Abstract
Background
Epidermal growth factor receptor (EGFR) is an attractive therapeutic target for a number of human tumors including non-small cell lung cancer (NSCLC). Most patients with NSCLC and somatic mutations have shown a dramatic initial clinical response to reversible EGFR inhibitors. The clinical efficacy of reversible inhibitors is, however, ultimately limited due to the emergence of drug resistance, which is usually conferred by the EGFR T790M mutation. Importantly, irreversible, synthetic small molecule inhibitors are currently evaluated and some of them have been shown to overcome the acquired resistance that is oftentimes observed in these patients. Thus far, irreversible EGFR inhibitors as a drug class have not received regulatory approval due in part to their poor effectiveness at clinically achievable concentrations. Therefore, there is an urgent need to discover and develop novel, potent irreversible inhibitors against the EGFR T790M mutation.
Material and methods
In the following study, we report a novel “hybrid strategy” to identify irreversible EGFR inhibitors with active scaffolds starting with the identification and extraction of a common chemical reactive feature and a pharmacophore feature. The chemical reactive feature was elucidated by investigating 138 currently known irreversible inhibitors at B3LYP/6-31G(d) level using the density function theory method. The pharmacophore feature was extracted from the same inhibitors using pharmacophore modeling. Based on these unique features, two constraints were set while calibrating the protocols of in silico screening. Compounds bearing these specific features were obtained from the National Cancer Institute diversity database to form our subsequent library. Finally, a structure based virtual screening against the library was conducted using standard protocols validated in our lab.
Results
Twenty-eight candidate compounds that demonstrated antitumor activity and that had novel scaffolds different from commonly known quinazoline/quinoline analogs were obtained. The interaction modes between three representative candidates and our model system are similar to that between the model system and the reference compound T-001, which has previously been reported to be one of the most potent of the 138 irreversible inhibitors.
Conclusion
The hybrid strategy starting with the extraction of common features is an effective approach to design potential irreversible inhibitors with novel scaffolds and therefore to obtain lead molecules in the selection process. These candidates possessing unique scaffolds have a strong likelihood to act as further starting points in the preclinical development of potent irreversible T790M EGFR inhibitors.
Introduction
As key regulators of critical cellular processes, the ErbB protein family or epidermal growth factor receptor (EGFR) family has received much attention for several decades.Citation1–Citation8 The human EGFR family consists of four members: EGFR [(Human Epidermal Growth Factor Receptor) HER1/ErbB1], HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4).Citation2,Citation3,Citation9–Citation11 They are structurally related receptor tyrosine kinases (RTKs) sharing a similar molecular architecture.Citation3,Citation10,Citation12–Citation14 Each of them comprises identical extracellular ligand-binding regions, a single hydrophobic transmembrane segment, and a cytoplasmic region. The extracellular region contains four sub-domains (I–IV)Citation12–Citation14 and the cytoplasmic region comprises a conserved protein tyrosine kinase (TK) catalytic domain as well as a carboxy terminal tail with tyrosine autophosphorylation sites.Citation2,Citation3
It is well recognized that ErbB members share remarkable homology in their endocellular TK domains, but are distinct in their extracellular component and carboxy terminal tails.Citation13 The ectodomain structure of ErbB2, for example, is radically different from the others.Citation14 ErbB2 has a fixed conformation that resembles the ligand-activated state of EGFR and ErbB3. Within the extracellular region of ErbB2, a unique sub-domain I–III interaction buries the ligand binding site and makes the site not accessible for interaction.Citation14 As such, ErbB2 lacks a ligand-binding domain to interact with a growth factor ligand. Although the intracellular TK domain of ErbB receptors is highly conserved, the kinase domain of ErbB3 has a substitution in critical amino acids, which results in no ErbB3 intrinsic kinase activity.Citation3,Citation13,Citation15,Citation16 ErbB2 and ErbB3 are non-autonomous TKs. They form heterodimeric complexes with other ErbBs that are capable of generating potent downstream signaling. In contrast, the other two members are autonomous. When bound to ligand growth factors, the receptor dimerization is induced and intracellular protein TK is activated with subsequent initiation of numerous downstream signaling events, which ultimately leads to cell proliferation, migration, and differentiation.Citation3,Citation13 Aberrant ErbB receptor activation and their intracellular signaling pathways and mutations in RTKs have been causally linked to cancers, diabetes, inflammation, severe bone disorders, arteriosclerosis, and angiogenesis.Citation12,Citation13,Citation17
Among all of the four members, EGFR was the first receptor protein-TK to be sequencedCitation10 and linked directly to human tumors.Citation3,Citation14 Not only is it involved in development of numerous types of human cancersCitation3,Citation14,Citation18 but it is even the host cofactor for hepatitis C virus entry as well.Citation19 It has been intensely pursued as a therapeutic target.Citation4,Citation10,Citation14,Citation17,Citation20 One of the most clinically advanced strategies toward aberrant ErbB receptor activation is small-molecule inhibition of the protein TK catalytic domainCitation13,Citation14,Citation21–Citation24 in which small-molecule TK inhibitors (TKIs) compete with adenosine triphosphate (ATP) in the TK domain. The first generation TKIs (erlotinib, gefitinib,Citation4 and lapatinib, for instance) are reversible small-molecule inhibitors. They prevent autophosphorylation of the EGFR TK by competing with ATP.Citation20 But cellular oncogenic transformation caused by different EGFR mutations results in differential sensitivity to the first generation TKIs.Citation24,Citation25 Although most patients with NSCLC and somatic mutations showed a dramatic initially clinical response to EGFR reversible inhibitors, they gradually developed acquired resistance to these drugsCitation26 due to the EGFR T790M mutation.Citation4,Citation6,Citation7,Citation16,Citation21,Citation24,Citation27 This mutation leads to a change in the 3-dimensional structure of the TK domain and prevents reversible inhibitors from binding to EGFR.Citation11,Citation23,Citation24 In this situation, various second generation EGFR TKIs are currently being evaluated. In contrast to the first generation TKIs that join to the catalytic site in the EGFR TK domain through classic competitive binding with ATP, second generation TKIs such as HKI-272 (neratinib),Citation15 EKB-569 (pelitinib),Citation28 BIBW2992 (tovok),Citation15,Citation20,Citation29 and PF00299804 (dacomitinib),Citation15,Citation20,Citation30 which are shown in , form covalent bonds to a conserved cysteine residueCitation5 within the active site. The obvious advantage of these compounds over the first generation entities is the ability to prevent the development of acquired resistance or overcome acquired resistance in patients previously treated with the first generation of drugs.Citation4,Citation5,Citation11,Citation17,Citation24,Citation26,Citation28,Citation31 Although clinical trials with a number of such compounds have shown promising results, irreversible EGFR inhibitors as a drug class have not received regulatory approval.Citation17,Citation28 In fact, some of them did not demonstrate effectiveness to overcome this mutant at clinically achievable concentrations.Citation16,Citation25,Citation31,Citation32 It is clear that much work remains to be done on the road to develop second generation EGFR TKIs.
Figure 1 Structures of irreversible TK inhibitor.Citation15,Citation20,Citation28–Citation30 The colored ellipsoid marks the common features.
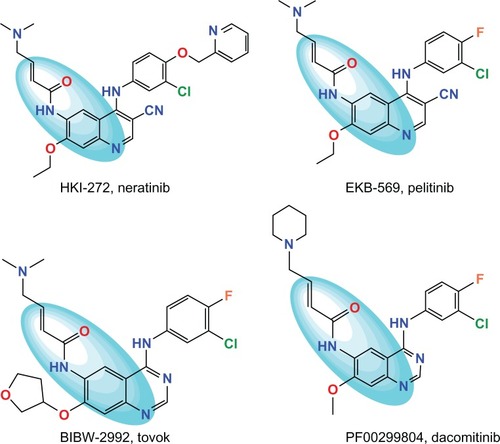
It was shown by our detailed review that the main group of currently known EGFR TKIs are almost all based on a quinazoline/quinoline scaffold.Citation28,Citation31,Citation33–Citation37 It is believed that the quinazoline/quinoline scaffold may not be the most potent or specific for inhibiting T790M EGFR.Citation4 Therefore, to discover and develop novel, potent irreversible inhibitors against the EGFR T790M is quite important. Compounds with cores different from the quinazoline/quinoline scaffold and possessing inhibitory activity against T790M EGFR could represent promising starting points as lead compounds. Undoubtedly, 2nd generation inhibitors against T790M EGFR must have some common features and it is therefore important to find those compounds by revealing and utilizing these common features.
In this study, we report a novel hybrid strategy, which combines quantum chemical calculation, pharmacophore modeling, and structure-based drug design, to identify irreversible inhibitors with novel scaffolds. It started with the extraction of a common chemical reactive feature and a pharmacophore feature. Compounds bearing those specific common features were obtained from the National Cancer Institute (NCI) database (release 3 files) and were docked to the binding site of the model system with a calibrated protocol. Finally, 28 compounds that demonstrated antitumor activity and bear novel scaffolds different from commonly known quinazoline/quinoline analogs were identified. For three representative candidates, their binding modes to the model system were predicted.
Material and methods
As an attractive target, EGFR TK plays a crucial role in cancer therapy. Although several inhibitors of EGFR TK have been clinically validated for the treatment of patients with cancers during the past several years,Citation26 the search for novel and more potent irreversible inhibitors against this target is still meaningful and considerably challenging. Our aim is to identify irreversible inhibitors with novel scaffolds against T790M EGFR starting with the identification and extraction of the common features from currently known irreversible inhibitors. The schematic representation chart of our workflow is shown in .
Figure 2 Schematic diagram of workflow for identifying potential EGFR-T790M inhibitors with novel scaffolds.
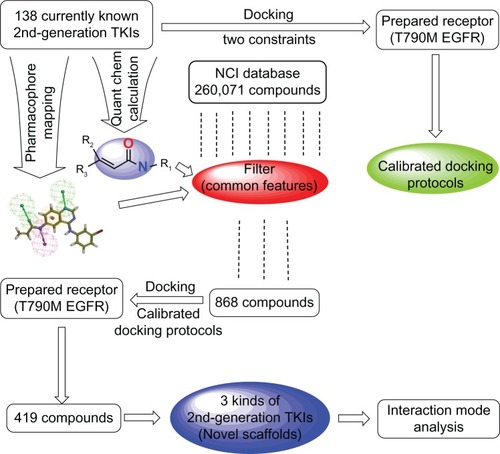
Extraction of common features
In view of the properties of currently known irreversible inhibitors that can form covalent bonds with a conserved cysteine residue within the active site of EGFR, we presumed that there must exist some common features among them. Obviously, the features must at least include a chemical reactive feature and a pharmacophore feature. The former is related to the forming of covalent bonds and the latter is essential for a drug’s biological activity.
Reactive site of irreversible inhibitors
At first, 138 TKIs ( in supplementary material) with biological activity values (IC50) ranging from 0.002 to 13.570 μM were selected from published documents.Citation34–Citation37 It was noted that almost all of them contain either an acrylamide group or a propargyl amide group.
The exact location of Michael receptor in irreversible TKIs was determined by optimizing structures of 138 TKIs and calculating their reactive sites. The structures were drawn using MarvinSketch (ChemAxon Ltd., Budapest, Hungary).Citation38 Next, their geometries were obtained at the same operating environment by calculating the lowest energy conformer using a MMFF94 force field.Citation39 Later, these geometries were further optimized with density functional theory (DFT) method at B3LYP/6-31G(d) level by employing Gaussian 03.Citation40 Finally, the Natural Bond Orbital (NBO) charge distribution of molecules was calculated using the B3LYP/6-31G(d)//B3LYP/6-31G(d) method. For a molecule containing N electrons, independent calculations were made for the corresponding N-electron molecule and (N + 1)-electron molecule with the same molecular geometry.Citation41 The variation of NBO charges on an atom of an (N + 1)-electron molecule relative to the N-electron molecule reflects the site reactivity of nucleophilic attack.Citation41 It is obvious that the atom with the largest variation of NBO charge is most probably the reactive site of a molecule that involves a Michael addition reaction with the sulfur atom of Cysteine (Cys)797 of EGFR.
Pharmacophore feature
For all of the 138 currently known 2nd generation TKIs, their common pharmacophore feature was extracted as follows.
At first, conformations of these 138 compounds were generated with Omega 2.2.5 (OpenEye Scientific Software Inc., Santa Fe, NM, USA)Citation42 using the geometry optimized at B3LYP/6-31G(d) level with DFT method. Then, pharmacophore models were built by employing Discovery Studio (Accelrys Inc., San Diego, CA, USA)Citation43 with compound T-001 (),Citation37 a reference compound that is one of the most potent irreversible TKIs among these 138 compounds. By manually adjusting the number and types of the function groups and through mapping all conformations of these 138 TKIs onto the pharmacophore models, the common pharmacophore feature of these inhibitors was finally identified and extracted. As shown in , the common pharmacophore feature of these 2nd generation inhibitors consists of one hydrogen bond donor, one aromatic hydrophobic center, and two hydrogen bond acceptors.
Figure 3 The common pharmacophore feature generated from compound T-001Citation37 (A) and 138 tyrosine kinase inhibitors (TKIs)Citation34–Citation37 superimposed on the feature (B). Aromatic hydrophobic center is represented by the orange wire mesh ball. Hydrogen bond accepter is depicted with the green arrow and wire mesh balls while the hydrogen bond donor is described by the pink arrow and wire mesh balls.
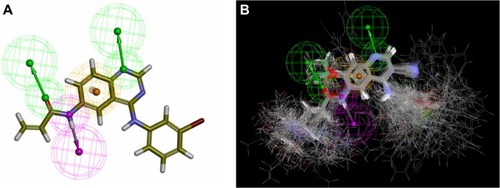
After that, another 21 irreversible inhibitorsCitation4,Citation34–Citation37 coupled with two reversible inhibitorsCitation4 ( in supplementary materials) were used to test whether these extracted features are effective enough to identify potential irreversible inhibitors.
Docking with constraints
As a reliable, cost-effective, and time-saving technique, structure based virtual screening (SBVS) has shown a powerful ability to discover lead compounds.Citation32 In the SBVS pipeline, preparation of receptor, determination of docking protocols and preparation of a compound library are the committed steps.
Preparation of receptor
TK cSrc was chosen as the model system of the mutant EGFR and its 3-dimensional structure was obtained from the Protein Data Bank (PDB ID: 2QQ7).Citation44 Water molecules in the crystallographic complex were stripped. Preparation of receptor for docking was done using FRED RECEPTOR 2.2.5 (OpenEye Scientific Software Inc., Santa Fe, NM, USA).Citation45 Four hundred different molecule sensors which were stored internally by the program were used to describe the binding site for docking. The size of the box is 6,140 cubic Angstroms. The inner contour and outer contour filters, which were detected with 400 sensor molecules, are 92 cubic and 1,442 cubic angstroms, respectively.
Identification of docking protocols
As many as 252 docking strategies were tested to find a satisfactory way upon which TKIs with potent activities could be enriched at the top of high-score ranking.
At first, conformers of the 138 TKIs were created via Omega 2.2.5Citation42 and the number of conformers for each TKI was limited up to 2,000. Then, in total, 252 schedules were tested for obtaining optimal docking strategy. At the conformer searching stage, four scoring functions including Shapegauss,Citation46 Plp,Citation47 Chemgauss2,Citation48 Chemgauss3Citation48 were chosen for exhaustive searching of conformers. At the optimization stage, seven scoring functions (Chemgauss2,Citation48 Chemgauss3,Citation48 Chemscore,Citation48 Shapegauss,Citation46 Plp,Citation47 OEChemscore, and ScreenscoreCitation49) were hired for rigid optimization of poses. For each one of the above 28 combinations, nine scoring functions (those seven used in rigid optimization plus ZapbindCitation50 and consensus score) were used for evaluating the interaction between the TKIs and the active site of EGFR. Scoring functions employed for identifying optimal docking protocols are listed in .
Table 1 Scoring functions employed for identifying optimal docking protocols at different stagesTable Footnote*
In the process of docking currently known irreversible TKIs, two constraints were added. The first one was that each potential irreversible TKI should provide a hydrogen acceptor to form a hydrogen bond with the hinge region M793. There is strong evidence that inhibitors make a hydrogen bond to the hinge region atom N-H in M793 in the protein kinase family.Citation4,Citation5,Citation8,Citation17,Citation28,Citation51,Citation52 The other constraint was to require an alkyne carbon or an alkene carbon being within 4 ± 1 angstrom range of the sulfur atom of the conserved Cys797 residue, which would allow for covalent bond formation.Citation53 This is supported by our quantum chemical calculations that demonstrated that the reactive site of the known irreversible TKIs locates at the β-carbon of the acrylamide or propargyl amide group.
Preparation of the molecule library
As described above, common features of currently known irreversible TKIs were extracted (–). Commonly, each of those TKIs has an acrylamide group or a propargyl amide group. At the same time, they all bear one hydrogen bond donor, one aromatic hydrophobic center, and two hydrogen bond acceptors. We expected that potential irreversible TKIs with novel scaffolds would have such features. These features were used as a filter (). Compounds in the NCI database (260,071) were filtered using these features. Those that do not meet these features were eliminated. After filtering, only 868 compounds in the NCI database were remained. Then, reference compound T-001 and another three irreversible inhibitorsCitation4 with novel scaffolds mixed into these molecules. Finally, conformations of these compounds were generated using Omega 2.2.5Citation42 and the maximum number of conformations was set to 2,000.
Constraint virtual screening and interaction mode analysis
With the optimal docking protocols mentioned above and two added constraints, 868 compounds, from the NCI database and bearing the common features, were docked to the binding site of a model system using FRED program version 2.2.5.Citation45 Potential irreversible TKIs were obtained from the top 200 of the SBVS result. For the reference compound T-001Citation37 and the screened potential irreversible TKIs with representative scaffolds, the interaction between them and the model system was analyzed and visualized using DS visualizer 3.1 (Accelrys Inc., San Diego, CA,USA).Citation43
Results and discussion
Common features
The irreversible TKIs can form covalent bonds with a conserved cysteine residue (Cys797) within the active site of T790M EGFR. Apparently, as shown in and in supplementary material, currently known 2nd generation TKIsCitation34–Citation37 contain either an acrylamide group or a propargyl amide group.
Quantum chemical calculations revealed that the reactive site of the known irreversible TKIs, which form a covalent bond to the conserved Cys797 within T790M EGFR, locates at the β-carbon of the acrylamide or propargyl amide group ( and ). This is the theoretical basis for us to set one of the constraints while conducting SBVS.
Table 2 NBO charge distribution and the relevant change of charge distribution in the N-electron and (N + 1)-electron molecule for N electrons systems T-005 and T-008
Figure 4 The electrophilic reactive site of some known irreversible TKIsCitation34–Citation37 calculated at B3LYP/6-31G(d) level with DFT method by employing Gaussian 03Citation40 and the effect of replacement on the bioactivity of irreversible TKI. Electrophilic center was highlighted by the red color and the nucleophilic center was painted with a blue color. Red arrow points to the β-carbon of the acrylamide group or propargyl amide group.
Abbreviations: DFT, density functional theory; TKI, tyrosine kinase inhibitor.
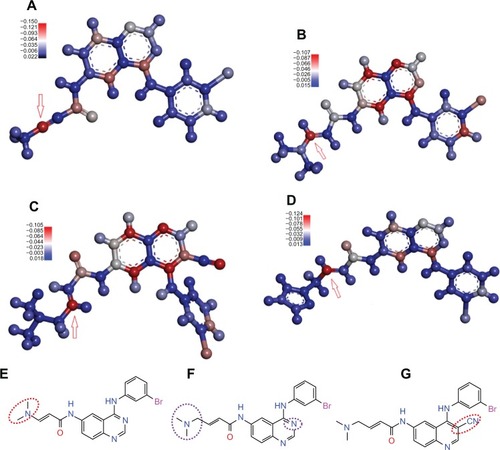
It is interesting to note that often the β-carbon of acrylamide group or propargyl amide group in the currently known irreversible TKIs is the most electrophilic, as shown in . While results of NBO analysis show that the β-carbon is no longer the most electrophilic atom when an electron-withdrawing atom directly links to the β-carbon () or the nitrogen atom at position three of the quinazoline group is replaced by a carbon linked with a cyano group (). The β-carbon becomes one of the most electrophilic atoms in the molecule. Even though it still keeps the tendency to attack the nucleophilic thiol group of the conserved Cys797 in T790M EGFR, the bioactivity of the molecule is much less than its analog ().
Except for the chemical reactive feature mentioned above, the currently known irreversible TKIs have a common pharmacophore feature that was extracted previously. As shown in , the common pharmacophore feature can be described to include one hydrogen bond donor, one aromatic hydrophobic center, and two hydrogen bond acceptors. It is known that pharmacophore refers to the 3-dimensional arrangement of functional groups essential to a drug’s biological activity.Citation54,Citation55 A diverse set of lead compounds, which potentially possess desired biological activity but have totally different chemical scaffolds, often share a common pharmacophore.Citation56
In a sense, compounds sharing these common chemical reactive and pharmacophore features could be guaranteed to form covalent bonds to the conserved Cys797. At the same time, they possess the desired biological activity and have different chemical scaffolds as well.
Selection of model system
There are many 3-dimensional structures of T790M EGFR determined using X-ray crystallography. We choose TK cSrc (PDB ID: 2QQ7)Citation44 as the model system of the mutant EGFR based on the following considerations. At first, the resolution of 2QQ7 is 2.38 angstrom and the crystal structure is complexed with an irreversible inhibitor. It was believed that when the resolution value of crystallographic data is lower than 2.5 angstrom the modeling of details in the protein structures is much more subjective than desired.Citation57,Citation58 It was also confirmed that selecting structures with poor resolution may produce false predicted conformations.Citation59 Thus, as a rule of thumb, data worse than 2.5 angstrom resolution should not be considered reliable for virtual library screening applications.Citation60
In addition, cSrc is a tractable target for the structural investigation of irreversible EGFR. It has many advantagesCitation44 for the study of homologous protein kinases over EGFR. The kinase domains between EGFR and cSrc share high identityCitation44 in structure and sequence. The sequence identity between their ATP binding sites is as high as 70%. Most importantly, cSrc has been validated as a model system for the structural investigation of irreversible EGFR inhibitors.Citation33
Constrained docking against the NCI database
Having a proper pose within the binding site is indispensable for ligand being effective. In addition, adding constraints is an effective way to gain proper poses of ligands. Therefore, two constraints, which are discussed above, were added while docking molecules to the binding site of the model system.
With those two constraints, 138 known irreversible TKIs were docked to the binding site of the model system. Finally, an optimal strategy that provides the highest enrichment rate was identified from among 252 combinations. Scoring functions Chemgauss2,Citation48 Screenscore,Citation49 and ShapegaussCitation46 were used for exhaustive searching of conformers, rigid optimization of poses, and interaction evaluation, respectively. According to this strategy, five potent TKIs were enriched within the top six of the 138 compounds. These results are presented in .
Table 3 The IC50 value and ShapegaussCitation46 score of five potent TKIsCitation34–Citation37 enriched within the top six of 138 second-generation TKIs
Together with reference compound T-001 and another three irreversible inhibitors,Citation4 868 molecules retrieved from the NCI database containing 260,071 compounds were docked to the binding site of the model system according to the optimal docking strategy. Of these, 419 compounds successfully docked to the site.
By manually analyzing compounds among the top 200 compounds, 28 of them are found to have novel scaffolds different from the currently known quinoline/quinazoline core and demonstrated bioactivity against various types of tumors. The average antitumor activities (GI50) of these 28 compounds are in the order of magnitude of 10−5 μg/mL ().
Table 4 The ShapegaussCitation46 score and antitumor activities of the 28 potential irreversible TKIs plus four reference irreversible TKIsCitation4,Citation37
Validation of the hybrid strategy
The committed step in the pipeline of SBVS is to validate docking strategy.Citation61 In order to test whether the extracted common features could be used to identify potential irreversible inhibitors, 23 molecules were collected to work as a testing set. This group compromised compound T-001, another 17 irreversible inhibitorsCitation34–Citation37 with the quinoline scaffold, three irreversible inhibitorsCitation4 with a core different from quinoline, and two reversible inhibitors (gefitinib and erlotinib).Citation4 As expected, the common features worked as a filter: reversible inhibitors were filtered out due to the fact that they lack the chemical reactive feature and only irreversible inhibitors with the quinoline scaffold or other scaffolds remained. As shown in , they could be mapped onto the pharmacophore feature. It was demonstrated that the extracted common features could work as an effective filter to identify irreversible inhibitors whether they contain the quinoline scaffold or not.
Figure 5 In total, 21 irreversible TKIsCitation4,Citation34–Citation37 in the testing set (refer to Table S2 in supplementary materials) were mapped on the common pharmacophore feature generated from compound T-001.Citation37 T-001 is depicted by the stick and its carbon atoms are colored in green. WZ-3146Citation4 is depicted by the stick and its carbon atoms are colored pink; WZ-4002Citation4 is depicted by the stick and its carbon atoms are colored sky blue; WZ-8040Citation4 is depicted by the stick and its carbon atoms are colored blue.
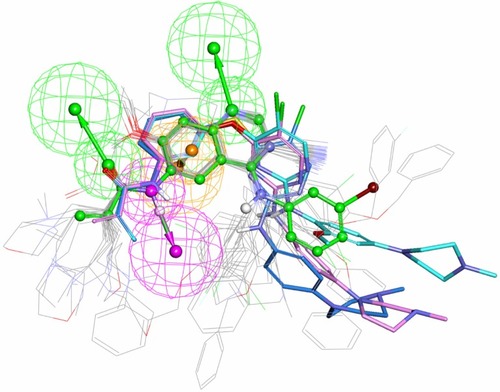
Before conducting a SBVS, four reference compounds (compound T-001,Citation37 WZ-3146,Citation4 WZ-4002,Citation4 and WZ-8040Citation4) with a scaffold different from quinoline/quinazoline were mixed into a molecule library that includes 868 compounds retrieved from the NCI database with the aid of a common feature filter. According to the optimal docking protocols, these molecules were docked to the binding site of the model system. As demonstrated in , the scorings of reference compound T-001 and the other three irreversible inhibitors with a scaffold different from quinoline/quinazoline are among the top 150 high score ranking results. These four irreversible inhibitors and the 28 potential inhibitors could be mapped onto the extracted pharmacophore feature ( and ). The results show that the hybrid strategy is an effective way to identify potent irreversible inhibitors bearing novel scaffolds different from quinoline/quinazoline analogs.
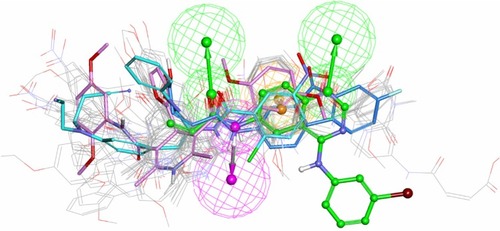
Figure 6 Potential irreversible TKIs that had been identified from the NCI database (refer to ) were mapped onto the common pharmacophore feature generated from reference compound T-001.Citation37 T-001 is depicted by the stick and its carbon atoms are colored green. Representative compounds NSC-621158, NSC-642576, and NSC-57443 are depicted by the stick and their carbon atoms are colored pink, sky blue, and blue, respectively.
Interaction mode analysis
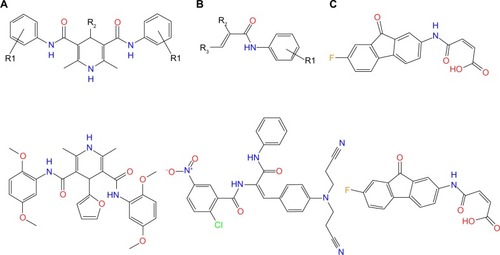
It is easy to identify that these 28 compounds belong to three types of scaffolds ( and in supplementary materials). Class A is a group of symmetric compounds. There are two symmetric acrylamide functional groups in this class of compound and the alkene moieties are parts of the dimethyl-1,4-dihydropyridine group. Class B is asymmetric. An aromatic ring is directly linked to the N atom of the acrylamide functional group in both class A and class B, while in class C, it is a fluorenone group that directly binds to the N atom of the acrylamide.
Figure 7 Three classes of potential irreversible TKIs with novel scaffolds and their corresponding representative molecules.
Abbreviations: NSC, Nomenclature Standards Committee; TKI, tyrosine kinase inhibitor.
From each class, we selected one representative molecule that had the highest score among its own class. Together with reference compound T-001,Citation37 the interaction modes between them and the mutant EGFR were analyzed with the aid of Discovery Studio.Citation43
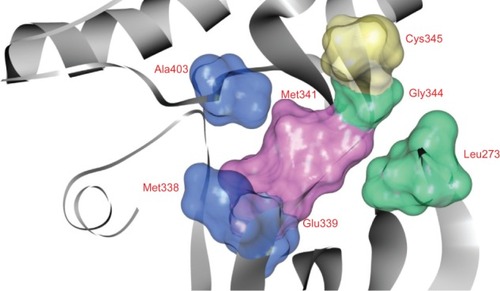
According to a pharmacophore modelCitation27,Citation51 of the ATP binding site in protein kinases, the conserved site is divided into five regions: (1) adenine region, which contains two amino acid residues, Glutamine (Gln)791 and Methionine (Met)793 on the hinge region corresponding to Glutamic acid (Glu)339 and Met341 in TK cSrc; (2) sugar pocket, which is the region extending from Cys797 (corresponding to Cys345 in TK cSrc) toward the solvent; (3) hydrophobic region I, which is composed of two residues Threonine (Thr)790 and Thr854, corresponding to Met338 and Alanine (Ala)403 in TK cSrc; (4) hydrophobic region II, which is formed by residues Leucine (Leu)718 and Glycine (Gly)796, corresponding to Leu273 and Gly344 in tyrosine kinase cSrc; and (5) phosphate binding region, which is largely solvent exposed. The ATP binding site in TK cSrc is shown in .
Figure 8 Pharmacophore modelCitation27,Citation51 of the ATP binding site in tyrosine kinase cSrc (PDB ID: 2QQ7).Citation44 This site is represented by the solid surface. Adenine region, sugar region, hydrophobic region I, and hydrophobic region II are colored pink, yellow, blue, and green, respectively.
As shown in , the four selected molecules similarly form hydrogen bonds with the N-H of residue Met341 and the β-carbon of acrylamide is near the S atom of Cys345. Surely, all of them have an interaction with the sugar region of the binding site. In hydrophobic region II, they interact with Leu273 and Gly344, while in the adenine region they only interact with Met341; Glu339 is far from them. Conversely, reference compound T-001,Citation37 compound A (NSC-621158), and compound B (NSC-642576) do not interact with residues in the hydrophobic region I. In contrast, compound C (NSC-57443) interacts with Met338 in this region ().
Figure 9 The illustrations showing the interaction between the model system and three representative compounds as well as between the reference compound T-001Citation37 and the model system. Residues are displayed in thin stick style. Carbon atoms of residues corresponding to adenine region, sugar region, hydrophobic region I, and hydrophobic region II are colored pink, yellow, blue, and light green, respectively. Compounds are represented by a thick stick and their carbon atoms are colored green. Interactions such as hydrogen bond, cation-π interaction, and π-σ interaction are highlighted.
Abbreviations: Ala, alanine; Gln, glutamine; Glu, glutamic acid; Met, methionine; Thr, threonine; Leu, leucine; Gly, glycine; Cys, cysteine.
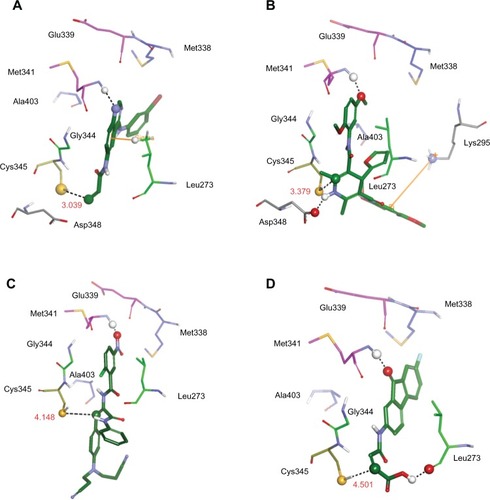
Specifically, it was noted that there is a π-σ interaction between T-001Citation37 and the side chain of Leu273 (). For compound A, a hydrogen bond interaction with the side chain of Asp348 is observed. Meanwhile, a cation-π interaction is noted between this compound and the side chain of Lys295 (). The hydrogen bond formed between compound B and N-H of Met341 is quite strong. The distance between the heavy atoms with high electro-negativity values is only 2.254 angstrom. The 3,3′-(phenylazanediyl) dipropanenitrile moiety of compound B extends to solvent (). Additional interaction between compound C and the hydrophobic region II was also observed. That is, a hydrogen bond forms between compound C and the carbonyl oxygen of Leu273 ().
Conclusion
In this work, we were trying to find irreversible TKIs with novel scaffolds. At first, quantum chemical calculations and pharmacophore modeling were employed to extract common features of 138 currently known irreversible TKIs. The β-carbon of acrylamide group or propargyl amide group was identified by quantum chemical calculations as the electrophilic reactive site or one of the sites in irreversible TKIs. Commonly, each such TKI bears one aromatic hydrophobic center, one hydrogen donor, and two hydrogen acceptors.
Next, optimal docking protocols were identified and validated. By adding two constraints and docking 138 currently known irreversible TKIs to the binding site of a model system, optimal docking protocols were identified from selecting the best one among 252 types of docking strategies. According to this optimal strategy, five potent irreversible TKIs were enriched within the top six of the 138 TKIs.
From the NCI diversity database (260,071 compounds), 868 molecules were retrieved with the aforementioned common features. These molecules plus another four irreversible inhibitors were then docked to the binding site of a model system by employing optimal docking protocols. Finally, 28 potential irreversible TKIs, which belong to three classes of novel scaffolds and exhibited antitumor activity, were gained from the top 200 with high score rankings. The interaction modes between three representative candidates and the model system are similar to that between the model system and the reference compound T-001, one of the most potent inhibitors among the identified 138 irreversible inhibitors. Still, there are subtle differences in their interactions with the model system, which contribute to their different inhibition activities.
The interaction scores of the four irreversible inhibitors are among the top 150 with high score rankings. Average antitumor activities (GI50) of the 28 compounds are in the order of magnitude of 10−5 μg·mL−1. The hybrid strategy starting with the extraction of common features is shown to be an effective approach to identify irreversible inhibitors with novel scaffolds. Candidates possessing unique scaffolds have a strong likelihood to act as further starting points in the preclinical development of potent irreversible inhibitors against T790M EGFR.
Supplementary tables
Table S3 The structures of 28 potential irreversible TKIs with three classes of novel scaffolds identified by docking molecules from NCI database (release 3 files) to the binding site of T790M EGFR using FRED 2.2.5Citation1 (OpenEye Scientific Software Inc., Santa Fe, NM, USA).
Acknowledgments
This work was supported by the National Key Programs of China during the 12th Five-Year Plan Period (Grant 2012ZX09103-101-017). We would like to acknowledge the OpenEye Scientific Software and ChemAxon for their academic licenses. The authors would like to acknowledge Dr Carl K Edwards for his review of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
- Shimizu N Behzadian MA Shimizu Y Genetics of cell surface receptors for bioactive polypeptides: binding of epidermal growth factor is associated with the presence of human chromosome 7 in human-mouse cell hybrids Proc Natl Acad Sci U S A 1980 77 6 3600 3604 6968072
- Schlessinger J Cell signaling by receptor tyrosine kinases Cell 2000 103 2 211 225 11057895
- Yarden Y Sliwkowski MX Untangling the ErbB signalling network Nat Rev Mol Cell Biol 2001 2 2 127 137 11252954
- Zhou W Ercan D Chen L Novel mutant-selective EGFR kinase inhibitors against EGFR T790M Nature 2009 462 7276 1070 1074 20033049
- Carmi C Cavazzoni A Vezzosi S Novel irreversible epidermal growth factor receptor inhibitors by chemical modulation of the cysteine-trap portion J Med Chem 2010 53 5 2038 2050 20151670
- Bai F Liu H Tong L Discovery of novel selective inhibitors for EGFR-T790M/L858R Bioorg Med Chem Lett 2012 22 3 1365 1370 22227214
- Li S Guo C Sun X Synthesis and biological evaluation of quinazoline and quinoline bearing 2,2,6,6-tetramethylpiperidine-N-oxyl as potential epidermal growth factor receptor(EGFR) tyrosine kinase inhibitors and EPR bio-probe agents Eur J Med Chem 2012 49 271 278 22309911
- Sato T Watanabe H Tsuganezawa K Identification of novel drug-resistant EGFR mutant inhibitors by in silico screening using comprehensive assessments of protein structures Bioorg Med Chem 2012 20 12 3756 3767 22607878
- Salomon DS Brandt R Ciardiello F Normanno N Epidermal growth factor-related peptides and their receptors in human malignancies Crit Rev Oncol Hematol 1995 19 3 183 232 7612182
- Roskoski R The ErbB/HER receptor protein-tyrosine kinases and cancer Biochem Biophys Res Commun 2004 319 1 1 11 15158434
- Gazdar AF Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors Oncogene 2009 28 Suppl 1 S24 S31 19680293
- Lemmon MA Schlessinger J Cell signaling by receptor tyrosine kinases Cell 2010 141 7 1117 1134 20602996
- Lurje G Lenz HJ EGFR signaling and drug discovery Oncology 2009 77 6 400 410 20130423
- Hynes NE Lane HA ERBB receptors and cancer: the complexity of targeted inhibitors Nat Rev Cancer 2005 5 5 341 354 15864276
- Doebele RC Oton AB Peled N Camidge DR Bunn PA New strategies to overcome limitations of reversible EGFR tyrosine kinase inhibitor therapy in non-small cell lung cancer Lung Cancer 2010 69 1 1 12 20092908
- Sequist LV Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer Oncologist 2007 12 3 325 330 17405897
- Yun CH Mengwasser KE Toms AV The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP Proc Natl Acad Sci U S A 2008 105 6 2070 2075 18227510
- Schneider MR Wolf E The epidermal growth factor receptor ligands at a glance J Cell Physiol 2009 218 3 460 466 19006176
- Lupberger J Zeisel MB Xiao F EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy Nat Med 2011 17 5 589 595 21516087
- Spicer JF Rudman SM EGFR inhibitors in non-small cell lung cancer (NSCLC): the emerging role of the dual irreversible EGFR/HER2 inhibitor BIBW 2992 Target Oncol 2010 5 4 245 255 20574858
- Rocha-Lima CM Soares HP Raez LE Singal R EGFR targeting of solid tumors Cancer Control 2007 14 3 295 304 17615536
- Okamoto I Epidermal growth factor receptor in relation to tumor development: EGFR-targeted anticancer therapy FEBS J 2010 277 2 309 315 19922468
- Ciardiello F Tortora G EGFR antagonists in cancer treatment New Engl J Med 2008 358 11 1160 1174 18337605
- Kobayashi S Ji H Yuza Y An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor Cancer Res 2005 65 16 7096 7101 16103058
- Greulich H Chen TH Feng W Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants PLoS Med 2005 2 11 e313 16187797
- Riely GJ Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer J Thorac Oncol 2008 3 6 Suppl 2 S146 S149 18520300
- Liu B Bernard B Wu JH Impact of EGFR point mutations on the sensitivity to gefitinib: insights from comparative structural analyses and molecular dynamics simulations Proteins 2006 65 2 331 346 16927343
- Wissner A Mansour TS The development of HKI-272 and related compounds for the treatment of cancer Arch Pharm (Weinheim) 2008 341 8 465 477 18493974
- Li D Ambrogio L Shimamura T BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models Oncogene 2008 27 34 4702 4711 18408761
- Engelman JA Zejnullahu K Gale CM PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib Cancer Res 2007 67 24 11924 11932 18089823
- Kwak EL Sordella R Bell DW Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib Proc Natl Acad Sci U S A 2005 102 21 7665 7670 15897464
- Godin-Heymann N Ulkus L Brannigan BW The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor Mol Cancer Ther 2008 7 4 874 879 18413800
- Blair JA Rauh D Kung C Structure-guided development of affinity probes for tyrosine kinases using chemical genetics Nat Chem Biol 2007 3 4 229 238 17334377
- Tsou HR Overbeek-Klumpers EG Hallett WA Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity J Med Chem 2005 48 4 1107 1131 15715478
- Wissner A Overbeek E Reich MF Synthesis and structure-activity relationships of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2) J Med Chem 2003 46 1 49 63 12502359
- Wissner A Brawner Floyd MB Rabindran SK Syntheses and EGFR and HER-2 kinase inhibitory activities of 4-anilinoquinoline-3-carbonitriles: analogues of three important 4-anilinoquinazolines currently undergoing clinical evaluation as therapeutic antitumor agents Bioorg Med Chem Lett 2002 12 20 2893 2897 12270171
- Tsou HR Mamuya N Johnson BD 6-Substituted-4-(3-bromophenylamino)quinazolines as putative irreversible inhibitors of the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor (HER-2) tyrosine kinases with enhanced ant-itumor activity J Med Chem 2001 44 17 2719 2734 11495584
- Marvin http://www.chemaxon.com [computer program] Version 5.6.0.1 ChemAxon Ltd 2011
- Halgren TA Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94 J Comput Chem 1996 17 5–6 490 519
- Gaussian 03 [computer program] Version Revision C.02 Wallingford CT Gaussian, Inc 2004
- Yang W Mortier WJ The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines J Am Chem Soc 1986 108 19 5708 5711 22175316
- Boström J Reproducing the conformations of protein-bound ligands: a critical evaluation of several popular conformational searching tools J Comput Aided Mol Des 2001 15 12 1137 1152 12160095
- Discovery Studio Modeling Environment [computer program] Version Release 3.1 San Diego Accelrys Software Inc 2012
- Michalczyk A Klüter S Rode HB Structural insights into how irreversible inhibitors can overcome drug resistance in EGFR Bioorg Med Chem 2008 16 7 3482 3488 18316192
- FRED program http://www.eyesopen.com [computer program] Version Version 2.2.5 OpenEye Scientific Software Inc Santa Fe, NM, USA 2012
- Nicholls A Grant JA Brown FK Gaussian docking functions Biopolymers 2003 68 1 76 90 12579581
- Verkhivker GM Bouzida D Gehlhaar DK Deciphering common failures in molecular docking of ligand-protein complexes J Comput Aided Mol Des 2000 14 8 731 751 11131967
- Eldridge MD Murray CW Auton TR Paolini GV Mee RP Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes J Comput Aided Mol Des 1997 11 5 425 445 9385547
- Stahl M Rarey M Detailed analysis of scoring functions for virtual screening J Med Chem 2001 44 7 1035 1042 11297450
- Grant JA Pickup BT Nicholls A A smooth permittivity function for Poisson-Boltzmann solvation methods J Comput Chem 2001 22 6 608 640
- Traxler P Furet P Strategies toward the design of novel and selective protein tyrosine kinase inhibitors Pharmacol Ther 1999 82 2–3 195 206 10454197
- Cavasotto CN Ortiz MA Abagyan RA Piedrafita FJ In silico identification of novel EGFR inhibitors with antiproliferative activity against cancer cells Bioorg Med Chem Lett 2006 16 7 1969 1974 16413185
- Rode HB Sos ML Grütter C Heynck S Simard JR Rauh D Synthesis and biological evaluation of 7-substituted-1-(3-bromophenylamino) isoquinoline-4-carbonitriles as inhibitors of myosin light chain kinase and epidermal growth factor receptor Bioorg Med Chem 2011 19 1 429 439 21130659
- Dror O Shulman-Peleg A Nussinov R Wolfson HJ Predicting molecular interactions in silico: I. A guide to pharmacophore identification and its applications to drug design Curr Med Chem 2004 11 1 71 90 14754427
- Güner OF History and evolution of the pharmacophore concept in computer-aided drug design Curr Top Med Chem 2002 2 12 1321 1332 12470283
- Xiang M Cao Y Fan W Chen L Mo Y Computer-aided drug design: lead discovery and optimization Comb Chem High Throughput Screen 2012 15 4 328 337 22221065
- Davis AM Teague SJ Kleywegt GJ Application and limitations of X-ray crystallographic data in structure-based ligand and drug design Angew Chem Int Ed Engl 2003 42 24 2718 2736 12820253
- Tuccinardi T Docking-based virtual screening: recent developments Comb Chem High Throughput Screen 2009 12 3 303 314 19275536
- Plewczynski D Łazniewski M von Grotthuss M Rychlewski L Ginalski K VoteDock: consensus docking method for prediction of protein-ligand interactions J Comput Chem 2011 32 4 568 581 20812324
- Schapira M Abagyan R Totrov M Nuclear hormone receptor targeted virtual screening J Med Chem 2003 46 14 3045 3059 12825943
- Ravindranathan KP Mandiyan V Ekkati AR Bae JH Schlessinger J Jorgensen WL Discovery of novel fibroblast growth factor receptor 1 kinase inhibitors by structure-based virtual screening J Med Chem 2010 53 4 1662 1672 20121196
References
- Wissner A Brawner Floyd MB Rabindran Sridhar K Syntheses and EGFR and HER-2 kinase inhibitory activities of 4-anilinoquinoline-3-carbonitriles: analogues of three important 4-anilinoquinazolines currently undergoing clinical evaluation as therapeutic antitumor agents Bioorg Med Chem Lett 2002 12 20 2893 2897 12270171
- Wissner A Overbeek E Reich MF Synthesis and Structure-Activity Relationships of 6,7-Disubstituted 4-Anilinoquinoline-3-carbonitriles. The Design of an Orally Active, Irreversible Inhibitor of the Tyrosine Kinase Activity of the Epidermal Growth Factor Receptor (EGFR) and the Human Epidermal Growth Factor Receptor-2 (HER-2) J Med Chem 2003 46 1 49 63 12502359
- Tsou H-R Overbeek-Klumpers EG Hallett WA Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity J Med Chem 2005 48 4 1107 1131 15715478
- Tsou H-R Mamuya N Johnson BD 6-Substituted-4-(3-bromophenylamino)quinazolines as Putative Irreversible Inhibitors of the Epidermal Growth Factor Receptor (EGFR) and Human Epidermal Growth Factor Receptor (HER-2) Tyrosine Kinases with Enhanced Antitumor Activity J Med Chem 2001 44 17 2719 2734 11495584
References
- Wissner A Brawner Floyd MB Rabindran Sridhar K Syntheses and EGFR and HER-2 kinase inhibitory activities of 4-anilinoquinoline-3-carbonitriles: analogues of three important 4-anilinoquinazolines currently undergoing clinical evaluation as therapeutic antitumor agents Bioorg Med Chem Lett 2002 12 20 2893 2897 12270171
- Wissner A Overbeek E Reich MF Synthesis and Structure-Activity Relationships of 6,7-Disubstituted 4-Anilinoquinoline-3-carbonitriles. The Design of an Orally Active, Irreversible Inhibitor of the Tyrosine Kinase Activity of the Epidermal Growth Factor Receptor (EGFR) and the Human Epidermal Growth Factor Receptor-2 (HER-2) J Med Chem 2003 46 1 49 63 12502359
- Tsou H-R Overbeek-Klumpers EG Hallett WA Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity J Med Chem 2005 48 4 1107 1131 15715478
- Tsou H-R Mamuya N Johnson BD 6-Substituted-4-(3-bromophenylamino)quinazolines as Putative Irreversible Inhibitors of the Epidermal Growth Factor Receptor (EGFR) and Human Epidermal Growth Factor Receptor (HER-2) Tyrosine Kinases with Enhanced Antitumor Activity J Med Chem 2001 44 17 2719 2734 11495584
- Zhou W Ercan D Chen L Novel mutant-selective EGFR kinase inhibitors against EGFR T790M Nature 2009 462 7276 1070 1074 20033049
References
- FRED program. http://www.eyesopen.com [computer program] Version Version 2.2.5 OpenEye Scientific Software Inc Santa Fe, NM, USA 2012
- Nicholls A Grant JA Brown FK Gaussian Docking Functions Biopolymers 2003 68 76 90 12579581