
Abstract
Hepatic fibrosis (HF) is a pathological process of structural and functional impairment of the liver and is a key component in the progression of chronic liver disease. There are no specific anti-hepatic fibrosis (anti-HF) drugs, and HF can only be improved or prevented by alleviating the cause. Autophagy of hepatic stellate cells (HSCs) is closely related to the development of HF. In recent years, traditional Chinese medicine (TCM) has achieved good therapeutic effects in the prevention and treatment of HF. Several active ingredients from TCM (AITCM) can regulate autophagy in HSCs to exert anti-HF effects through different pathways, but relevant reviews are lacking. This paper reviewed the research progress of AITCM regulating HSCs autophagy against HF, and also discussed the relationship between HSCs autophagy and HF, pointing out the problems and limitations of the current study, in order to provide references for the development of anti-HF drugs targeting HSCs autophagy in TCM. By reviewing the literature in PubMed, Web of Science, Embase, CNKI and other databases, we found that the relationship between autophagy of HSCs and HF is currently controversial. HSCs autophagy may promote HF by consuming lipid droplets (LDs) to provide energy for their activation. However, in contrast, inducing autophagy in HSCs can exert the anti-HF effect by stimulating their apoptosis or senescence, reducing type I collagen accumulation, inhibiting the extracellular vesicles release, degrading pro-fibrotic factors and other mechanisms. Some AITCM inhibit HSCs autophagy to resist HF, with the most promising direction being to target LDs. While, others induce HSCs autophagy to resist HF, with the most promising direction being to target HSCs apoptosis. Future research needs to focus on cell targeting research, autophagy targeting research and in vivo verification research, and to explore the reasons for the contradictory effects of HSCs autophagy on HF.
Introduction
Hepatic fibrosis (HF) is a pathological process of hepatic structural dysfunction, the essence of which is excessive deposition of diffuse extracellular matrix (ECM) in the liver.Citation1 The progression of HF can lead to cirrhosis, portal hypertension and even liver cancer, and ultimately to liver failure, which carries a risk of significant morbidity and mortality. The prevalence of advanced fibrosis and cirrhosis was 2.85% and 0.87%, respectively, in a study of 5,757,335 people in the general population and high-risk groups in China.Citation2 In a large population-based study conducted in the United States, the prevalence of cirrhosis in the general population was 0.27%, accounting for more than 600.000 patients.Citation3 Cirrhosis is also a major risk for hepatocellular carcinoma, which causes about 700,000 deaths worldwide each year.Citation4 Therefore, reversion of HF is of great significance for the prognosis of chronic liver disease (CLD). Despite recent advances in the understanding of the pathogenesis of HF, specific anti-hepatic fibrosis (anti-HF) drugs are lacking and the search for effective anti-HF agents remains a hot area.
Activation of hepatic stellate cells (HSCs) is a central event of HF development and a therapeutic target. Activated HSCs enhance ECM migration and deposition, and can release pro-inflammatory and pro-fibrotic factors, ultimately leading to HF.Citation5 Autophagy plays a role in maintaining cellular and tissue homeostasis.Citation6,Citation7 Numerous studies have found that dysregulation of autophagy can lead to the occurrence and development of CLD, and that HF as a repair response to CLD is regulated and affected by autophagy. In HF, the effects of autophagy are cell-type-specific due to the diversity of cells in the liver’s internal environment (including hepatocytes, HSCs, cholangiocytes, immune cells and other cells). Studies have shown that autophagy of HSCs exerts different effects on the development of HF by inhibiting or promoting HSCs activation.Citation8 Therefore, it is of great significance to study anti-HF drugs targeting HSCs autophagy.
In recent years, traditional Chinese medicine (TCM) has shown great potential in the prevention and treatment of HF, but the mechanism of action is usually unclear, which greatly limits its clinical promotion. Autophagy of HSCs is one of the key pathways or targets of various active ingredients from TCM (AITCM) to improve HF, but there is a lack of review. To clarify the efficacy of AITCM against HF and the specific molecular mechanism of targeting HSCs autophagy, we set the keywords as“‘liver fibrosis’ or ‘hepatic fibrosis’ and ‘traditional Chinese medicine’”, and searched databases such as PubMed, Web of Science, Embase and China National Knowledge Infrastructure (CNKI). In this paper, we will firstly introduce the pathogenesis of HF based on HSCs autophagy, and then review the research progress of AITCM regulating HSCs autophagosomes against HF.
Pathogenesis of HF
HF is caused by hepatotoxic injuries (such as viral hepatitis, fatty liver disease) and cholestatic injuries (such as biliary atresia) due to obstruction of bile flow.Citation1 The process of HF mainly involves long-term chronic parenchymal injury, sustained activation of inflammatory response and oxidative stress, massive deposition of ECM, and fibrous scar formation, which together destroy the normal structure and function of the liver. Following liver injury, the death of hepatocytes and the infiltration of immune cells activate the cascade of inflammatory and fibrotic signals. Hepatocytes apoptosis and release of damage-associated patterns (DAMPs) by hepatocytes not only activate HSCs directly but also induce recruitment and activation of lymphocytes and macrophages that promote the transdifferentiation of HSCs into myofibroblasts (MFs) by producing pro-inflammatory and pro-fibrotic cytokines.Citation9,Citation10
HF is sustained by the activation of MFs, which are a heterogeneous population of proliferating, migrating and pro-fibrotic cells, and are the major source of ECM in fibrotic livers.Citation11 HSCs are the major source of MFs in the injured liver and their activation is a central event in the biological processes of HF.Citation12 In the normal liver, HSCs reside in the subendothelial space of Disse, contain retinoids and vitamin A, and are quiescent. Under conditions of chronic liver injury, HSCs are activated into MF-like cells (HSCs/MFs or MFs derived from HSCs). Activated HSCs upregulate the expression of α-smooth muscle actin (α-SMA) and other MFs intracellular microfilaments, and synthetize large amounts of ECM components, particularly fibrillar collagens, to produce the fibrous scar.Citation1,Citation13 In addition, a complex network of cytokine-mediated signaling pathways regulates the activation of HSCs and fibrosis progression. Among the cytokines that play a key role are mainly transforming growth factor-beta (TGF-β), platelet-derived growth factor (PDGF) and inflammatory factors.Citation14
HSCs Autophagy and HF
The relationship between autophagy of HSCs and HF is currently controversial: the majority view is that HSCs autophagy can promote fibrosis, but it should not be overlooked that there are indeed studies demonstrating that inducing autophagy in HSCs can resist fibrosis.
HSCs Autophagy Promoting HF
Autophagy has been shown to regulate lipid droplets (LDs) metabolism, by degrading LDs to free fatty acids, which then undergo β-oxidation to generate ATP, providing the energy necessary for the activation of HSCs.Citation15 Hong et alCitation16 found that Atg2A was located in LDs and knockdown of Atg2A inhibited autophagic flux, which consequently suppressed LX-2 cells activation and LDs degradation. Many studies have shown that HSCs autophagy can promote HF. Autophagy was enhanced in HSCs isolated from mouse with HF induced by carbon tetrachloride (CCL4) and thioacetamide (TAA), and in HSCs isolated from HBV-infected human liver.Citation15 Spontaneous and induced activation of LX-2 cells enhanced autophagy, and inhibition of autophagy inhibited HF.Citation17,Citation18 Similarly, autophagy showed pro-fibrotic effect in JS1 and RAT HSC-T6 cell lines.Citation19,Citation20 Down-regulation of autophagy treated with autophagy inhibitors 3-methyladenine (3-MA) or Atg5 siRNA, Atg7 siRNA and MAP1LC3 siRNA resulted in attenuation of HSCs activation and fibroblastic properties, confirming the pro-fibrotic effect of HSCs autophagy from another perspective.Citation15,Citation21
HSCs Autophagy Resisting HF
On the other hand, however, some studies have shown that the induction of autophagy in HSCs can resist HF. Apoptosis and senescence are both ways to eliminate activated HSCs. Inducing autophagy in HSCs can promote their apoptosis or senescence, and then reduce activated HSCs, resulting in anti-HF effect. Resveratrol (RSV)Citation22 induced autophagy and apoptosis in JS1 cells in a dose-dependent manner, thereby inhibiting its activation. When autophagy was inhibited by chloroquine (CQ) or 3-MA, this effect could be alleviated, further demonstrating its anti-HF effect by inducing autophagy. Inducing autophagy in HSCs can also reduce type I collagen accumulation and inhibit the extracellular vesicles release to attenuate HF. Overexpression of phospholipase D1 (PLD1)Citation23 induced autophagy in HSCs to reduce type I collagen accumulation, whereas inhibition of its autophagy enhanced type I collagen expression. Further, Gao et al showed that autophagy in HSCs inhibited the release of fibrotic extracellular vesicles to attenuate HF.Citation24 In addition, induction of autophagy can also degrade pro-fibrotic factors to resist fibrosis. In primary mouse HSCs (mHSCs), impaired autophagy significantly promotes their activation, migration and proliferation, as well as the accumulation of pro-fibrotic factors. In contrast, restoration of mHSCs autophagy would alleviate fibrosis by degrading pro-fibrotic factors and inhibiting mHSCs activation.Citation25
Intervention of HSCs Autophagy by AITCM
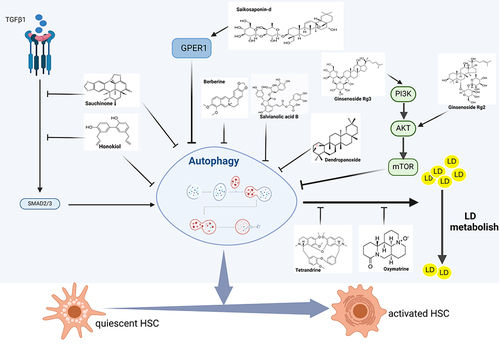
Basic experiments have confirmed that a variety of AITCM can inhibit or induce HSCs autophagy and exert anti-fibrotic effects through different pathways. In this paper, we found 18 kinds of AITCM for anti-HF ().
Table 1 List of Active Ingredients from TCM (AITCM) with Anti-Hepatic Fibrosis (Anti-HF) Effects
Inhibiting HSCs Autophagy to Attenuate HF
Several studies have shown that autophagy is signaling pathway-dependent, particularly the initiation phase of autophagy. Autophagy is induced a wide range of extra- and intracellular stresses including nutrient starvation, the absence of growth factors, and hypoxia.Citation46,Citation47 The mTOR (mammalian target of Rapamycin) kinase may be the central signaling molecule in determining the levels of autophagy in cells, being a signaling control point downstream of nutrient starvation, growth factor receptor signaling, hypoxia, ATP levels and insulin signaling, and it may mediate its effects on autophagy by inhibiting the ATG1/ULk1/2 complex at the earliest stages in phagophore formation.Citation48,Citation49 Various stress signals regulate the activity of mTORC1 (mTOR complex 1) mainly through classical signaling pathways such as PI3K/AKT, Ras-cAMP-PKA, AMPK, and Ras/Raf/MEK/ERK.Citation46,Citation50 TGF-β1 transduces signaling through its receptors on the surface of HSCs via SMAD signaling and the non-SMAD pathway.Citation51 SMAD signaling directly affects Beclin1/ATGL7 leading to autophagy. In the non-SMAD pathway, TGF-β1 regulates mTORC1 activity through PI3K / AKT, ERK or JNK pathways, thereby affecting autophagy.Citation52,Citation53
Inhibition of Autophagy to Attenuate HSCs Activation by Blocking the TGF-β1/Smad Signaling Pathway
TGF-β1 signaling is initiated by binding to the type II receptor. Subsequently, the receptor forms a dimer with its type I receptor and binds to Smad2 and Smad3; the complex is phosphorylated and released into the cytoplasm, where it binds to Smad4. They then translocate to the nucleus and regulate the transcription of α-SMA and pre-collagen I to promote the activation and proliferation of HSCs.Citation54 TGF-β has been reported to upregulate autophagy,Citation55 and the inhibition of the TGF-β1/Smad 3 signaling cascade response can suppress autophagy.Citation56,Citation57 Sauchinone (SAU)Citation26 was a lignan found in Saururus chinensis that attenuated CCl4-induced HF and HSCs activation in mouse. In vitro studies showed that it inhibited TGF-β1-induced Smad2/3 phosphorylation and transcript levels of matrix metalloproteinase-2(MMP-2) and autophagy in LX-2 cells, and suppressed HSCs activation. However, target validation was not performed in this study. Therefore, whether the antifibrotic effect is achieved by inhibiting autophagy and the specific molecular mechanism need to be further confirmed. Honokiol (HKL)Citation27 was isolated from Magnolia grandiflora, which can alleviate CCL4-induced HF in mouse. In LX-2 cells, it reduced HSCs activation and collagen I expression by blocking TGF-β1/Smad signaling and autophagy. However, this study only confirmed the effect of HKL on HSCs autophagy and activation in vitro, and lacked HSCs cell-specific research in mouse liver tissue.
Inhibition of Autophagy to Attenuate HSCs Activation Through Activating Akt/mTOR Pathway
As a key regulator in the initiation phase of autophagy, mTOR inhibits autophagy and is a negative regulator of autophagy.Citation58 The AKT/mTOR pathway is the classic pathway of negative regulation of autophagy.Citation59 Akt is a serine/threonine kinase that inhibits autophagy by blocking the activity of the mTOR inhibitor TSC2, resulting in the activation of mTOR.Citation60 A number of factors are upstream of this pathway, generating signaling cascades such as AMPK/AKT/mTOR and PI3K/AKT/mTOR. Ginsenoside Rg3 (G-Rg3),Citation28 a saponin with a high content in red ginseng, significantly attenuated TAA-induced HF, inhibited autophagy, and activated the PI3K/Akt/mTOR signaling cascade in the liver of mouse. In vitro (HSC-T6 cells), G-Rg3 also activated the PI3K/AKT/mTOR signaling pathway and dose-dependently inhibited lipopolysaccharide (LPS)-stimulated autophagy and activation of HSC-T6 cells, which was attenuated by the PI3K inhibitor LY294002. And rapamycin (Rapa), an mTOR inhibitor, induced HSC-T6 autophagy and activation, and G-Rg3 reversed this effect. In this study, target validation was carried out, indicating that G-Rg3 could inhibit autophagy in HSC-T6 cells to attenuate HF through activation of the PI3K/AKT/mTOR signaling pathway. Ginsenoside Rg2 (G-Rg2),Citation29 a protopanaxatriol saponin, can ameliorate HF induced by a choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) in mouse and activate AKT/mTOR signaling pathway. In vitro (HSC-T6 cells), G-Rg2 activated the AKT/mTOR signaling cascade reaction and reversed LPS-induced autophagy and activation. After intervention with Rapa or MK2206 (Akt inhibitor), the above effects of G-Rg2 were attenuated, confirming the targeting relationship of G-Rg2 with autophagy and Akt/mTOR signaling pathway, indicating that the antifibrosis effect of G-Rg2 was achieved through the activation of Akt/mTOR pathway to inhibit autophagy. G protein-coupled estrogen receptor1 (GPER1), a novel estrogen receptor (ER) involved in estrogen-mediated regulation of gene transcription and signaling pathways, can regulate autophagy via the Akt pathway. Studies have reported that G1 (a specific activator of GPER1) can inhibit autophagy by modulating the Akt-mediated pathway.Citation61,Citation62 In addition, estrogen could directly interfere with the development of HF by inhibiting ER-mediated HSCs activation.Citation63 Saikosaponin-d (SSd)Citation30 was the major active component of Radix bupleuri. In CCL4-induced mouse, SSd attenuated HF and increased the expression of ER-β and GPER1 in liver tissues. In primary mHSCs and LX-2 cells, SSd increased GPER1 expression and inhibited cellular autophagy and activation, and this effect was further verified by G1 versus G15 (a GPER1 antagonist), confirming that SSd attenuated HF by modulating the GPER1/autophagy pathway.
In addition to the above pathways, Salvianolic acid B (Sal B),Citation31 was the major active water-soluble component of the Chinese medicinal herb Salviae Miltiorrhizae Radix. Sal B inhibited TGF-β1-induced autophagy and activation of HSCs in vitro. Pretreatment with the autophagy inhibitors CQ and 3-MA or silencing ATG7 enhanced the above effects, whereas overexpression of the autophagy agonist Rapa or ATG5 exerted an opposite effect, confirming that Sal B inhibited HSCs activation by suppressing TGF-β1-induced HSCs autophagy. And in a protein chip, Sal B could down-regulate ERK, p38 and JNK pathway- related proteins, but target validation was not performed in this study, and it was not yet possible to explain the specific molecular mechanism of Sal B to inhibit autophagy and activation. Berberine (BBR)Citation32 was widely distributed in various medicinal plants such as Coptis chinensis, which can reduce liver injury in vivo (CCL4-treated mouse) and in vitro (PDGF-BB-treated LX-2 cells). In vitro, it can reduce the levels of autophagy-related proteins and inhibit the activation of LX-2 cells, but the specific mechanism remains to be further studied. Dendropanoxide (DPX),Citation33 the active ingredient of Dendropanax morbifera Léveille, was a sesquiterpene alkaloid. It can inhibit autophagy and activation of LX-2 cells in vitro and attenuate their fibroblastic properties, but the specific mechanism has not been studied in detail.
Inhibition of Autophagy to Reduce the Degradation of LDs to Attenuate HSCs Activation
In normal liver, HSCs are quiescent with a large amount of LDs present within them, and the loss of LDs is a feature of HSCs activation during liver injury.Citation64 Under conditions of liver injury, cytoplasmic LDs in HSCs are recognized by the autophagic process and thus degraded by lysosomes.Citation65 The free fatty acids released by LDs degradation are considered to be the energy source for HSCs to transdifferentiate into MFs and then generate fibrotic components,Citation66 suggesting that LDs degradation is closely related to HSCs activation. Tetrandrine (Tet) was a bisbenzylisoquinoline alkaloid isolated from the Chinese medicinal herb Stephania tetrandra, which induced lipids accumulation and attenuates HSCs activation by blocking autophagy in HSC-T6 cells.Citation34 This was the first description of a small molecule that can induce HSCs LDs accumulation by blocking autophagy. Oxymatrine (OM),Citation35 an alkaloid extracted and isolated from the leguminous plant ginseng, can also increase the number of intracellular LDs in LX-2 cells, inhibit autophagy, and attenuate its fibroblastic properties. However, no target validation was carried out in this study, and further research is needed. The above studies suggest that inhibiting HSCs autophagy may reduce the degradation of LDs and induce lipid accumulation, thus attenuating the activation of HSCs, which may be an effective way to prevent and control HF.
The above studies provide a reference for the development of anti-HF drugs in TCM by inhibiting HSCs autophagy (). However, the study of HSCs autophagy is mostly cultured in vitro, rather than exploring the AITCM in vivo or isolated HSCs, which has some limitations and cannot provide direct evidence.
Figure 1 Mechanisms involved in the treatment of Hepatic fibrosis (HF) with active ingredients from TCM (AITCM) by inhibiting autophagy in hepatic stellate cells (HSCs). Created with BioRender.com.
Induction of HSCs Autophagy to Attenuate HF
Contrary to the above views, it has recently been found that some AITCM exert anti-HF effects by inducing HSCs autophagy ().
Figure 2 Mechanisms involved in the treatment of Hepatic fibrosis (HF) with active ingredients from TCM (AITCM) by inducing autophagy in hepatic stellate cells (HSCs). Created with BioRender.com.
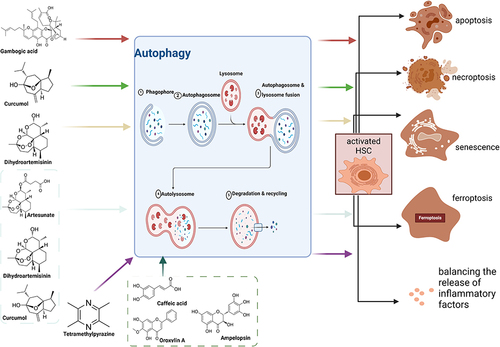
Induction of Autophagy to Promote Apoptosis
Apoptosis is one way to reduce the activation of HSCs. By inducing autophagy, it can promote the apoptosis of HSCs, thereby reducing activated HSCs and reversing HF.Citation67 Gambogic acid (GA),Citation36 the main active component of Garcinia cambogia, significantly ameliorated dimethylnitrosamine (DMN) and bile duct ligation (BDL)-induced HF in rats. In vitro, GA inhibited the proliferation and activation of HSC-T6 and LX-2 cells. The effects on autophagy and apoptosis can be divided into two phases: apoptosis was inhibited when GA induced autophagy at an early stage (12 h), but the accumulation of autophagy over the following 24 h eventually led to apoptosis. Necroptosis is a non-apoptotic form of cell death. Curcumol,Citation37 a sesquiterpenoid extracted from Guangxi Curcuma, attenuated HF in CCL4-induced mouse by targeting Sirt1 to regulate HSCs autophagy and necroptosis, and specific knockdown of Sirt1 in HSCs attenuated this effect, validating the targeting role of Sirt1. In vitro, it inhibited the activation of HSCs by promoting RIP1/RIP3-mediated necroptosis, which required the autophagy-related protein Atg5 and autophagosomes, and knockdown of Atg5 significantly reversed this effect. Curcumol regulated autophagy by activating Sirt1 and promoting Atg5 deacetylation; its induction of autophagy and necroptosis in HSCs was significantly reduced after Sirt1 downregulation. This study demonstrated that curcumol could activate Sirt1 to promote Atg5 deacetylation, thereby inducing HSCs autophagy and necroptosis, which attenuated HF.
Induction of Autophagy to Promote Cellular Senescence
Cellular senescence is also a way to remove activated HSCs and attenuate HF. Senescent activated HSCs reduced the secretion of ECM components, enhanced immune surveillance, and promoted the reversion of HF.Citation39,Citation68, Dihydroartemisinin (DHA)Citation39 induced HSCs senescence and attenuated fibrosis in vivo (CCL4-induced rats and mouse) and in vitro (primary rat HSCs). GATA6 was an important target in this process, and its accumulation promoted the induction of HSCs senescence by DHA, while knockdown of GATA6 attenuated this effect. DHA increased autophagosome generation and autophagic flux in activated HSCs. Autophagy depletion reduced GATA6 accumulation, while autophagy induction showed synergistic effects with DHA. This study performed target validation and demonstrated that DHA could induce senescence of activated HSCs through autophagy-dependent GATA6 accumulation, thereby attenuating HF.
Induction of Autophagy to Promote Ferroptosis
Ferroptosis is a novel mode of cell death regulation caused by iron overload, redox homeostasis disruption and increased lipid peroxidation,Citation69 which is closely associated with HF. Autophagy plays an important role in ferroptosis by regulating cellular iron homeostasis and cellular ROS generation.Citation70 ArtesunateCitation41 was a sesquiterpene lactone extracted and isolated from artemisinin. It induced ferroptosis of activated HSCs in vivo and in vitro, and blocking ferroptosis inhibited the antifibrotic effect induced by artesunate. Artesunate promoted ferritinophagy of activated HSCs in vitro, whereas inhibition of ferritinophagy with CQ suppresses its pro-ferroptosis and antifibrotic effects. This study showed that artesunate promoted HSCs ferroptosis and exerted the antifibrotic effect by inducing ferritinophagy. DHACitation40 alleviated HF in CCL4-induced mouse. It inhibited HSCs activation via inducing ferroptosis, which required the activation of autophagy and m6A methylation. - Inhibition of autophagy treated with 3-MA significantly eliminated DHA-induced ferroptosis in HSCs, further demonstrating that DHA can promoted ferroptosis by inducing autophagy to resist HF. CurcumolCitation38 promoted ferroptosis and ferric ion release in HSC-T6 cells, leading to iron overload, which in turn induced ferroptosis and elimination of activated HSCs. NCOA4 and FTH1 were the targets of curcumol-induced ferritin autophagy, which reduced the expression of FTH1 and induced the expression of NCOA4, which was reversed by the effect of the autophagy inhibitor 3-MA, validating the target. However, this study was not performed in vivo.
Induction of Autophagy to Regulate Inflammatory Factors
HSCs are highly sensitive to pro-inflammatory cytokines and can be activated by an inflammatory response.Citation71 And pro-inflammatory and pro-fibrotic cytokines released by activated HSCs can enhance the response of inflammatory and fibrotic tissues.Citation5 Tetramethylpyrazine (TMP)Citation42 was a natural alkaloid that exerted the anti-HF effect in vivo (CCL4-induced rats) and in vitro (PDGF-BB-stimulated HSCs-T6) by inhibiting the Akt-mTOR pathway to promote autophagy of HSCs-T6, thereby balancing the release of inflammatory cytokines and degradation of ECM. The therapeutic effect of TMP was enhanced by the autophagy inducer rapamycin, while the autophagy inhibitor 3-MA, the PI3K pathway inhibitor LY 294002 and the AKT pathway agonist SC79 had the opposite effect, validating the target.
In addition, although some studies have shown that induction of autophagy can resist HF, they have not elucidated the further effects after autophagy induction. Oroxylin ACitation43 was a natural monoflavonoid isolated from Scutellariae Radix that reduced liver injury and HSCs activation in vivo (CCL4-induced mouse) and in vitro (PDGF-BB-stimulated primary mHSCs). In both models, Oroxylin A promoted autophagy in activated HSCs and its antifibrotic effects were attenuated by the use of autophagy inhibitors in vitro. Similarly, Ampelopsin (Amp)Citation44 attenuated HSCs activation in CCL4-treated mouse tissues and cultured activated primary mHSCs. It induced HSCs autophagy and inhibited the SIRT1/TGF-β1/ Smad3 pathway in vitro, and inhibition of autophagy treated with 3-MA reduced its antifibrosis effect. Caffeic acid phenethyl ester (CAPE)Citation45 inhibited HF and HSCs activation by inhibiting TGF-β1/Smad3 signal transduction in vivo (CCL4-induced rats) and in vitro (HSC-T6 cells). In vitro studies also showed that CAPE induced autophagy and inhibited AKT/mTOR signaling pathway in HSC-T6 cells..
Conclusion and Perspective
The relationship between autophagy of HSCs and HF is currently controversial. The majority view is that HSCs autophagy can promote fibrosis. HSCs autophagy may promote HF by consuming LDs to provide energy for their activation. However, in contrast, some studies have shown that inducing autophagy in HSCs can resist HF by stimulating their apoptosis or senescence, reducing type I collagen accumulation, inhibiting the extracellular vesicles release, degrading pro-fibrotic factors and other mechanisms. We speculate that the reason for the contradictory effects is that the degree of autophagy is related to the activation state of HSCs. When autophagy is at a basic level, HSCs reside in a quiescent state,Citation65,Citation72 and when liver injury occurs, autophagy is enhanced to promote HSCs activation, thereby promoting HF.Citation15,Citation66 When autophagy exceeds to the standard threshold, it induces apoptosis or senescence to remove activated HSCs, and it reduces the synthesis of type I collagen and degrades pro-fibrotic factors, thereby inhibiting fibrosis.Citation23,Citation24,Citation39,Citation67,Citation68,Citation73 It is the uncertainty of the relationship between HSCs autophagy and HF that leads to a certain contradiction in the development of anti-HFs drugs targeting HSCs autophagy. In this paper, 18 kinds of AITCM were reviewed, some of which inhibit HSCs autophagy to resist HF, while others induce HSCs autophagy to resist HF.
The most promising direction for the development of AITCM that inhibit HSCs autophagy and thus antifibrosis is to target LDs. There is sufficient evidence that HSCs autophagy can degrade LDs and provideenergy for their activation, which is crucial to the progression of fibrosis. Moreover, the AITCM Tet and OM can reduce the degradation of LDs by inhibiting HSCs autophagy, thereby reducing HSCs activation. Therefore, this may be a potential approach for the development of antifibrotic drugs. However, drug development targeting this pathway should take into account the timeframe: intervention in pre-disease liver injury, as autophagy in HSCs may not respond to treatment in the later stages of the disease. While, the most promising direction for the development of AITCM that promote HSCs autophagy and thus anti-fibrosis is to target HSCs apoptosis. Apoptosis is one of the major pathways for inactivation of activated HSCs, and studies have shown that approximately 50% of activated HSCs undergo apoptosis during fibrosis regression.Citation74,Citation75 Autophagy beyond a certain threshold may lead directly to autophagic cell death or to the execution of apoptotic cell death via common regulators.Citation72,Citation76–78 In addition, it has been reported that GA and curcumol can stimulate apoptosis by inducing HSCs autophagy to deplete activated HSCs. The development of drugs targeting this pathway will again need to consider timing. However, there are still many shortcomings in the studies on the anti-hepatic fibrosis of AITCM targeting HSCs autophagy: 1) Most studies of HSCs autophagy have been conducted in vitro rather than exploring the effects of the AITCM in experimental animals. 2) The design of experimental protocols is relatively crude, and some studies are limited to the phenomenal level without targeted validation of the mechanism of action. 3) There are a large number of preclinical studies, but no high-level randomized controlled clinical trials.
In conclusion, there are great prospects for finding antifibrotic drugs targeting HSCs autophagy in TCM. Future research should focus on the following directions: 1. cell targeting research, antifibrotic drugs should precisely target HSCs; 2. autophagy targeting research, antifibrotic drugs should target autophagy of cells; 3. in vivo verification studies, more animal experiments or clinical studies are needed to provide a basis for clinical application. Besides, why autophagy of HSCs has contradictory effects on HF and under what conditions autophagy should be promoted or inhibited to resist HF should also be a major subject of future study.
Disclosure
The authors report no conflicts of interest in this work.
Data Sharing Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Additional information
Funding
References
- Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18(3):151–166. doi:10.1038/s41575-020-00372-7
- Man S, Deng Y, Ma Y, et al. Prevalence of liver steatosis and fibrosis in the general population and various high-risk populations: a nationwide study with 5.7 million adults in China. Gastroenterology. 2023;165(4):1025–1040. doi:10.1053/j.gastro.2023.05.053
- Scaglione S, Kliethermes S, Cao G, et al. The epidemiology of cirrhosis in the United States: a population-based study. J Clin Gastroenterol. 2015;49:690–696. doi:10.1097/MCG.0000000000000208
- McGlynn KA, Petrick JL, London WT. Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis. 2015;19(2):223–238. doi:10.1016/j.cld.2015.01.001
- Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. doi:10.1146/annurev-pathol-011110-130246
- Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42. doi:10.1016/j.cell.2018.09.048
- Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020;383(16):1564–1576. doi:10.15252/embj.2021108863
- Hung TM, Hsiao CC, Lin CW, et al. Complex cell type-specific roles of autophagy in liver fibrosis and cirrhosis. Pathogens. 2020;9(3):225. doi:10.3390/pathogens9030225
- Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17(5):306–321. doi:10.1038/nri.2017.11
- Barron L, Wynn TA. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am J Physiol Gastrointest Liver Physiol. 2011;300(5):G723–G728. doi:10.1152/ajpgi.00414.2010
- Parola M, Pinzani M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med. 2019;65:37–55. doi:10.1016/j.mam.2018.09.002
- Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi:10.1152/physrev.00013.2007
- Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411. doi:10.1038/nrgastro.2017.38
- Roehlen N, Crouchet E, Baumert TF. Liver fibrosis: mechanistic concepts and therapeutic perspectives. Cells. 2020;9(4):875. doi:10.3390/cells9040875
- Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142(4):938–946. doi:10.1053/j.gastro.2011.12.044
- Hong Y, Li S, Wang J, et al. In vitro inhibition of hepatic stellate cell activation by the autophagy-related lipid droplet protein ATG2A. Sci Rep. 2018;8:9232. doi:10.1038/s41598-018-27686-6
- Wang Z, Tao Y, Qiu T, et al. Taurine protected As₂O₃-induced activation of hepatic stellate cells through inhibiting PPARα-autophagy pathway. Chem Biol Interact. 2019;300:123–130. doi:10.1016/j.cbi.2019.01.019
- Deng J, Huang Q, Wang Y, et al. Hypoxia-inducible factor-1alpha regulates autophagy to activate hepatic stellate cells. Biochem Biophys Res Commun. 2014;454:328–334. doi:10.1016/j.bbrc.2014.10.076
- Huang TJ, Ren JJ, Zhang QQ, et al. IGFBPrP1 accelerates autophagy and activation of hepatic stellate cells via mutual regulation between H19 and PI3K/AKT/mTOR pathway. Biomed Pharmacother. 2019;116:109034. doi:10.1016/j.biopha.2019.109034
- Yang R, Hu Z, Zhang P, et al. Probucol ameliorates hepatic stellate cell activation and autophagy is associated with farnesoid X receptor. J Pharmacol Sci. 2019;139:120–128. doi:10.1016/j.jphs.2018.12.005
- Li J, Zeng C, Zheng B, et al. HMGB1-induced autophagy facilitates hepatic stellate cells activation: a new pathway in liver fibrosis. Clin Sci. 2018;132(15):1645–1667.
- Zhang J, Ping J, Jiang N, et al. Resveratrol inhibits hepatic stellate cell activation by regulating autophagy and apoptosis through the SIRT1 and JNK signaling pathways. J Food Biochem. 2022;46(12):e14463. doi:10.1111/jfbc.14463
- Seo H-Y, Jang B-K, Jung Y-A, et al. Phospholipase D1 decreases type I collagen levels in hepatic stellate cells via induction of autophagy. Biochem Biophys Res Commun. 2014;449:38–43. doi:10.1016/j.bbrc.2014.04.149
- Gao J, Wei B, de Assuncao TM, et al. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J Hepatol. 2020;73:1144–1154. doi:10.1016/j.jhep.2020.04.044
- Zhang XW, Zhou JC, Peng D, et al. Disrupting the TRIB3-SQSTM1 interaction reduces liver fibrosis by restoring autophagy and suppressing exosome-mediated HSC activation. Autophagy. 2020;16:782–796. doi:10.1080/15548627.2019.1635383
- Lee JH, Jang EJ, Seo HL, et al. Sauchinone attenuates liver fibrosis and hepatic stellate cell activation through TGF-β/Smad signaling pathway. Chem Biol Interact. 2014;224:58–67. doi:10.1016/j.cbi.2014.10.005
- Kataoka S, Umemura A, Okuda K, et al. Honokiol acts as a potent anti-fibrotic agent in the liver through inhibition of TGF-β1/Smad signaling and autophagy in hepatic stellate Cells. Int J Mol Sci. 2021;22(24):13354. doi:10.3390/ijms222413354
- Liu X, Mi X, Wang Z, et al. Ginsenoside Rg3 promotes regression from hepatic fibrosis through reducing inflammation-mediated autophagy signaling pathway. Cell Death Dis. 2020;11(6):454. doi:10.1038/s41419-020-2597-7
- He Z, Chen S, Pan T, et al. Ginsenoside Rg2 ameliorating CDAHFD-induced hepatic fibrosis by regulating AKT/mTOR-mediated autophagy. J Agric Food Chem. 2022;70:(6)1911–1922. doi:10.1021/acs.jafc.1c07578
- Chen Y, Que R, Zhang N, et al. Saikosaponin-d alleviates hepatic fibrosis through regulating GPER1/autophagy signaling. Mol Biol Rep. 2021;48(12):7853–7863. doi:10.1007/s11033-021-06807-x
- Jiang N, Zhang J, Ping J, et al. Salvianolic acid B inhibits autophagy and activation of hepatic stellate cells induced by TGF-β1 by downregulating the MAPK pathway. Front Pharmacol. 2022;13:938856. doi:10.3389/fphar.2022.938856
- Yuehao Tan, Can Li, Fengmei Deng, et al. Berberine relieves liver fibrosis in mice by inhibiting autophagy of hepatic stellate cells. J Chengdu Med Coll. 2023;18(01):33–38. doi:10.3969/j.issn.1674-2257.2023.01.007
- Park YJ, Kim DM, Choi HB, et al. Dendropanoxide, a triterpenoid from Dendropanax morbifera, ameliorates hepatic fibrosis by inhibiting activation of hepatic stellate cells through autophagy inhibition. Nutrient. 2021;14(1):98. doi:10.3390/nu14010098
- Miyamae Y, Nishito Y, Nakai N,et al. Tetrandrine induces lipid accumulation through blockade of autophagy in a hepatic stellate cell line. Biochem Biophys Res Commun. 2016;477(1):40–46. doi:10.1016/j.bbrc.2016.06.018
- Ma ZH, Zhang JY, Yang L, et al. Oxymatrine inhibits hepatic stellate cell (HSC) autophagy during HSC activation induced by arsenic. Chinese J Pathophysiol. 2019;35(09):1662–1667. doi:10.3969/j.issn.1000-4718.2019.09.020
- Yu Z, Jv Y, Cai L, et al. Gambogic acid attenuates liver fibrosis by inhibiting the PI3K/AKT and MAPK signaling pathways via inhibiting HSP90. Toxicol Appl Pharmacol. 2019;371:63–73. doi:10.1016/j.taap.2019.03.028
- Sun S, Li Z, Huan S, et al. Modification of lysine deacetylation regulates curcumol-induced necroptosis through autophagy in hepatic stellate cells. Phytother Res. 2022;36(6):2660–2676. doi:10.1002/ptr.7483
- Zheng Y, Zhao T, Wang J, et al. Curcumol alleviates liver fibrosis through inducing autophagy and ferroptosis in hepatic stellate cells. FASEB J. 2022;36(12):e22665. doi:10.1096/fj.202200933RR
- Zhang Z, Yao Z, Zhao S, et al. Interaction between autophagy and senescence is required for dihydroartemisinin to alleviate liver fibrosis. Cell Death Dis. 2017;8(6): e2886. doi:10.1038/cddis.2017.255
- Shen M, Guo M, Li Y, et al. m6A methylation is required for dihydroartemisinin to alleviate liver fibrosis by inducing ferroptosis in hepatic stellate cells. Free Radic Biol Med. 2022;182:246–259. doi:10.1016/j.freeradbiomed.2022.02.028
- Kong Z, Liu R, Cheng Y, et al. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed Pharmacother. 2019;109:2043–2053. doi:10.1016/j.biopha.2018.11.030
- Hu Z, Su H, Zeng Y, et al. Tetramethylpyrazine ameliorates hepatic fibrosis through autophagy-mediated inflammation. Biochem Cell Biol. 2020;98(3):327–337. doi:10.1139/bcb-2019-0059
- Chen W, Zhang Z, Yao Z, et al. Activation of autophagy is required for Oroxylin A to alleviate carbon tetrachloride-induced liver fibrosis and hepatic stellate cell activation. Int Immunopharmacol. 2018;56:148–155. doi:10.1016/j.intimp.2018.01.029
- Ma JQ, Sun YZ, Ming QL, et al. Ampelopsin attenuates carbon tetrachloride-induced mouse liver fibrosis and hepatic stellate cell activation associated with the SIRT1/TGF-β1/Smad3 and autophagy pathway. Int Immunopharmacol. 2019;77:105984. doi:10.1016/j.intimp.2019.105984
- Yang N, Dang S, Shi J, et al. Caffeic acid phenethyl ester attenuates liver fibrosis via inhibition of TGF-β1/Smad3 pathway and induction of autophagy. Biochem Biophys Res Commun. 2017;486(1):22–28. doi:10.1016/j.bbrc.2017.02.057
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi:10.1146/annurev-genet-102808-114910
- Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20(3):460–473. doi:10.1089/ars.2013.5371
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12. doi:10.1002/path.2697
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–1109. doi:10.1038/ncb1007-1102
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6(9):729–734. doi:10.1038/nrc1974
- Peng D, Fu M, Wang M, et al. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer. 2022;21(1):104. doi:10.1186/s12943-022-01569-x
- Siapoush S, Rezaei R, Alavifard H, et al. Therapeutic implications of targeting autophagy and TGF-β crosstalk for the treatment of liver fibrosis. Life Sci. 2023;329:121894. doi:10.1016/j.lfs.2023.121894
- Zhang J, Jiang N, Ping J, et al. TGF β1 induced autophagy activates hepatic stellate cells via the ERK and JNK signaling pathways. Int J Mol Med. 2021;47(1):256–266. doi:10.3892/ijmm.2020.4778
- Inagaki Y, Okazaki I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56(2):284–292. doi:10.1136/gut.2005.088690103
- Chen M, Liu J, Yang L, et al. AMP-activated protein kinase regulates lipid metabolism and the fibrotic phenotype of hepatic stellate cells through inhibition of autophagy. FEBS Open Bio. 2017;7(6):811–820. doi:10.1002/2211-5463.12221
- Feng J, Chen K, Xia Y, et al. Salidroside ameliorates autophagy and activation of hepatic stellate cells in mice via NF-κB and TGF-β1/Smad3 pathways. Drug Des Devel Ther. 2018;12:1837–1853. doi:10.2147/DDDT.S162950
- Tong H, Yin H, Hossain MA, et al. Starvation-induced autophagy promotes the invasion and migration of human bladder cancer cells via TGF-β1/ Smad3-mediated epithelial-mesenchymal transition activation. J Cell Biochem. 2019;120(4):5118–5127. doi:10.1002/jcb.27788
- Paquette M, El-Houjeiri L, Pause A, et al. mTOR pathways in cancer and autophagy. Cancers. 2018;10(1):18. doi:10.3390/cancers10010018
- Zhao F, Wang J, Lu He, et al. Neuroprotection by walnut-derived peptides through autophagy promotion via Akt/mTOR signaling pathway against oxidative stress in PC12 cells. J Agric Food Chem. 2020;68:11 3638–3648. doi:10.1021/acs.jafc.9b08252
- Vucicevic L, Misirkic M, Janjetovic K, et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2011;7(1):40–50. doi:10.4161/auto.7.1.13883
- Prossnitz ER, Barton M. Estrogen biology: new insights into GPER function and clinical opportunities. Mol Cell Endocrinol. 2014;389(1–2):71–83. doi:10.1016/j.mce.2014.02.002
- Pei H, Wang W, Zhao D, et al. G protein-coupled estrogen receptor 1 inhibits angiotensin II-induced cardiomyocyte hypertrophy via the regulation of PI3K-Akt-mTOR signalling and autophagy. Int J Biol Sci. 2019;15(1):81–92. doi:10.7150/ijbs.28304
- McCarty MF, Barroso-Aranda J, Contreras F. Genistein and phycocyanobilin may prevent hepatic fibrosis by suppressing proliferation and activation of hepatic stellate cells. Med Hypotheses. 2009;72(3):330–332. doi:10.1016/j.mehy.2008.07.045
- Shao C, Xu H, Sun X, et al. New perspectives on Chinese medicine in treating hepatic fibrosis: lipid droplets in hepatic stellate cells. Am J Chin Med. 2023;51(6): 1413–1429. doi:10.1142/S0192415X23500647
- Thoen LFR, Guimarães ELM, Dollé L, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353–1360. doi:10.1016/j.jhep.2011.07.010
- Hernández-Gea V, Friedman SL. Autophagy fuels tissue fibrogenesis. Autophagy. 2012;8(5):849–850. doi:10.4161/auto.19947
- Kamm DR, McCommis KS. Hepatic stellate cells in physiology and pathology. J Physiol. 2022;600(8):1825–1837. doi:10.1113/JP281061
- Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–667. doi:10.1016/j.cell.2008.06.049
- Xiang X, Gao J, Su D, et al. The advancements in targets for ferroptosis in liver diseases. Front Med. 2023;10:1084479. doi:10.3389/fmed.2023.1084479
- Gao M, Monian P, Pan Q, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–1032. doi:10.1038/cr.2016.95
- Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61(3):1066–1079. doi:10.1002/hep.27332
- Wang K. Autophagy and apoptosis in liver injury. Cell Cycle. 2015;14(11):1631–1642. doi:10.1080/15384101.2015.1038685
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi:10.1016/j.cell.2007.12.018
- Iredale JP, Benyon RC, Pickering J, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102(3):538–549. doi:10.1172/JCI1018
- Kisseleva T, Cong M, Paik Y, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci USA. 2012;109(24):9448–9453. doi:10.1073/pnas.1201840109
- Galluzzi L, Vicencio JM, Kepp O, et al. To die or not to die: that is the autophagic question. Curr Mol Med. 2008;8(2):78–91. doi:10.2174/156652408783769616
- Reimers K, Choi CY, Bucan V, et al. The Bax Inhibitor-1 (BI-1) family in apoptosis and tumorigenesis. Curr Mol Med. 2008;8(2):148–156. doi:10.2174/156652408783769562
- Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017;121:27–42. doi:10.1016/j.addr.2017.05.007