009 8 10 783 793 19794442