
Abstract
Purpose
Zoledronate (ZA) stands as a highly effective antiresorptive agent known to trigger medication-related osteonecrosis of the jaw (MRONJ). Its clinical dosages primarily encompass those used for oncologic and osteoporosis treatments. While inflammation is recognized as a potential disruptor of mucosal healing processes associated with ZA, prior research has overlooked the influence of varying ZA dosages on tissue adaptability. Therefore, a deeper understanding of the specific mechanisms by which inflammation exacerbates ZA-induced MRONJ, particularly when inflammation acts as a risk factor, remains crucial.
Methods
Cell proliferation and migration of human oral keratinocytes (HOK) was analyzed after treatment with different doses of ZA and/or lipopolysaccharide (LPS) to assess their possible effect on mucosal healing of extraction wounds. Mouse periodontitis models were established using LPS, and histological changes in extraction wounds were observed after the administration of oncologic dose ZA. Hematoxylin and eosin (HE) staining and immunofluorescence were used to evaluate mucosal healing.
Results
In vitro, LPS did not exacerbate the effects of osteoporosis therapeutic dose of ZA on the proliferation and migration of HOK cells, while aggravated these with the oncologic dose of ZA treatment by inducing mitochondrial dysfunction and oxidative stress via regulating SIRT1 expression. Furthermore, SIRT1 overexpression can alleviate this process. In vivo, local injection of LPS increased the nonunion of mucous membranes in MRONJ and decreased the expression of SIRT1, PGC-1α, and MnSOD.
Conclusion
Inflammation aggravates oncologic dose of ZA-induced mitochondrial dysfunction and oxidative stress via a SIRT1-dependent pathway, enhancing the risk of impaired mucosal healing in MRONJ. Our study implies that inflammation becomes a critical risk factor for MRONJ development at higher ZA concentrations. Elucidating the mechanisms of inflammation as a risk factor for mucosal non-healing in MRONJ could inform the development of SIRT1-targeted therapies.
Introduction
Medication-related osteonecrosis of the jaw (MRONJ) is a potentially severe, adverse event.Citation1 In 2022, the American Association of Oral and Maxillofacial Surgeons (AAOMS) updated its 2014 guidelines with significant changes to the staging and definition of MRONJ. It was defined as patients with no history of jaw radiation therapy and metastatic disease of the jaw who were currently or previously treated with anti-bone absorptive or anti-angiogenic agents and had detected bone or bone exposure through any fistula in the maxillofacial region for eight weeks.Citation2 Soft tissue toxicity, one of MRONJ’s pathogenic hypotheses, can lead to non-healing of the mucosa, thereby prolonging the jawbone’s exposure to the oral microbiota, which in turn promotes secondary infections and exacerbates osteonecrosis.Citation3–7 Currently, zoledronate (ZA), the third generation of nitrogen-bisphosphonate (BP), clinically used as an anti-resorptive agent for the treatment of osteoporosis and tumor bone metastasis, is considered to be one of the important sources of mucosal toxicity and is the most common pathogenic drug of MRONJ.Citation8–10
Drug concentration is also a key factor affecting mucosal toxicity, and ZA can reduce the metabolic activity of human oral mucosal cells in a dose-dependent manner at clinically relevant doses.Citation9 The clinical dose and frequency of ZA use vary according to therapeutic purposes; patients with metastatic bone disease typically receive high-dose intravenous administration every 3 to 4 weeks, and patients with osteoporosis should receive 4 mg or 5 mg of ZA annually.Citation3,Citation11 Clinically, intravenous ZA is administered with increased frequency and bioavailability in oncology patients compared to oral dosing in osteoporosis patients.Citation12,Citation13 Thus, ZA doses were significantly lower in osteoporosis patients than in cancer patients.Citation14 In addition, differences in drug concentration may affect the elasticity of the mucosa to environmental stimuli, thereby transforming non-risk factors at low concentrations into risk factors at high concentrations, further aggravating mucosal toxicity.Citation15 Therefore, clarifying the risk of mucosal injury caused by environmental factors at different doses of ZA will bring important inspiration for the precise prevention and treatment of MRONJ in clinical practice.
In the periodontal microenvironment, inflammation is one of the most common irritants. It has been shown that inflammation is considered as a risk factor for ZA-induced mucosal nonunion of MRONJ.Citation16 However, existing studies define inflammation as a risk factor . It was not shown whether inflammation was a risk factor at different concentrations. Instead, the effect of inflammation on MRONJ was directly determined under the condition of an oncologic dose of ZA. Considering that differences in ZA dose may affect the degree of mucosal tolerance to environmental stimuli, there may be a shift in inflammation from a low-risk factor to a high-risk factor at different clinical doses of ZA. Therefore, it is of clinical interest to investigate the effect of inflammation on mucosal healing disorders induced by different doses of ZA. Besides, keratinocytes are one type of cell involved in the intricate physiological process of oral mucosal healing.Citation17 Following teeth extraction, oral keratinocytes near the margin of the periodontal wound proliferated quickly and migrated to the surface of the teeth extraction wound, playing a vital role in the restoration of epithelial tissue.Citation18–20 Therefore, exploring the effect of inflammation on the proliferation and migration of oral keratinocytes under different doses of ZA provides a reference for clarifying whether inflammation can be used as a risk factor to aggravate the mucosal nonunion of MRONJ induced by different doses of ZA.
After clarifying the conditions for inflammation as a high-risk factor for MRONJ, it is of great clinical significance to explore the specific mechanisms by which inflammation exacerbates mucosal nonunion. It has been demonstrated that mucosal healing is hampered by oxidative stress caused by excessive reactive oxygen species (ROS).Citation19,Citation21 Oxidative stress could be induced by BP therapy,Citation22 and ROS could also be produced by chronic local inflammation and associated infection mechanisms.Citation23,Citation24 Several scholars have also explored oxidative stress as a potential pathophysiological mechanism of MRONJ through animal and clinical studies.Citation25–27 Thus, inflammation as a risk factor exacerbating ZA-induced mucosal healing abnormalities may be closely related to oxidative stress. Since mitochondria are the primary regulators of REDOX balance, impairment in their function would cause REDOX imbalance and consequent oxidative stress.Citation28,Citation29 According to studies, using BP could induce mitochondrial dysfunction, DNA damage, oxidative stress, and apoptosis in osteoblasts.Citation30 Furthermore, the inflammatory factor TNF-α increased mitochondrial mass and oxygen consumption upon activation.Citation31 Therefore, by damaging mitochondrial function, ZA and inflammation may exacerbate cellular oxidative stress. However, the effects of both on mitochondrial function and the specific regulatory mechanisms are still unclear. NAD (+)-dependent histone deacetylase sirtuin 1 (SIRT1) could reduce mitochondrial ROS formation and inflammation by increasing the production of peroxisome-proliferator-activated receptor-gamma co-activator-1 (PGC-1α) protein and reducing its acetylation.Citation32–35 Therefore, in this study, after clarifying the identity of inflammation as a low-risk factor or high-risk factor for mucosal non-healing of MRONJ under different therapeutic doses of ZA, we further investigated whether inflammation as a high-risk factor aggravated high dose of ZA-induced keratinocyte proliferation and migration disorders and MRONJ mucosal non-healing through mediating oxidative stress and mitochondrial dysfunction in a SIRT1-dependent manner.
Materials and Methods
Cell Culture and Drug Treatment
Human oral keratinocytes (HOK) cell line was purchased from Shanghai Spectrometer Biotechnology Co., Ltd. (Shanghai, China). The cells were cultured in DMEM containing 10% FBS (Gibco, Grand Island, NY, USA), and 1% penicillin-streptomycin at 37°C in humidified atmosphere of 5% CO2 and 95% air. To mimic the in vivo inflammatory microenvironment, we treated HOK cells with lipopolysaccharide (LPS) (5, 10, 20, 50, 100 ng/mL; InvivoGene, CA, USA), and the cell viability was determined according to CCK-8, and a final concentration of 10 ng/mL was used in the present study. Briefly speaking, in DMEM medium containing 10% FBS, two different doses of ZA (Aclasta, Novartis, CH) were combined with LPS to co-culture HOK cells for 48 hours. After literature review and pre-experimentation, we determined 2.5 μM as the ZA dose for osteoporosis treatmentCitation36 and 10 μM as the ZA dose for tumor treatment.Citation30 Moreover, to detect the effect of oxidative stress on HOK, 5 mM ROS inhibitor N-acetylcysteine (NAC)Citation37 or ZA (10 μΜ)+LPS (10 ng/mL) or NAC (5 mΜ)+ZA (10 μΜ)+LPS (10 ng/mL) were added to the medium and treated for 48 hours. In addition, to verify the relationship between SIRT1 and mitochondrial dysfunction, resveratrol (Res, 75 mM, Proteintech, CM01142) was added to the culture medium to modulate SIRT1 expression.Citation38
Cell Counting Kit-8 (CCK-8)
HOK cells were seeded at a density of 2×103 cells/well in a 96-well plate and allowed to adhere to the plate overnight. Subsequently, the cells were treated with varying concentrations of LPS (5, 10, 20, 50, and 100 ng/mL) for 48 hours to identify optimal concentrations. Additionally, HOK cells were stimulated for 48 hours combining either the osteoporosis therapeutic dose or the oncologic dose of ZA with LPS in the culture medium. To determine cell viability, CCK-8 fluid (Med ChemExpress, China) was diluted according to the manufacturer’s instructions. Briefly, each well received 10 µL of the CCK-8 solution, and the plates underwent an hour-long incubation at 37°C. An optical density measurement was made at 450 nm using a BioRad microplate reader (Bio-Rad, USA; Model 680).
Wound Healing Assay
HOK cells with exponential growth were seeded into 6-well plates at a density of 6×105/well and cultured to 90% confluence. Use the 200 μL pipette tip to make a vertical scratch on the surface of the plate. Cells were then incubated in 2% FBS-DMEM and then treated and incubated according to the protocol. The cells were cultured in the LPS group alone, the ZA group at different doses, and the ZA combined with LPS at different doses. In addition, to explore the relationship between oxidative stress and cell migration, the control group, the 5 mM NAC group, the 10 μM ZA+10 ng/mL LPS group, and the 5 mM NAC+ZA+LPS group were set. The control group, the 75 μM Res group, the 10 μM ZA+10 ng/mL LPS group, and the 75 μM Res+ZA+LPS group were set up to verify the relationship between SIRT1 and cell migration. Photographs were taken under the microscope (Olympus BX53, Tokyo, Japan) 0, 24, and 48 hours after scratches, and the Image-Pro Plus 6.0 software (Media Cybernetics) was used for analysis to calculate the percentage of healing.
Western Blot Assay
A cell scraper was used to scrape the cells into the EP tube, and RIPA Lysis Buffer (Cwbio, Beijing, China) was used to lyse them. This mixture also contained protease inhibitors and phosphatase inhibitors, in a ratio of 98:1:1. Each EP tube received 1/4 liter of 5×SDS loading buffer, which was then heated at 97°C for 5 minutes after the protein concentration was determined using the BCA protein assay (Beyotime, Beijing, China). After being separated by electrophoresis on a 10% SDS-PAGE gel, the protein samples were transferred to a polyvinylidene fluoride (PVDF) membrane. These membranes were then incubated for an hour at room temperature with a 0.1% Tween-20 solution that was Tris-buffered and contained 5% BSA. Anti-GAPDH (10,494-1-Ap), anti-PCNA (NA03-200UGCN, Calbiochem), and anti-SIRT1 (13,161-1-AP, Proteintech) antibodies were incubated on the membrane overnight at 4°C. The next day, after rewarming for an hour, washed three times with PBS. Goat anti-rabbit IgG H&L (HRP) (1:2000 dilution; ab6721, Abcam) or goat anti-mouse IgG+IgM+IgA H&L (HRP) (1:2000 dilution; ab102448, Abcam) incubated the membrane at room temperature for one hour. Immune response bands were observed using an enhanced chemiluminescence reagent (B500024, Proteintech). Images were produced using a gel imaging system (General Electric Company Amersham Imager 600). Grayscale values were measured, quantified, and normalized to GAPDH levels using Media Cybernetics’ Image-Pro Plus 6.0 program.
Detection of ROS
We measured intracellular ROS levels using the fluorescent dye DCFH-DA (S0033, Beyotime). HOK cells were treated with drugs; the control group, the 10 μM ZA group alone, the 10 ng/mL LPS group, and the 10 μM ZA+10 ng/mL LPS combined group were set up to detect the level of ROS production in cells. To detect the reversal effect of NAC on oxidative stress caused by ZA+LPS, the control group, the 5 mM NAC group, the 10 μM ZA+10 ng/mL LPS group, and the 5 mM NAC+ZA+LPS group were set up. In addition, to verify the relationship between SIRT1 and cellular oxidative stress, the control group, the 75 μM Res group, the 10 μM ZA+10 ng/mL LPS group, and the 75 μM Res+ZA+LPS group were set up. After the treatment of HOK cells for 48 hours, the cells of each group were stained with 10 μM DCFH-DA at 37°C for 30 minutes in a dark environment. The cells were then washed three times with DMEM without fetal bovine serum. Images were obtained using a fluorescence microscope (DMi8 Automatic, Leica Microsystems CMS GmbH, Germany). The field of view was randomly selected for photography. The fluorescence intensity was then assessed using Image-Pro Plus 6.0 software (Media Cybernetics).
Proliferation Assay – EdU Staining
HOK cells (5×103) were plated on a 48-well plate. The cells had been treated for 48 hours before being incubated at 37°C for 2 hours with an EdU working solution. The cells were permeabilized with 0.3% Triton X-100 for 15 minutes following 15 minutes of fixation with 4% paraformaldehyde (PFA). After that, the cells were exposed to Azide 594 at room temperature for 30 minutes while being kept out of the light. DAPI then incubated the cells for 5 minutes. After the cells were stained with EdU and examined with a fluorescence microscope (DMi8 Automatic, Leica Microsystems CMS GmbH, Germany), the percentage of EdU-positive cells was calculated using the ratio of the number of EdU-positive cells to the total number of cells.
JC-1 Mitochondrial Membrane Potential (MMP) Assay
The variation in mitochondrial transmembrane potential (ΔΨm) was measured using an enhanced MMP assay kit with JC-1 (C2006, Beyotime). Simply speaking, treated HOK cells were exposed to JC-1 dye for 20 minutes before being captured in the fluorescence microscope image. When JC-1 assembles into a polymer in a healthy mitochondrial matrix, red fluorescence will be the result. In contrast, as the fall of MMP, as assessed by a fluorescence microscope (DMi8 Automatic, Leica Microsystems CMS GmbH, Germany), JC-1 effluxes to the cytoplasm and emits monomers with green fluorescence.
Distribution of Mitochondria Measured
Treated cells were incubated in DMEM with 100 nM Mito-Tracker Green (C1048, Beyotime) for 30 minutes at 37°C in the dark. After washing, cells were stained and observed under a fluorescence microscope (DMi8 Automatic, Leica Microsystems CMS GmbH, Germany). The fluorescence intensity was then assessed using Image-Pro Plus 6.0 software (Media Cybernetics).
Immunofluorescence Staining
The HOK cells were fixed with 4% PFA, washed in PBS, permeabilized with 0.5% Triton X-100, and blocked with 5% BSA. After that, cells were treated overnight at 4°C with the corresponding primary diluted antibody. A fluorescent secondary antibody was then added to indicate the primary antibody after the primary antibodies had been washed in 0.1% Tween-20. Images were captured with fluorescence microscopy (Automated Model DMi8, Leica Microsystems CMS GmbH, Germany), and the nucleus was stained with DAPI.
Immersing paraffin-embedded sections in 0.3% H2O2 for 30 minutes and blocking them in 1% BSA-PBS for 20 minutes was necessary for immunofluorescence staining. Sections were incubated overnight at 4°C with the following mixed antibodies: anti-SIRT1 (13,161-1-AP, Proteintech), anti-PGC-1α (66,369-1-lg, Proteintech), anti-COL1 (ab34710, Abcam) and anti-MnSOD (24,127-1-AP, Proteintech). The sections were exposed to a combination of green fluorescent goat anti-rabbit IgG H&L (Alexa Fluor 647) (1:200; ab150081, Abcam) and red fluorescent goat anti-mouse IgG H&L (Alexa Fluor 488) (1:200; ab150119, Abcam) antibodies at room temperature in darkness for 1 hour. After three washes in PBS, the samples underwent nuclear staining using 4′,6-diamidino-2-phenylindole (ab104139, Abcam) for a period of 5 minutes. Images were captured using inverted fluorescence microscope (DMi8 Automatic, Leica Microsystems CMS GmbH, Germany). The fluorescence intensities were analyzed with Image-Pro Plus 6.0 software (Media Cybernetics).
MRONJ Model Establishment and Tissue Preparation
Thirty eight-week-old C57BL/6J male mice were purchased from Jinan Pengyue Experimental Animal Breeding Co., Ltd. (Jinan, China) and fed with a 12-hour light/dark cycle in a standard environment. All animal testing procedures are conducted following the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. The experimental animals were approved by the Affiliated Hospital of Shandong University School of Stomatology’s Institutional Animal Care and Use Committee (IACUC) (No. 20220711). To investigate the effect of the inflammation-exacerbated oncologic dose of ZA on mucosal healing in MRONJ disease, C57BL/6J mice were randomly divided into 3 groups (n=10): control, ZA-treated, and ZA+LPS-treated groups. Mice in the control group were given saline solution as blank control. The ZA and ZA+LPS groups were injected intraperitoneally with ZA 200 μg/kg/weekCitation39 for eight weeks. Simultaneously, the periodontitis animal model was established by local injection of 1 mg/ mL LPS (10 μL) into the gingival groove between the right maxillary first and second molar teeth, twice a week. After eight weeks of administration, mice were anesthetized with 1% pentobarbital sodium (40 mg/kg), and the right maxillary first and second molar teeth were removed under anesthesia. After teeth extraction, all groups were given medication as before. Two weeks after teeth extraction,Citation40 all mice were anesthetized, and their hearts were infused. The maxillas were collected for histological analysis.
Staining with Hematoxylin and Eosin (HE)
Histological alterations were assessed using HE staining. After dewaxing and hydration, the slices were immersed in hematoxylin for 15 minutes, washed in distilled water, immersed in eosin for 7 minutes, rewashed in distilled water, and finally dehydrated and mounted. Subsequently, the sections were observed, and digital images were taken under an optical microscope (BX-53; Olympus Corporation, Tokyo, Japan).
Statistical Analysis
QQ plots were used to assess data distribution, and when the scatter plot was close to a straight line, it can be considered that the data set was approximately normally distributed. All experiments were independently repeated at least 3 times. Data was presented as mean±standard error of the mean (SEM). Unpaired two-tailed Student’s t-tests were used to compare the means of the two groups, and one-way ANOVA with the Bonferroni post hoc test for multiple comparisons was used to compare the means of the multiple comparisons. Differences were considered statistically significant at P value <0.05 and indicated by “*”, P <0.01 were indicated by “**”, P <0.001 were indicated by “***”, and P < 0.0001 were indicated by “****”.
Results
Inflammation Exacerbates the Inhibition of HOK Proliferation and Migration Promoted by the Oncologic Dose of ZA Treatment, but Not Aggravates This Effect with the Osteoporosis Therapeutic Dose of ZA Treatment
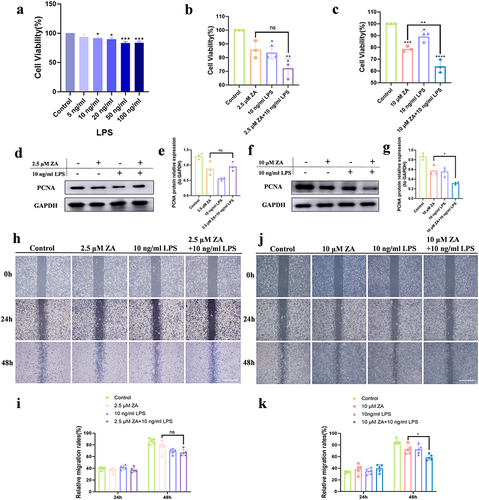
To investigate the effects of inflammation and different doses of ZA on the proliferation and migration of HOK cells, CCK-8 was used to measure the viability of HOK cells. Following a 48-hour stimulation period with varying doses of LPS, HOK cell viability showed a dose-dependent decrease (). The combination of the osteoporosis therapeutic dose of ZA and LPS had no statistically significant difference in the cell viability of HOK compared with the osteoporosis therapeutic dose of ZA alone (). For HOK, there was a statistical difference between the oncologic dose of ZA combined with LPS and ZA alone. The oncologic dose of ZA reduced the viability of HOK cells, and inflammation enhanced this inhibitory effect (). Western blot analysis showed that the combination of the osteoporosis therapeutic dose of ZA and LPS did not affect PCNA expression than the ZA group in HOK cells ( and ), while LPS with the oncologic dose of ZA significantly inhibited its level compared with ZA-treated alone ( and ). There was no difference in cell migration rates between the osteoporosis therapeutic dose ZA combined with LPS group and the osteoporosis therapeutic dose ZA dosing group ( and ). The oncologic dose ZA combined with LPS group had a wider distance between the two sides of the scratch and a larger middle white space compared to the oncologic dose ZA group (). Wound healing assays showed that inflammation exacerbated the inhibitory effect of the oncologic dose of ZA on HOK migration ().
Figure 1 Inflammation exacerbated the inhibition of HOK proliferation and migration with oncologic dose of ZA treatment, but did not aggravate this with the osteoporosis therapeutic dose of ZA treatment. (a) Cell viability of HOK treated with gradient concentration LPS for 48 hours. (b) Effect of the osteoporosis therapeutic dose of ZA (2.5 μM) and LPS (10 ng/mL) on HOK cell viability for 48 hours. (c) Effects of the oncologic dose of ZA (10 μM) and LPS on HOK cell viability for 48 hours. (d) Representative Western blot of PCNA. (e) Western blot analysis. (f) Western blot analysis of PCNA. (g) Quantification of Western blot results. (h) Wound healing assays in HOK cells were simulated with 2.5 μM ZA or/and 10 ng/mL LPS at 24, and 48 hours. Scale bar, 500 μm. (i) The analysis of wound healing assay. (j) Wound healing assays in HOK cells were simulated with 10 μM ZA or/and 10 ng/mL LPS at 0, 24, and 48 hours. Scale bar, 500 μm. (k) The analysis of the wound-healing assay. Data were given as the mean±SD. *P < 0.05. **P < 0.01. ***P < 0.001. ****P < 0.0001.
Inflammation Exacerbates the Inhibition of HOK Proliferation and Migration Induced by the Oncologic Dose of ZA Through Up-Regulation of Oxidative Stress
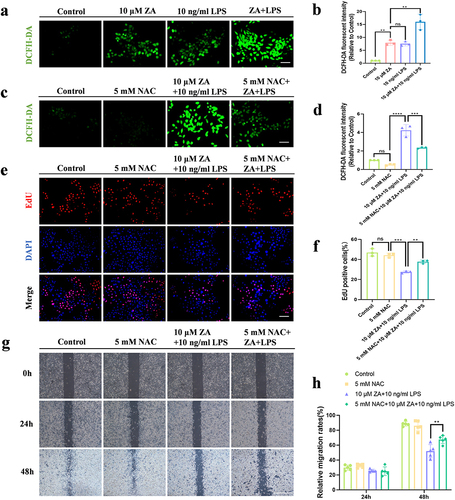
Our results showed that LPS had no significant effect on the proliferation and migration of HOK cells in the treatment of the osteoporosis therapeutic dose of ZA. Conversely, when inflammation was present, the inhibitory effect of oncologic dose ZA on the proliferation and migration of HOK cells became more significant. Therefore, we further explored the specific mechanism of LPS exacerbating the effect of oncologic dose ZA. We evaluated ROS levels to investigate whether inflammation exacerbates the inhibition of HOK cell proliferation and migration induced by the oncologic dose of ZA through oxidative stress. Using DCFH-DA detection, we found that the expression of green fluorescence in each group was gradually enhanced (), and ROS generation in the ZA+LPS group was significantly increased compared with that in the ZA group (). Compared to the ZA+LPS group, there was a significant decrease in the level of oxidative stress after the combination of NAC ( and ). In addition, the proliferation assay (EdU staining) was used to detect cell proliferation. The number of red fluorescent cells in the ZA+LPS group was less than in the ZA group, and the ZA+LPS+NAC group was higher than that in the ZA+LPS group (). There was significant difference in the number of positive cells between the ZA+LPS group and the NAC+ZA+LPS group (). In addition, scratch experiments showed that NAC promoted the migration of HOK cells and narrowed the cell spacing, and the inhibition of migration in the ZA+LPS group was also relieved to a certain extent, and the cell spacing became smaller (). Inflammation enhanced the inhibitory effect of the oncologic dose of ZA on HOK cells migration, and the addition of NAC mitigated this inhibitory effect with statistical significance (). These results suggest that inflammation exacerbates the inhibition of HOK proliferation and migration induced by the oncologic dose of ZA through upregulating oxidative stress.
Figure 2 Inflammation exacerbated the inhibition of HOK proliferation and migration induced by the oncologic dose of ZA through up-regulation of oxidative stress. (a) Representative images of intracellular ROS fluorescence in response to 10 μM ZA or/and LPS. Scale bar, 100 μm. (b) Quantitative analysis of cellular ROS levels. (c) Representative images of intracellular ROS fluorescence in response to 5mM NAC or/and ZA+LPS. Scale bar, 100 μm. (d) Statistical analysis of intracellular ROS levels. (e) Representative images of the proliferation assay (EdU staining) of HOK cells under different treatments. Scale bar, 100 μm. (f) The bar graph showed the percentage of EdU-positive cells. (g) HOK cell wound healing experiments were simulated for 0, 24 and 48 hours according to the group. Scale bar, 500 μm. (h) The analysis of Wound-healing assay. Data were given as the mean±SD. ns: nonsense. **P < 0.01. ***P < 0.001. ****P < 0.0001.
Inflammation Regulates Mitochondrial Dysfunction in a SIRT1-Dependent Manner, Exacerbating Oxidative Stress Induced by the Oncologic Dose of ZA
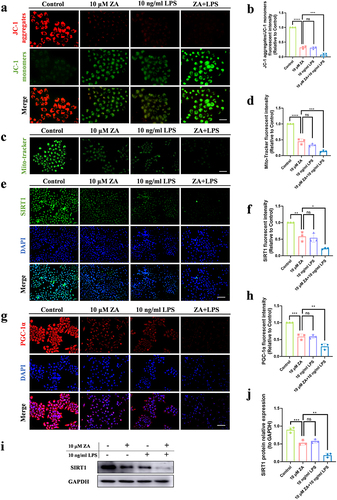
Damaged mitochondria are the main source of intracellular ROS. Consequently, we measured MMP using the JC-1 assay to detect mitochondrial function. The MMP was depolarized more in the ZA group than in the control group (), and the inflammation intensified the reduction of MMP induced by the oncologic dose of ZA (). Furthermore, mitochondrial counts were detected using Mito-Tracker Green, which showed that LPS treatment exacerbated ZA-induced mitochondrial reduction ( and ). To explore the specific mechanism of SIRT1, the expression level of SIRT1 was detected by cellular immunofluorescence. Compared with the ZA group, the expression of SIRT1 in the ZA+LPS group was significantly reduced ( and ). Considering that SIRT1 can up-regulate its downstream molecule PGC-1α to improve mitochondrial biosynthesis, kinetics, and function, cellular immunofluorescence of PGC-1α was applied and its level in ZA+LPS group was significantly weakened ( and ). In addition, the protein expression of SIRT1 also showed a decreased trend ( and ). These results suggest that LPS exacerbates mitochondrial dysfunction and reduces the expression of SIRT1 and PGC-1α in HOK treated with the oncologic dose of ZA.
Figure 3 Inflammation exacerbated mitochondrial dysfunction caused by oncologic dose of ZA by modulating SIRT1. (a) JC-1 fluorescence staining showed MMP in the control group, the 10 μM ZA group, the 10 ng/mL LPS group, and the ZA+LPS group. Scale bar, 50 μm. (b) Statistical analysis of MMP in each group. (c) Representative images of mitochondrial distribution in each group. Scale bar, 100 μm. (d) Fluorescence intensity and statistical analysis of Mito-Tracker Green staining in each group were displayed. (e) Cellular immunofluorescence images of SIRT1. Scale bar, 100 μm. (f) Quantitative analysis of the fluorescence intensity of SIRT1. (g) Immunofluorescence staining of PGC-1α in HOK cells was observed under an inverted fluorescence microscope. Scale bar, 100 μm. (h) Quantitative analysis of PGC-1α cell immunofluorescence. (i) Western blot was used to detect SIRT1 protein levels in HOK cells after drug treatment 48 hours. (j) Quantitative analysis of SIRT1 protein bands was performed. Data were expressed as mean±SD. ns: nonsense. *P < 0.05. **P < 0.01. ***P < 0.001. ****P < 0.0001.
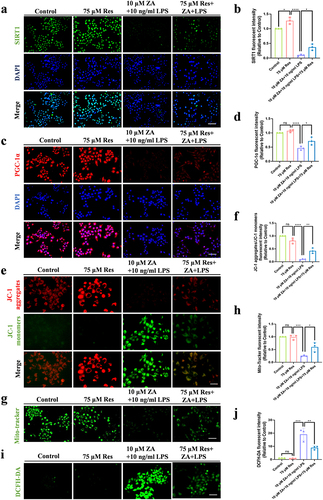
To illustrate the function of SIRT1 in vitro, Res, an agonist of SIRT1, was added to the ZA+LPS group to overexpress SIRT1. Cellular immunofluorescence showed that Res alleviated the inhibition of SIRT1 by ZA+LPS ( and ). We looked at PGC-1α expression levels to verify changes in the SIRT1 targeting protein. PGC-1α fluorescence was weakened by ZA+LPS and regained following treatment with Res ( and ). Simultaneously, changes in the MMP were detected, confirming the positive impact of SIRT1 on HOK mitochondrial dysfunction ( and ). The Mito-Tracker Green assay revealed that overexpression of SIRT1 increased the number of mitochondria than the ZA+LPS group ( and ). DCFH-DA assay showed that Res treatment alleviated ZA+LPS-induced oxidative stress ( and ). These findings imply that LPS intensifies ROS production through down-regulating SIRT1 expression in damaged mitochondria of HOK, induced by the oncologic dose of ZA.
Figure 4 Res alleviated mitochondrial dysfunction and oxidative stress caused by inflammation and the oncologic dose of ZA by regulating SIRT1. (a) The expression of SIRT1 in HOK cells after drug treatment was detected by immunofluorescence assay. Scale bar, 100 μm. (b) Fluorescence quantitative analysis of SIRT1. (c) HOK cell PGC-1α expression was detected by immunofluorescence after drug treatment. Scale bar, 100 μm. (d) Fluorescence quantitative analysis of PGC-1α. (e) Fluorescence staining of JC-1 showed the MMP of each group. Scale bar, 50μm. (f) Statistical fluorescence analysis of MMP. (g) Representative images of mitochondrial distribution in each group. Scale bar, 100 μm. (h) The fluorescence intensity of Mito-Tracker Green staining was shown. (i) The oxidative stress levels in HOK cells were detected by DCFH-DA staining. Scale bar, 100 μm. (j) Statistical analysis of intracellular ROS levels. Data were expressed as mean±SD. ns: nonsense. *P < 0.05. **P < 0.01. ***P < 0.001. ****P < 0.0001.
Inflammation Aggravates the Inhibitory Effect of the Oncologic Dose of ZA on the Proliferation and Migration of HOK by Regulating SIRT1 Signaling
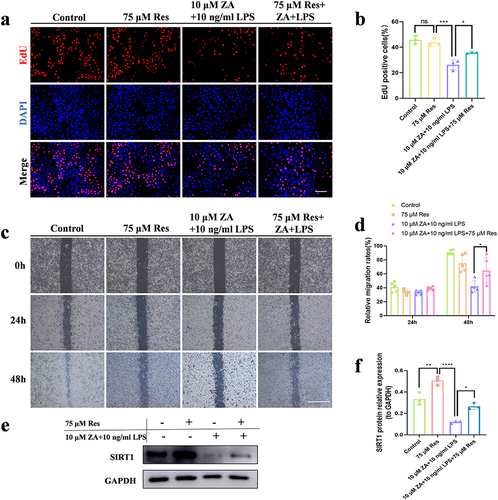
Proliferation Assay (EdU staining) was used to identify cell proliferation and investigate whether in vitro SIRT1 regulation of mitochondria impacts HOK cell proliferation and migration. The inhibitory effect of ZA+LPS on cell proliferation was alleviated after Res treatment ( and ). Scratch experiments demonstrated that intercellular migration was promoted after treatment of HOK cells with Res and ZA+LPS ( and ). Western blot verified the overexpression of SIRT1 after Res treatment (). The expression level of SIRT1 in the ZA+LPS+Res group was significantly upregulated compared with that in the ZA+LPS group (). This suggests that inflammation exacerbates the inhibition of HOK proliferation and migration induced by the oncologic dose of ZA through regulating SIRT1 signaling.
Figure 5 Inflammation aggravated the inhibitory effect of the oncologic dose of ZA on the proliferation and migration of HOK by regulating SIRT1 signaling. (a) Representative images of the proliferation assay (EdU staining) of HOK cells under different treatments. Scale bar, 100 μm. (b) The bar chart showed the percentage of EdU-positive cells. (c) The HOK cell scratch experiment was simulated for 0, 24, and 48 hours in each groups, respectively. Scale bar, 500 μm. (d) Statistical analysis of scratch experiments. (e) Western blot detection of SIRT1 protein levels in HOK cells after drug treatment. (f) Quantitative analysis of the SIRT1 protein. The data were given as the mean±SD. ns: nonsense. *P < 0.05. **P < 0.01. ***P < 0.001. ****P < 0.0001.
Inflammation as a Risk Factor Aggravates MRONJ Mucosal Non-healing Induced by an Oncologic Dose of ZA Through Mediating Mitochondrial Dysfunction and Oxidative Stress in a SIRT1-Dependent Manner
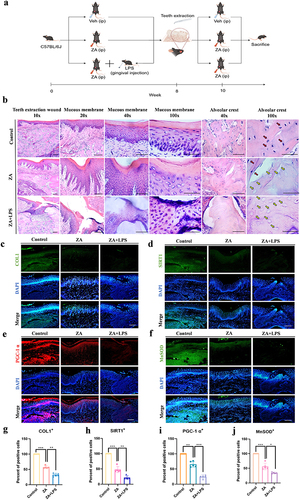
To investigate the role of the inflammatory microenvironment in the development of MRONJ at an oncologic dose of ZA, periodontitis models with a local injection of LPS were used. Mice were intraperitoneally injected with ZA and locally injected with LPS around the gums between the first and second maxillary teeth. After eight weeks, the maxillary first and second molars were extracted and then all groups were given medication as before for another two weeks (). Two weeks after teeth extraction, maxillary examination showed complete wound healing in all mice in the control group, and the incidence of incomplete mucosal healing and bone exposure in mice treated with ZA and LPS was higher than that in the ZA group. The incidence of incomplete mucosal healing and bone exposure was lower in 30% (3/10) of ZA-treated mice, whereas ZA+LPS-treated mice had a higher incomplete mucosal healing and bone exposure of 50% (5/10). The tissue repair process in the teeth extraction site and the overcoating mucosa of the ZA+LPS group was seriously impaired, with increased inflammatory areas, damaged epithelial integrity, and epithelial discontinuity (). HE staining illustrated a woven bone and prominent periosteum at the alveolar ridge in the control group (). By contrast, a woven bone filling with some empty osteocyte lacunae was observed in the ZA group. However, bone healing was impaired in the ZA+LPS group, and necrotic areas with more empty osteocyte lacunae and cortical bone destruction were observed. A marked inflammatory infiltrate was observed adjacent to the osteonecrosis area. During normal wound healing, the cells around the wound migrate to the wound area and promote wound healing by increasing collagen synthesis and secretion, including collagen type I (COL1). We discovered that the COL1 fluorescence in the ZA and ZA+LPS groups had decreased through tissue immunofluorescence (). To investigate the signaling pathway further, the tissue immunofluorescence assay was also used to detect the fluorescence content of SIRT1, and the expression of SIRT1 in the ZA+LPS group was lower than that in the ZA group (), which was consistent with the results in vitro. Considering whether the healing of teeth extraction wounds are related to mitochondrial function, we detected the expression of PGC-1α at the teeth extraction sites, and the results showed that the expression of PGC-1α in the ZA+LPS group was lower than that in the ZA group (). In addition, MnSOD, an indicator related to oxidative stress, was also detected, and tissue immunofluorescence showed that in the ZA group, MnSOD at the site of teeth extraction was higher than that in the ZA+LPS group (). The statistical analysis results are shown in .
Figure 6 Inflammation as a risk factor aggravated MRONJ mucosal non-healing induced by the oncologic dose of ZA through mediating mitochondrial dysfunction and oxidative stress in a SIRT1-dependent manner. (a) Schematic drawing of the procedures conducted in this study. By Figdraw. (b) HE staining at the teeth-extracted sites in MRONJ mice models. Empty bone lacunas with no osteocytes were pointed by yellow arrows. Normal osteocytes in bone lacunas were indicated by red arrows. Scale bars, 200 μm, 100 μm, 50 μm or 20 μm. (c) The representative immunofluorescence staining images of COL1 in teeth-extracted sites. Scale bar, 50 μm. (d) Representative images of SIRT1 immunofluorescence staining in teeth-extracted sites. Scale bar, 50 μm. (e) Tissue immunofluorescence image of PGC-1α detected at teeth extraction wound site. Scale bar, 50 μm. (f) Representative images of MnSOD immunofluorescence staining in teeth-extracted sites. Scale bar, 50 μm. (g–j) Statistical analysis of immunofluorescence in tissue sections. Data were presented as mean±SD. *P < 0.05. **P < 0.01. ***P < 0.001.
Discussion
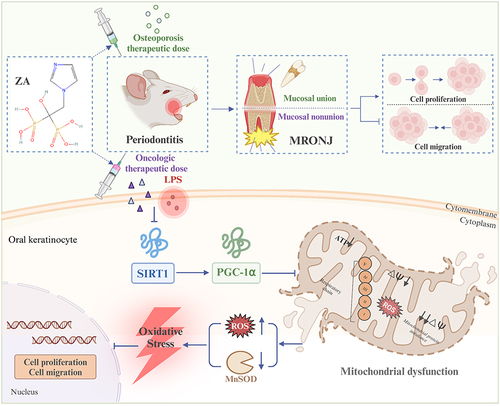
In this study, we comprehensively considered that the difference in ZA dose might have a different effect on the position of inflammation as a key risk factor for MRONJ mucosal nonunion by influencing mucosal adaptation to the periodontal inflammatory microenvironment. We investigated the related function of inflammation by administering osteoporosis or the oncologic dose of ZA and the related mechanisms via which it poses a risk. It was found that inflammation did not aggravate the effect of HOK proliferation and migration caused by osteoporosis therapeutic doses of ZA. On the other hand, the role of inflammation became more pronounced when the dose of ZA was increased to oncologic therapeutic levels. In this case, inflammation significantly increases the risk of MRONJ mucosal healing damage by mediating mitochondrial dysfunction and oxidative stress (). This suggests that inflammation becomes a significant risk factor for exacerbating the onset and progression of MRONJ under higher doses of ZA.
Figure 7 Inflammation exacerbated the inhibition of HOK proliferation and migration promoted by the oncologic dose of ZA treatment, but not aggravated this with the osteoporosis therapeutic dose of ZA treatment. Inflammation exacerbated the impaired mucosal healing of MRONJ extraction wounds induced by the oncologic dose of ZA. One possible mechanism may be that inflammation, as a high-risk factor, exacerbates the inhibition of HOK proliferation and migration induced by the oncologic dose of ZA by modulating mitochondrial dysfunction and the resulting oxidative stress in a SIRT1-dependent manner. There are five proteins in the inner mitochondrial membrane: complex I (NADH dehydrogenase), complex II (succinate dehydrogenase), complex III (cytochrome c reductase), complex IV (cytochrome c oxidase), and complex V (ATP synthase). Created with BioRender.com.
Mucosal nonunion is a major complication of MRONJ, and MRONJ is clinically diagnosed mainly through mucosal non-healing and maxillofacial bone baring for more than 8 weeks.Citation41 When the mucosal damage in the oral cavity fails to heal, it could affect the blood supply to the surface of the bone, leading to a possible secondary infection and osteonecrosis.Citation42 Relevant studies have shown that oral mucosal damage could occasionally be the first sign of MRONJ.Citation42 As key players in the mucosal healing process, the alterations in the proliferation and migration of HOK cells would affect how well mucosal damage was healing.Citation43 One of the theories explaining the etiology of MRONJ focuses on the negative effects bisphosphonate drugs have on soft tissues, especially keratinocytes. Keratinocytes are crucial for oral wound healing and soft tissue regeneration due to their high cellular activity and migratory capacity. They migrate to affected areas, a vital step in wound closure. However, nitrogen-containing BPs, notably pamidronate and ZA, have been shown to exert a profound negative influence on the cellular viability, migratory ability, and apoptotic behavior of HOK cells.Citation44 This inhibitory effect on HOK cells may contribute to the delayed healing observed in MRONJ patients, thus increasing the risk of developing this condition.
It has been noted that ZA therapeutic doses for tumors and osteoporosis could cause MRONJ.Citation45,Citation46 Furthermore, using ZA would negatively impact HOK cell migration and proliferation,Citation47 resulting in mucosal toxicity and the development of mucosal nonunion following teeth extraction.Citation48 It has been demonstrated that ZA treatments may increase the risk of MRONJ, and therefore the decision-making needs to be weighed against the pros and cons of ZA to ensure that the patients are fully aware of the risks and that patients take preventive measures. Meanwhile, preventive oral care is crucial, and the risk of MRONJ can be effectively reduced through regular oral examinations and appropriate treatment.Citation6,Citation49
The risk factors associated with MRONJ include oral infections and related inflammatory conditions such as untreated dental caries, pulp infections, and periodontal diseases. These conditions can lead to the extraction of teeth that may become irreparable, thereby increasing the risk of developing MRONJ.Citation50,Citation51 While current studies have suggested that inflammation is a risk factor for mucosal nonunion in MRONJ, but these studies have neglected the effect of ZA dose on tissue adaptability. Therefore, it is not comprehensive to directly determine inflammation as a risk factor based on the background of oncologic dose ZA. Thus, we determined the proliferation and migration capacity of HOK cells through CCK-8 assay, the PCNA content analysis, and wound healing assays, and discovered that inflammation only serves to intensify the inhibition of HOK cell migration and proliferation induced by an oncologic dose of ZA; at the osteoporosis therapeutic dose of ZA, inflammation did not exacerbate the effect of HOK cell migration or proliferation. Given these findings, patients receiving ZA as an oncologic dose should be more cautious about teeth extraction when facing dental problems due to inflammation. Additionally, several studies have indicated that retaining infected teeth poses a greater risk of developing MRONJ than extracting them.Citation52 Consequently, minimizing the need for tooth extraction and mitigating the risk of MRONJ becomes paramount. The most critical factor in achieving this is the regular conduct of oral examinations every three months prior to and during ZA treatment, along with the implementation of appropriate measures to address oral health concerns.
In addition, it is not clear what mechanisms make inflammation a risk factor for MRONJ. To gain a deeper understanding of how inflammation exacerbates MRONJ in the context of oncologic ZA dose, we conducted a detailed investigation. Several previous studies have shown evidence that ZA can increase the degree of oxidative stress in cells.Citation53 It was commonly recognized that elevated oxidative stress was the primary indicator of an inflammatory environment and that oxidative stress was crucial for tissue healing.Citation54 Given the diverse effects of oxidative stress on keratinocytes reported in various studies,Citation55–57 we hypothesized that oxidative stress might be a crucial factor linking inflammation to MRONJ pathogenesis. To test this hypothesis, we employed NAC, a synthetic derivative of the endogenous amino acid L-cysteine and a precursor of glutathione. NAC is known for its significant role in regulating oxidative stress and modulating various pathophysiological processes intimately associated with disease, including mitochondrial dysfunction, cell apoptosis, and inflammatory responses. Additionally, it indirectly affects the production of neurotransmitters such as glutamate and dopamine.Citation58 Therefore, in order to explore the relationship between oxidative stress and HOK proliferation and migration, we added the ROS inhibitor NAC. Not only were the oxidative stress levels in HOK cells caused by ZA+LPS+NAC decreased, but so was the cells’ capacity to proliferate and migrate increased.
Given the close relationship between mitochondrial dysfunction and oxidative stress,Citation59 in further studies, we explored the pathway through which inflammation affects oxidative stress due to mitochondrial dysfunction in HOK cells in the case of the oncologic dose of ZA. The SIRT1 protein, which affected multiple biological processes by deacetylating a variety of proteins, played an important role in regulating inflammation.Citation60 To improve mitochondrial biogenesis and lessen mitochondrial dysfunction, SIRT1 could activate a range of transcription factors.Citation60 A previous study had demonstrated that menaquinone-4 alleviates the oxidative damage induced by ZA through SIRT1-FoxO1 signaling pathway.Citation30 SIRT1 modifies mitochondrial dysfunction to mediate alterations in oxidative stress through PGC-1α.Citation61 Based on previous research,Citation30 we hypothesized that inflammation influences mitochondrial dysfunction through the SIRT1-PGC-1α pathway at the oncologic dose of ZA, exacerbating oxidative stress in HOK cells. We observed a significant decrease in mitochondrial number and function, an increase in oxidative stress level, and a significant decrease in PGC-1α in the ZA+LPS group compared to the other groups.
Res, as an activator of SIRT1, exhibits various beneficial effects on wound healing.Citation62 Studies have demonstrated that Res treatment attenuates high glucose-induced mitochondrial oxidative stress, an effect that is dependent on the expression of SIRT1 and the antioxidant transcription factor Nrf2.Citation63 Furthermore, it enhances autophagy mediated by the cAMP signaling pathway to alleviate mitochondrial dysfunction and oxidative stress.Citation64 To further confirm the relationship between SIRT1 and mitochondrial dysfunction, we then used the SIRT1 agonist Res.Citation65 The addition of Res significantly increased the expression level of ZA+LPS-induced SIRT1, resulting in an augmented mitochondrial count, improved mitochondrial function, and a comparable elevation in PGC-1α expression level when compared to other experimental groups. The above findings confirmed that under the oncologic dose of ZA, inflammation affected mitochondrial dysfunction through the SIRT1-PGC-1α pathway, thereby increasing the level of oxidative stress in HOK cells.
Subsequently, we further explored the effects of SIRT1 expression regulated by the oncologic dose of ZA and inflammation on HOK proliferation and migration. Studies have shown that SIRT1 could effectively improve the wound-healing process by affecting the cell viability of epithelial keratinocytes.Citation66 Additionally, in vivo experiments have shown marked improvements in wound healing following the application of Res, attributed to its antioxidant activity.Citation67 In vitro research has demonstrated that Res exerts a protective effect on fibroblasts by reducing oxidative stress levels. Its antioxidant properties stabilize cell proliferation, enhance migration quality, and preserve ultrastructural integrity.Citation68,Citation69 This finding is consistent with our observations in this study. Because, when the SIRT1 agonist Res used in combination with the inflammatory stimulus and oncologic dose of ZA, our results revealed significant enhancements in the proliferation and migration of HOK cells (). Thus, the inhibitory effect of the inflammation-exacerbated oncologic dose ZA on HOK cell proliferation and migration may be related to the SIRT1 signaling pathway mechanism.
We used in vivo animal models to further confirm the unique mechanism by which inflammation exacerbates the inhibitory effect of the oncologic dose of ZA on the mucosal healing process after teeth extraction. Consistent with the findings in vitro, our data demonstrated that inflammation exacerbated the mucosal healing disorder and MRONJ mediated by ZA of the oncologic dose. Simultaneously, SIRT1 expression levels in mucosal tissues decreased, as did the expression of mitochondrial genesis-related factor PGC-1α and the antioxidant enzyme MnSOD in mitochondria. Therefore, inflammation may mediate mitochondrial dysfunction and oxidative stress in a SIRT1-dependent manner, exacerbating mucosal nonunion in teeth extraction wounds induced by ZA of oncologic dose, thereby increasing the risk of MRONJ. Therefore, it is a possible therapeutic target of MRONJ to alleviate mitochondrial dysfunction, oxidative stress, and mucosal healing disorders by regulating SIRT1 signal transduction. As previous studies found that menaquinone-4 prevents MRONJ by inhibiting osteoblast apoptosis induced by cell stress mediated by the SIRT1 signal.Citation30
In some previous studies, reduced bone remodeling and infection or inflammation were considered to be the leading causes of MRONJ pathogenesis.Citation70 In healthy conditions, the mucosal immune system protects the oral cavity against various microorganisms and maintains immune homeostasis.Citation71 Anti-resorption drugs had direct toxicity to mucosal soft tissues, and the application of anti-resorption drugs, bacterial infection, and mucosal barrier damage jointly increased the risk of MRONJ.Citation36 Periodontal disease was the most prevalent chronic oral inflammation, which could seriously compromise the integrity of the oral mucosal barrier.Citation72,Citation73 When the periodontal disease was extraordinarily serious and teeth extraction was required, the local inflammatory response would lead to mucosal non-healing after teeth extraction in tumor patients treated with ZA, and the possibility of MRONJ would increase.Citation16 Research has indicated a dose-response association between the clinically relevant doses of ZA and the incidence of MRONJ.Citation74 However, whether inflammation as a risk factor promotes higher doses of ZA-induced MRONJ has not been extensively documented. Our findings suggest that inflammation does not exacerbate the inhibition of HOK cell proliferation and migration by the osteoporosis therapeutic dose of ZA. Conversely, inflammation exacerbated the inhibition of HOK cell proliferation and migration by the oncologic dose of ZA, increasing the risk of mucosal nonunion. Furthermore, we were able to verify the specific mechanism by which inflammation, as a risk factor, exacerbates mucosal nonunion caused by oxidative stress and mitochondrial dysfunction induced by the oncologic dose of ZA by regulating SIRT1 pathways. The addition of the SIRT1 agonist Res resulted in a reduction of intracellular mitochondrial dysfunction and oxidative stress, an increase in SIRT1 and PGC-1α expression, and an improvement in the migration and proliferation capacity of HOK cells.
Our study revealed that when administered at osteoporosis therapeutic dose, the risk of inflammation is relatively minor; however, at oncologic dose, inflammation becomes a critical factor in exacerbating mucosal non-healing in MRONJ. This observation suggests that the relatively low risk of inflammation in the presence of therapeutic dose for osteoporosis may not be a major concern. Conversely, in the setting of oncologic dose of ZA, the elevated dose of ZA renders inflammation a highly significant risk factor. Thus, when administering oncologic dose of ZA, it is imperative to carefully consider the impact of inflammation and implement corresponding preventative measures to mitigate the occurrence of MRONJ. For osteoporosis patients with periodontitis, attention should be paid to the damaging effect of ZA itself on the mucous membrane. In contrast, for tumor patients, regular dental examinations and preventive treatments every three months may be more effective in managing the risks associated with ZA therapy. The intricate interplay between oxidative stress, inflammation, and impaired wound healing in MRONJ underscores the complexity of this condition. Based on inflammation as a risk factor for MRONJ, controlling oxidative stress levels or providing gene therapy to up-regulate SIRT1 pathways might be therapeutic targets for reducing or blocking MRONJ in patients treated with ZA for tumor-complicating inflammation.
In conclusion, we found that inflammation plays an important role in mucosal nonunion when oncologic dose of ZA is used rather than osteoporosis therapeutic dose of ZA. Inflammation becomes a high-risk factor for the occurrence of MRONJ through increasing mitochondrial dysfunction and oxidative stress via the SIRT1-dependent way. This study clarifies the conditions under which inflammation is established as a high-risk factor for MRONJ and further investigates the specific mechanisms by which inflammation exacerbates mucosal nonunion of MRONJ. Our study also highlights the variability and complexity of inflammatory effects at different therapeutic doses. This suggests that a combination of factors, including the dose of ZA, the degree of inflammation, and other potential risk factors, will need to be considered in future studies of the relationship between ZA treatment and MRONJ. Through an in-depth study of the interactions and mechanisms between these factors, we can gain a more comprehensive understanding of the pathogenesis of MRONJ and provide a scientific basis for developing more effective preventive and therapeutic measures.
Ethical Approval
Animal care and experimental procedures were performed by the Guidelines for Animal Experimentation of Shandong University, with approval from the Affiliated Hospital of Shandong University School of Stomatology’s Institutional Animal Care and Use Committee (IACUC).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no conflicts of interest in this work.
Additional information
Funding
References
- Nicolatou-Galitis O, Kouri M, Papadopoulou E, et al. Osteonecrosis of the jaw related to non-antiresorptive medications: a systematic review. Support Care Canc. 2019;27(2):383–394. doi:10.1007/s00520-018-4501-x
- Ruggiero SL, Dodson TB, Aghaloo T, Carlson ER, Ward BB, Kademani D. American association of oral and maxillofacial surgeons’ position paper on medication-related osteonecrosis of the jaws-2022 update. J Oral Maxillof Surg. 2022;80(5):920–943. doi:10.1016/j.joms.2022.02.008
- Yamashita J, McCauley LK. Antiresorptives and osteonecrosis of the jaw. J Evid Based Dent Pract. 2012;12(3 Suppl):233–247. doi:10.1016/S1532-3382(12)70046-5
- Reid IR, Bolland MJ, Grey AB. Is bisphosphonate-associated osteonecrosis of the jaw caused by soft tissue toxicity? Bone. 2007;41(3):318–320. doi:10.1016/j.bone.2007.04.196
- Kyrgidis A, Vahtsevanos K, Koloutsos G, et al. Bisphosphonate-related osteonecrosis of the jaws: a case-control study of risk factors in breast cancer patients. J Clin Oncol. 2008;26(28):4634–4638. doi:10.1200/JCO.2008.16.2768
- Hoff AO, Toth BB, Altundag K, et al. Frequency and risk factors associated with osteonecrosis of the jaw in cancer patients treated with intravenous bisphosphonates. J Bone Mineral Res. 2008;23(6):826–836. doi:10.1359/jbmr.080205
- Zandi M, Dehghan A, Janbaz P, Malekzadeh H, Amini P. The starting point for bisphosphonate-related osteonecrosis of the jaw: alveolar bone or oral mucosa? A randomized, controlled experimental study. J Craniomaxillofac Surg. 2017;45(1):157–161. doi:10.1016/j.jcms.2016.10.015
- Voss PJ, Stoddart M, Ziebart T, et al. Zoledronate induces osteonecrosis of the jaw in sheep. J Craniomaxillofac Surg. 2015;43(7):1133–1138. doi:10.1016/j.jcms.2015.04.020
- Bullock G, Miller C, McKechnie A, Hearnden V. Synthetic hydroxyapatite inhibits bisphosphonate toxicity to the oral mucosa in vitro. Materials. 2020;13(9):2086. doi:10.3390/ma13092086
- Hoff AO, Toth B, Hu M, Hortobagyi GN, Gagel RF. Epidemiology and risk factors for osteonecrosis of the jaw in cancer patients. Ann N Y Acad Sci. 2011;1218:47–54. doi:10.1111/j.1749-6632.2010.05771.x
- Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. New Engl J Med. 2007;356(18):1809–1822. doi:10.1056/NEJMoa067312
- Skerjanec A, Berenson J, Hsu C, et al. The pharmacokinetics and pharmacodynamics of zoledronic acid in cancer patients with varying degrees of renal function. J Clin Pharmacol. 2003;43(2):154–162. doi:10.1177/0091270002239824
- Shiraki M, Tanaka S, Suzuki H, Ueda S, Nakamura T. Safety, pharmacokinetics, and changes in bone metabolism associated with zoledronic acid treatment in Japanese patients with primary osteoporosis. J Bone Mineral Metabol. 2017;35(6):675–684. doi:10.1007/s00774-016-0806-3
- Weiss HM, Pfaar U, Schweitzer A, Wiegand H, Skerjanec A, Schran H. Biodistribution and plasma protein binding of zoledronic acid. Drug Metabol Disposit. 2008;36(10):2043–2049. doi:10.1124/dmd.108.021071
- Troeltzsch M, Zeiter S, Arens D, et al. Chronic periodontal infection and not iatrogenic interference is the trigger of medication-related osteonecrosis of the jaw: insights from a large animal study (PerioBRONJ Pig Model). Medicina. 2023;59(5). doi:10.3390/medicina59051000
- Soundia A, Hadaya D, Esfandi N, et al. Zoledronate impairs socket healing after extraction of teeth with experimental periodontitis. J Dent Res. 2018;97(3):312–320. doi:10.1177/0022034517732770
- Elsubeihi ES, Heersche JN. Quantitative assessment of post-extraction healing and alveolar ridge remodelling of the mandible in female rats. Arch Oral Biol. 2004;49(5):401–412. doi:10.1016/j.archoralbio.2003.12.003
- Calenic B, Greabu M, Caruntu C, Tanase C, Battino M. Oral keratinocyte stem/progenitor cells: specific markers, molecular signaling pathways and potential uses. Periodontol. 2015;69(1):68–82. doi:10.1111/prd.12097
- Ko KI, Sculean A, Graves DT. Diabetic wound healing in soft and hard oral tissues. Transl Res. 2021;236:72–86. doi:10.1016/j.trsl.2021.05.001
- Amler MH. The time sequence of tissue regeneration in human extraction wounds. Oral Surg Oral Med Oral Pathol. 1969;27(3):309–318. doi:10.1016/0030-4220(69)90357-0
- Guo S, Dipietro LA. Factors affecting wound healing. J Dent Res. 2010;89(3):219–229. doi:10.1177/0022034509359125
- Koçer G, Nazıroğlu M, Çelik Ö, et al. Basic fibroblast growth factor attenuates bisphosphonate-induced oxidative injury but decreases zinc and copper levels in oral epithelium of rat. Biol Trace Elem Res. 2013;153(1–3):251–256. doi:10.1007/s12011-013-9659-y
- Awodele O, Olayemi SO, Nwite JA, Adeyemo TA. Investigation of the levels of oxidative stress parameters in HIV and HIV-TB co-infected patients. J Infect Dev Ctries. 2012;6(1):79–85. doi:10.3855/jidc.1906
- Lebreton F, van Schaik W, Sanguinetti M, et al. AsrR is an oxidative stress sensing regulator modulating Enterococcus faecium opportunistic traits, antimicrobial resistance, and pathogenicity. PLoS Pathog. 2012;8(8):e1002834. doi:10.1371/journal.ppat.1002834
- Bagan J, Sáez GT, Tormos MC, et al. Oxidative stress in bisphosphonate-related osteonecrosis of the jaws. J Oral Pathol Med. 2014;43(5):371–377. doi:10.1111/jop.12151
- Tamaoka J, Takaoka K, Hattori H, et al. Osteonecrosis of the jaws caused by bisphosphonate treatment and oxidative stress in mice. Exp Ther Med. 2019;17(2):1440–1448. doi:10.3892/etm.2018.7076
- Taniguchi N, Osaki M, Onuma K, et al. Bisphosphonate-induced reactive oxygen species inhibit proliferation and migration of oral fibroblasts: a pathogenesis of bisphosphonate-related osteonecrosis of the jaw. J Periodontol. 2020;91(7):947–955. doi:10.1002/JPER.19-0385
- Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015;6:472–485. doi:10.1016/j.redox.2015.09.005
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–950. doi:10.1152/physrev.00026.2013
- Cui Y, Zhang W, Yang P, Zhu S, Luo S, Li M. Menaquinone-4 prevents medication-related osteonecrosis of the jaw through the SIRT1 signaling-mediated inhibition of cellular metabolic stresses-induced osteoblast apoptosis. Free Radic Biol Med. 2023;206:33–49. doi:10.1016/j.freeradbiomed.2023.06.022
- Davizon-Castillo P, McMahon B, Aguila S, et al. TNF-α-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood. 2019;134(9):727–740. doi:10.1182/blood.2019000200
- Waldman M, Cohen K, Yadin D, et al. Regulation of diabetic cardiomyopathy by caloric restriction is mediated by intracellular signaling pathways involving ‘SIRT1 and PGC-1α’. Cardiovasc Diabetol. 2018;17(1):111. doi:10.1186/s12933-018-0754-4
- Singh CK, Chhabra G, Ndiaye MA, Garcia-Peterson LM, Mack NJ, Ahmad N. The role of sirtuins in antioxidant and redox signaling. Antioxid Redox Signal. 2018;28(8):643–661. doi:10.1089/ars.2017.7290
- Vega RB, Horton JL, Kelly DP. Maintaining ancient organelles: mitochondrial biogenesis and maturation. Circ Res. 2015;116(11):1820–1834. doi:10.1161/CIRCRESAHA.116.305420
- Iwabu M, Yamauchi T, Okada-Iwabu M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature. 2010;464(7293):1313–1319. doi:10.1038/nature08991
- Xu Q, Zhan P, Li X, et al. Bisphosphonate-enoxacin inhibit osteoclast formation and function by abrogating RANKL-induced JNK signalling pathways during osteoporosis treatment. J Cell & Mol Med. 2021;25(21):10126–10139. doi:10.1111/jcmm.16949
- Jiang Y, Yang P, Li C, et al. Periostin regulates LPS-induced apoptosis via Nrf2/HO-1 pathway in periodontal ligament fibroblasts. Oral Dis. 2023;29(5):2188–2204. doi:10.1111/odi.14189
- Qu B, Gong K, Yang H, et al. SIRT1 suppresses high glucose and palmitate-induced osteoclast differentiation via deacetylating p66Shc. Molec Cellul Endocrinol. 2018;474:97–104. doi:10.1016/j.mce.2018.02.015
- Aguirre JI, Castillo EJ, Kimmel DB. Preclinical models of medication-related osteonecrosis of the jaw (MRONJ). Bone. 2021;153:116184. doi:10.1016/j.bone.2021.116184
- Zhao N, Li QX, Wang YF, et al. Anti-angiogenic drug aggravates the degree of anti-resorptive drug-based medication-related osteonecrosis of the jaw by impairing the proliferation and migration function of gingival fibroblasts. BMC Oral Health. 2023;23(1):330. doi:10.1186/s12903-023-03034-7
- Nicolatou-Galitis O, Schiødt M, Mendes RA, et al. Medication-related osteonecrosis of the jaw: definition and best practice for prevention, diagnosis, and treatment. Oral Surg Oral Med Oral Pathol Oral Radiol. 2019;127(2):117–135. doi:10.1016/j.oooo.2018.09.008
- Chang J, Hakam AE, McCauley LK. Current understanding of the pathophysiology of osteonecrosis of the Jaw. Curr Osteoporos Rep. 2018;16(5):584–595. doi:10.1007/s11914-018-0474-4
- Hujiahemaiti M, Sun X, Zhou J, et al. Effects of quercetin on human oral keratinocytes during re-epithelialization: an in vitro study. Arch Oral Biol. 2018;95:187–194. doi:10.1016/j.archoralbio.2018.08.004
- Pabst AM, Ziebart T, Koch FP, Taylor KY, Al-Nawas B, Walter C. The influence of bisphosphonates on viability, migration, and apoptosis of human oral keratinocytes--in vitro study. Clin Oral Investigat. 2012;16(1):87–93. doi:10.1007/s00784-010-0507-6
- Limones A, Sáez-Alcaide LM, Díaz-Parreño SA, Helm A, Bornstein MM, Molinero-Mourelle P. Medication-related osteonecrosis of the jaws (MRONJ) in cancer patients treated with denosumab VS. zoledronic acid: a systematic review and meta-analysis. Med Oral Patol Oral Cir Bucal. 2020;25(3):e326–e336. doi:10.4317/medoral.23324
- Park KM, Cheong J, Pang NS, Kim KD, Lee JS, Park W. Medication-related osteonecrosis of the jaw using periodontitis-induced rat before tooth extraction. BMC Oral Health. 2023;23(1):561. doi:10.1186/s12903-023-03200-x
- Tamari T, Elimelech R, Cohen G, et al. Endothelial Progenitor Cells inhibit jaw osteonecrosis in a rat model: a major adverse effect of bisphosphonate therapy. Sci Rep. 2019;9(1):18896. doi:10.1038/s41598-019-55383-5
- Kozutsumi R, Kuroshima S, Kaneko H, Sasaki M, Ishisaki A, Sawase T. Zoledronic acid deteriorates soft and hard tissue healing of murine tooth extraction sockets in a dose-dependent manner. Calcif Tissue Internat. 2022;110(1):104–116. doi:10.1007/s00223-021-00890-9
- Vahtsevanos K, Kyrgidis A, Verrou E, et al. Longitudinal cohort study of risk factors in cancer patients of bisphosphonate-related osteonecrosis of the jaw. J Clin Oncol. 2009;27(32):5356–5362. doi:10.1200/JCO.2009.21.9584
- Bilezikian JP. Osteonecrosis of the jaw--do bisphosphonates pose a risk? New Engl J Med. 2006;355(22):2278–2281. doi:10.1056/NEJMp068157
- Thumbigere-Math V, Michalowicz BS, Hodges JS, et al. Periodontal disease as a risk factor for bisphosphonate-related osteonecrosis of the jaw. J Periodontol. 2014;85(2):226–233. doi:10.1902/jop.2013.130017
- Soutome S, Otsuru M, Hayashida S, et al. Relationship between tooth extraction and development of medication-related osteonecrosis of the jaw in cancer patients. Sci Rep. 2021;11(1):17226. doi:10.1038/s41598-021-96480-8
- Kara M, Boran T, Öztaş E, Jannuzzi AT, Özden S, Özhan G. Zoledronic acid-induced oxidative damage and endoplasmic reticulum stress-mediated apoptosis in human embryonic kidney (HEK-293) cells. J Biochem Mol Toxicol. 2022;36(8):e23083. doi:10.1002/jbt.23083
- Bryan N, Ahswin H, Smart N, Bayon Y, Wohlert S, Hunt JA. Reactive oxygen species (ROS)--A family of fate deciding molecules pivotal in constructive inflammation and wound healing. Eur Cell Mater. 2012;24:249–265. doi:10.22203/eCM.v024a18
- Mohammed AI, Sangha S, Nguyen H, et al. Assessment of Oxidative Stress-Induced Oral Epithelial Toxicity. Biomolecules. 2023;13(8):1239. doi:10.3390/biom13081239
- Pierfelice TV, Lazarevic M, Mitic D, et al. Red light and 5% aminolaevulinic Acid (5%) inhibit proliferation and migration of dysplastic oral keratinocytes via ROS production: an in vitro study. Gels. 2023;9(8):604. doi:10.3390/gels9080604
- Rizwan H, Pal S, Sabnam S, Pal A. High glucose augments ROS generation regulates mitochondrial dysfunction and apoptosis via stress signalling cascades in keratinocytes. Life Sci. 2020;241:117148. doi:10.1016/j.lfs.2019.117148
- Raghu G, Berk M, Campochiaro PA, et al. The Multifaceted Therapeutic Role of N-Acetylcysteine (NAC) in Disorders Characterized by Oxidative Stress. Curr Neuropharmacol. 2021;19(8):1202–1224. doi:10.2174/1570159X19666201230144109
- Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. 2019;51(12):1–13. doi:10.1038/s12276-019-0355-7
- Yang Y, Liu Y, Wang Y, et al. Regulation of SIRT1 and Its Roles in Inflammation. Front Immunol. 2022;13:831168. doi:10.3389/fimmu.2022.831168
- Zhao Y, Zhang J, Zheng Y, et al. NAD(+) improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1α pathway. J Neuroinflammation. 2021;18(1):207. doi:10.1186/s12974-021-02250-8
- Christovam AC, Theodoro V, Mendonça FAS, Esquisatto MAM, Dos Santos GMT, Do Amaral MEC. Activators of SIRT1 in wound repair: an animal model study. Archives of Dermatological Res. 2019;311(3):193–201. doi:10.1007/s00403-019-01901-4
- Ungvari Z, Bagi Z, Feher A, et al. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Phys Heart Circulat Physiol. 2010;299(1):H18–24. doi:10.1152/ajpheart.00260.2010
- Chen ML, Yi L, Jin X, et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy. 2013;9(12):2033–2045. doi:10.4161/auto.26336
- Chen Y, Zhang H, Ji S, et al. Resveratrol and its derivative pterostilbene attenuate oxidative stress-induced intestinal injury by improving mitochondrial redox homeostasis and function via SIRT1 signaling. Free Radic Biol Med. 2021;177:1–14. doi:10.1016/j.freeradbiomed.2021.10.011
- Lee JH, Kim JS, Park SY, Lee YJ. Resveratrol induces human keratinocyte damage via the activation of class III histone deacetylase, Sirt1. Oncol Rep. 2016;35(1):524–529. doi:10.3892/or.2015.4332
- Gokce EH, Tuncay Tanrıverdi S, Eroglu I, et al. Wound healing effects of collagen-laminin dermal matrix impregnated with resveratrol loaded hyaluronic acid-DPPC microparticles in diabetic rats. Europ J Pharmaceut Biopharmac. 2017;119:17–27. doi:10.1016/j.ejpb.2017.04.027
- Eroğlu İ, Gökçe EH, Tsapis N, et al. Evaluation of characteristics and in vitro antioxidant properties of RSV loaded hyaluronic acid-DPPC microparticles as a wound healing system. Colloids Surf B. 2015;126:50–57. doi:10.1016/j.colsurfb.2014.12.006
- Kaleci B, Koyuturk M. Efficacy of resveratrol in the wound healing process by reducing oxidative stress and promoting fibroblast cell proliferation and migration. Dermatologic Therapy. 2020;33(6):e14357. doi:10.1111/dth.14357
- Goodman SB, Maruyama M. Inflammation, bone healing and osteonecrosis: from bedside to bench. J Inflamm Res. 2020;13:913–923. doi:10.2147/JIR.S281941
- Wu RQ, Zhang DF, Tu E, Chen QM, Chen W. The mucosal immune system in the oral cavity-An orchestra of T cell diversity. Int J Oral Sci. 2014;6(3):125–132. doi:10.1038/ijos.2014.48
- Hajishengallis G, Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. 2021;21(7):426–440. doi:10.1038/s41577-020-00488-6
- Meyle J, Chapple I. Molecular aspects of the pathogenesis of periodontitis. Periodontology. 2015;69(1):7–17. doi:10.1111/prd.12104
- Gaudet C, Odet S, Meyer C, et al. Reporting criteria for clinical trials on medication-related osteonecrosis of the Jaw (MRONJ): a review and recommendations. Cells. 2022;11(24):4097. doi:10.3390/cells11244097