Abstract
P-glycoprotein is one of the earliest known multidrug transporters and plays an important role in resistance to chemotherapeutic drugs. In this study, we detected levels of P-glycoprotein and its mRNA expression in a rat brain model of medically intractable epilepsy established by amygdala kindling and drug selection. We investigated whether inhibition of P-glycoprotein affects the concentration of antiepileptic drugs in cortical extracellular fluid. We found that levels of P-glycoprotein and its mRNA expression were upregulated in epileptic cerebral tissue compared with cerebral tissue from normal rats. The concentrations of two antiepileptic drugs, carbamazepine and phenytoin, were very low in the cortical extracellular fluid of rats with medically intractable epilepsy, and were restored after blockade of P-glycoprotein by verapamil. These results show that increased P-glycoprotein levels alter the ability of carbamazepine and phenytoin to penetrate the blood–brain barrier and reduce the concentrations of these agents in extracellular cortical fluid. High P-glycoprotein levels may be involved in resistance to antiepileptic drugs in medically intractable epilepsy.
Introduction
Although a number of new antiepileptic drugs have been launched over the past two decades, drug resistance remains a major problem. About 30% of patients are refractory to treatment with more than one antiepileptic drug. This so-called medically intractable epilepsy is often associated with a poor prognosis, ie, increased morbidity and mortality in patients.Citation1–Citation3 Therefore, it is important to investigate the mechanism of drug resistance in medically intractable epilepsy and develop new treatment strategies. Earlier research in the field of cancer has shown that the activity of P-glycoprotein and other ATP-binding cassette (ABC) transporters, such as multidrug resistance-associated proteins (MDRs), are directly related to drug resistance.Citation4,Citation5
MDR1-encoded P-glycoprotein is widely expressed in tissues with an excretory function, including the liver, kidneys, and cerebrum, as an energy-dependent efflux transporter, and is involved in barrier functioning, such as in the blood–brain barrier.Citation5 In normal tissues, P-glycoprotein is thought to participate in the protection of cells from toxins or xenobiotics. In the brain, P-glycoprotein is predominantly located on the membrane of capillary endothelial cells which form the blood–brain barrier. Overexpression of P-glycoprotein on endothelial cells of the blood–brain barrier would limit penetration of antiepileptic drugs into the brain and reduce drug concentrations in cerebral tissue, suggesting that P-glycoprotein may be involved in mechanisms of drug resistance in patients with medically intractable epilepsy.
There is accumulating evidence showing that P-glycoprotein is involved in resistance to antiepileptic drugs. Tishler et alCitation6 were the first to report that P-glycoprotein is overexpressed in epileptogenic brain tissue from patients with pharmacoresistant partial epilepsy. More recently, P-glycoprotein was reported to be overexpressed in endothelial cells of the blood–brain barrier in a kainate model of temporal lobe epilepsy in rats.Citation7 To confirm this phenomenon, expression of P-glycoprotein was studied by real-time reverse transcription polymerase chain reaction (RT-PCR) and Western blot technique in a rat model of medically intractable epilepsy created by amygdala kindling.Citation8 Previous research has confirmed that the concentration of phenytoin, a classic antiepileptic drug, in extracellular cortical fluid is significantly increased by inhibition of P-glycoprotein, indicating that phenytoin is a substrate for P-glycoprotein.Citation9 However, the opposite effect was observed for carbamazepine, another antiepileptic drug.Citation10,Citation11 To address this issue directly, we undertook a brain microdialysis experiment to study the impact of P-glycoprotein inhibition on penetration of antiepileptic drugs through the blood–brain barrier in rats with medically intractable epilepsy.
Materials and methods
Materials
Male Sprague Dawley rats (250–300 g) were obtained from the Shanghai Animal Center, Medical College of Fudan University, Shanghai, People’s Republic of China, and maintained in the animal facility at 20°C±2°C with a relative humidity of 60% and a 12-hour light and dark cycle for 5 days before the experiment. A total of 80 rats were used (four rats per cage). All rat experiments were carried out in accordance with the Guidelines for Animal Experiments of the Chinese Academy of Medical Sciences and with approval from the ethics committee for animal care at Jinshan Hospital.
Thirty-two rats were randomly assigned to either a control group (n=16) or to a group with medically intractable epilepsy (n=16) for detection of P-glycoprotein expression, and a further 48 rats were randomly assigned to a phenytoin group (n=24) or a carbamazepine group (n=24) for a microdialysis experiment. The carbamazepine and phenytoin groups were further divided into three subgroups: a control group (n=8) comprising normal rats that received intraperitoneal injection of antiepileptic drugs (carbamazepine 20 mg/kg or phenytoin 50 mg/kg, Sigma-Aldrich, St Louis, MO, USA); an epilepsy group (n=8) comprising kindled rats that received the same injections as the control group; and a verapamil group (n=8) comprising kindled rats that received 50 μL of verapamil (20 mmol/L, Sigma-Aldrich) at a rate of 2.5 μL per minute from the inflow tube into the cortex 30 minutes before intraperitoneal antiepileptic drug injection.
Establishment of a medically intractable epilepsy model
The amygdala kindling model was generated using a well established protocol.Citation12 Seizure severity was evaluated in accordance with the standards published by Racine.Citation13 Rats with three consecutive stage V seizures were considered to be successfully kindled and were injected intraperitoneally with phenytoin to screen for resistance. Successful creation of a medically intractable epilepsy model was deemed to have occurred if there was a reduction in the after discharge threshold. The after discharge threshold was determined using an ascending series procedure whereby electrical kindling stimulation (2 seconds, 60 Hz, biphasic square wave pulses) was started at 20 μA and increased in steps of 20 μA until an after discharge was triggered. The interval between stimulations was one minute. The after discharge threshold was defined as the minimum current intensity required to provoke a synchronous spike-and-wave pattern of at least 3 seconds in the electroencephalogram. Rats that did not respond to phenytoin were considered to have medically intractable epilepsyCitation14 and electrode-implanted animals that did not display electrical kindling stimulation were used as the control group.
RNA extraction and real-time RT-PCR
Total RNA was extracted from rat brain tissue using TRIzol reagent (Invitrogen, New York, NY, USA). Total RNA (1 μg) was reversely transcribed using a reverse transcription kit (MBI Fermentas, Burlington, OT, Canada), followed by polymerase chain reaction amplification. Primers for MDR1 and β-actin were synthesized by Shanghai Saibaisheng Company (Shanghai, People’s Republic of China). The primer sequences were: 5′-ACTCGGGAGCAGAAGTTTGA-3′ (forward) and 5′-GGAGCCACTGGACATTGAGT-3′ (reverse) for MDR1 (600 bp); and 5′-AACCCTAAGGC-CAACCGTGAAAAG-3′ (forward) and 5′-TCATGAG-GTAGTCTGTCAGGT-3′ (reverse) for β-actin (241 bp). Thirty cycles of PCR were performed. The products of PCR were then separated on 1.5% agarose gel and analyzed using a gel imaging system (GeneGenius®, Syngene Co, Ltd, Cambridge, UK).
Western blotting
Total protein was extracted from brain tissue using the following protocol. Equal amounts of protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Each membrane was incubated using rabbit anti-rat primary antibody (1:500) and then horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:2,000). Signals were determined using an ECL-Plus kit (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Brain microdialysis
Each rat was anesthetized by intraperitoneal injection of 1% pentobarbital sodium (Sigma-Aldrich) at 40 mg/kg. A microdialysis probe was implanted according to stereotaxic coordinates. The head of the rat was then fixed in a stereotaxic apparatus (RWD Life Science Co, Ltd, Shenzhen, People’s Republic of China) and a hole was drilled in the middle of the frontal cortex, 1 mm from the anterior fontanelle. A catheter was inserted via the hole into the cerebral cortex from the vertical line at a 30-degree angle to a depth of 3 mm and then fixed with dental cement. After connecting the microdialysis device, the catheter probe was inserted into the cerebral cortex of the rat. The catheter probe consists of a shaft with a semipermeable fiber membrane at its tip and was inserted into the cortex in its entirety. The microdialysis experiment was performed one week after surgical implantation of the probe.
The recovery efficiency of antiepileptic drugs was measured prior to each microdialysis. Briefly, the artificial cerebrospinal fluid rate with 20 μg/mL carbamazepine or phenytoin was injected using a microperfusion pump (KD100, KD Scientific Inc, Holliston, MA, USA) through the inflow tube, probe, and outflow tube at a constant flow rate (2.5 μL per minute), then balanced for 1 hour. Following three consecutive collections of 30 μL of effluent from each group, the concentration of effluent drug was determined. Recovery efficiency was calculated based on the concentration of the effluent drug over the standard concentration of drug. For microdialysis sampling, 25 μL of exchanged effluent dialysate was collected at different time periods after intraperitoneal injection of carbamazepine or phenytoin. The drug concentration in extracellular cortical fluid was normalized to the recovery efficiency; the drug concentration in cortical extracellular fluid was equal to the drug concentration in dialysate/recovery efficiency.
Measurement of drug concentration by high-performance liquid chromatography
Concentrations of carbamazepine and phenytoin in serum and dialysate were detected using high-performance liquid chromatography (Model 510, Beijing Syltech Scientific Instrument Co, Ltd, Beijing, People’s Republic of China). For measurement of serum drug levels, 200 μL of acetonitrile containing 10 μg of hexobarbital (as an internal standard) was added to 200 μL of serum. This mixture was then agitated for 15 seconds and centrifuged for 10 minutes. The supernatant (15 μL) was injected into the chromatography column. For measurement of the dialysate, 1 μL of acetonitrile containing hexobarbital was added to 20 μL of dialysate. The mixture was left to stand for 10 seconds, and a total of 15 μL was then injected into the chromatograph. A 20 μL sample was prepared according to the protocol. Chromatographic conditions were as follows: column ODS C18 (200 mm * 4 mm, 10 μm particle size, Lichrosorb RP); mobile phase, methanol/water (55:45, v:v); flow rate, 1.3 mL per minute; ultraviolet wavelength of detection, 210 nm; pressure of pump, 1,500 psi; and paper running speed, 0.5 cm per minute. Minimum detection was 0.4 μg/mL for phenytoin and 0.3 μg/mL for carbamazepine.
Statistical analysis
All statistical analyses were carried out using SigmaStat (Chicago, IL, USA). Pairwise comparison of P-glycoprotein expression and antiepileptic drug concentration between the groups was done using either the paired Student’s t-test or one-way analysis of variance as indicated. The data are presented as the mean ± standard deviation. Differences were considered to be statistically significant at P<0.05.
Results
Selection of phenytoin responders and nonresponders
Among the 20 kindled rats treated with phenytoin, eight were resistant (nonresponders) and nine showed variable responses to phenytoin (responders). The remaining three rats did not pass the drug selection phase due to variability in the after discharge threshold.
Upregulation of P-glycoprotein in cortex and hippocampus of epileptic rats
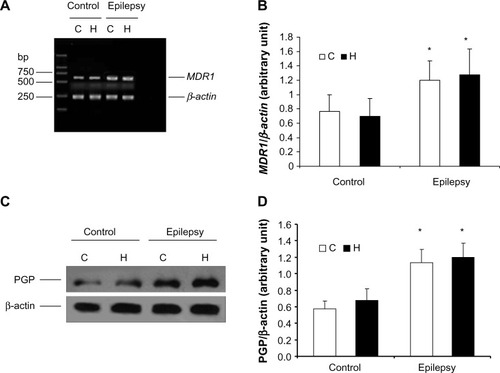
Real-time RT-PCR showed that MDR1 (P-glycoprotein mRNA) expression was higher in the cerebral tissue from epileptic rats than in that from normal rats (). Semiquantitative analysis by densitometry illustrated that the amount of MDR1 expression was significantly increased in the hippocampus and cortex of kindled rats (cortex, P=0.002; hippocampus, P=0.013 []). However, there was no difference between MDR1 levels in the cortex and those in the hippocampus in the control or epilepsy group, indicating that the increase in MDR1 levels were characteristic of epilepsy regardless of its distribution in the cerebra. In addition, P-glycoprotein levels were significantly increased in cerebral tissue from epileptic rats (n=8; cortex, P=0.0003; hippocampus, P=0.0001) as shown in .
Figure 1 Expression of P-glycoprotein and its mRNA in cerebral tissue. MDR1 was determined using real-time reverse transcription polymerase chain reaction and P-glycoprotein using Western blotting. RNA and total protein was extracted from the hippocampus (H) and cortex (C) of control rats and kindled rats. (A) Products of PCR were run on a 1% w/w agarose gel. β-actin (lower band, 241 bp) and MDR1 (upper band, 600 bp) are shown. (B) Semiquantitative analysis using gel after densitometry. The amount of MDR1 expression is represented by the ratio of MDR1 to β-actin (mean ± standard deviation, n=8) and normalized. *P<0.05 versus control group. (C) Western blotting of P-glycoprotein, and (D) quantitative analysis. The amount of P-glycoprotein expression was normalized to β-actin (mean ± standard deviation, n=8). *P<0.05 versus control group.
Inhibition of P-glycoprotein increased antiepileptic drug concentration in extracellular cortical fluid
Since the serum concentration of drugs is an indicator after the administration of drugs, we first detected the serum phenytoin concentration after intraperitoneal injection. After injection of phenytoin 50 mg/kg, plasma phenytoin levels peaked rapidly and were maintained at 15–45 minutes after administration. The serum phenytoin concentration was 12.29–17.35 μg/mL at 15–180 minutes post-injection (). Next, we measured concentrations of phenytoin in extracellular cortical fluid after it had passed through the blood–brain barrier. In the control rats, phenytoin rapidly crossed the blood–brain barrier into cerebral tissue, and was detectable in extracellular cortical fluid at 15 minutes after administration. The highest phenytoin concentration in extracellular cortical fluid was 1.24±0.23 μg/mL at 60 minutes post-injection (), with levels in the range of 0.59–1.24 μg/mL seen over 15–180 minutes. The ratio of the concentration of phenytoin in extracellular cortical fluid to that in serum was 3%–8%. In kindled animals, phenytoin could be detected in extracellular cortical fluid 15 minutes after administration, reaching a peak at 60 minutes, and then gradually decreasing thereafter. The trend of drug concentration in the cortical extracellular fluid of epilepsy was similar to that in normal group. However, compared with controls, phenytoin concentrations in the extracellular cortical fluid of kindled rats was significantly lower than in the control group at each time point over 30–180 minutes (P=0.038, 0.034, 0.001, 0.011, 0.010, 0.025, and 0.007, respectively, n=8) as shown in ).
Figure 2 Concentrations of carbamazepine and phenytoin in extracellular cortical fluid and serum.
Abbreviations: CBZ, carbamazepine; PHT, phenytoin; AED, antiepileptic drug.
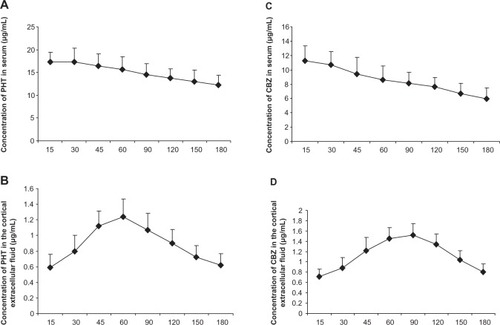
Figure 3 Concentrations of phenytoin (A) and carbamazepine (B) in extracellular cortical fluid as measured by high-performance liquid chromatography.
Abbreviations: CBZ, carbamazepine; PHT, phenytoin.
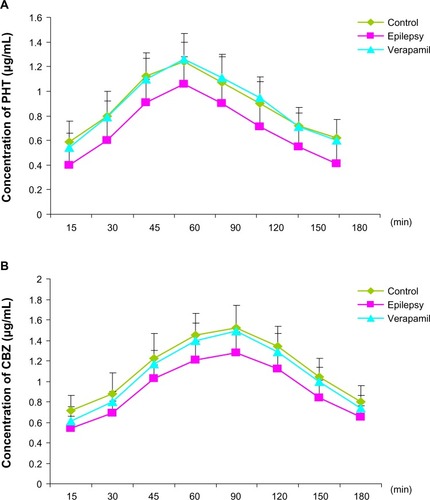
As with phenytoin, mean plasma carbamazepine levels peaked at 11.26±2.12 μg/mL 15 minutes after intraperitoneal injection at a 20 mg/kg dose, and then gradually decreased. Carbamazepine concentrations ranged from 5.94 to 11.26 μg/mL over 15–180 minutes (). In the control group, carbamazepine passed rapidly through the blood–brain barrier into cerebral tissue, as determined by carbamazepine levels in extracellular cortical fluid at 15 minutes after injection of the drug. These peaked at a mean of 1.52±0.22 μg/mL at 90 minutes post administration (), and ranged from 0.71 to 1.52 μg/mL over 15–180 minutes. The ratio of carbamazepine concentration in extracellular cortical fluid to that in serum ranged from 6% to 19%. In kindled animals, carbamazepine was detectable in extracellular cortical fluid at 15 minutes post administration, reaching a peak at 90 minutes, and then gradually decreased. The fluctuation trend in extracellular cortical fluid was similar in the epilepsy and control groups. However, the carbamazepine level found in extracellular cortical fluid from kindled rats was much lower than that in controls at each time point over 30–150 minutes (P=0.024, 0.014, 0.016, 0.006, 0.028, and 0.016, respectively, n=8), as shown in .
In addition, verapamil, a P-glycoprotein inhibitor, did not change the concentration trend of phenytoin and carbamazepine, but significantly increased antiepileptic drug concentrations in extracellular cortical fluid from rats with epilepsy at each time point in the phenytoin group (, n=8, P=0.024, 0.003, 0.015, 0.031, 0.028, 0.005, 0.008, and 0.016 versus epilepsy group, respectively) and at 45, 60, and 120 minutes in the carbamazepine group (, n=8, P=0.028, 0.016, and 0.036 versus epilepsy group, respectively). The phenytoin concentration in rats with epilepsy after treatment with verapamil was similar to that in control group, indicating that inhibition of P-glycoprotein alters the ability of antiepileptic drugs to penetrate the blood–brain barrier, restoring the accumulation of phenytoin in extracellular cortical fluid.
Discussion
P-glycoprotein is known as ABCB1 and belongs to the ATP-binding protein superfamily.Citation15 The characteristics of this molecule include two nucleotide-binding domains and three membrane-spanning domains, the function of which appears to involve an ATP-dependent drug pump. Organic anions and pharmaceutics are pumped across the plasma membrane.Citation16,Citation17 As a key multidrug transporter in the cerebrum, P-glycoprotein is involved in the functioning of the blood–cerebrospinal and blood–brain barriers, which can remove excess brain metabolites and prevent toxic exogenous substances from entering into the brain tissue and so help to maintain the stability of the brain environment.Citation18–Citation20 In normal conditions, expression of P-glycoprotein at both the protein and mRNA levels is low in the hippocampus and cortex, as we observed, suggesting that low P-glycoprotein is sufficient for cerebral homeostasis.Citation21,Citation22
However, in pathologic conditions, such as medically intractable epilepsy and glioma, P-glycoprotein expression in capillary endothelial cells and astrocytes is increased.Citation23–Citation25 Our research confirms that P-glycoprotein is highly expressed in the cerebrum of rats with medically intractable epilepsy, as detected by real-time PCR and Western blotting. Previously, cell lines have been used in vitro and gene knockout rats have been used in vivo; however, we used the rat amygdala kindling model which can effectively mimic uncontrolled seizures in humans.
The findings of our research are in agreement with those of other recent studies showing that high expression of P-glycoprotein may be involved in drug resistance.Citation26–Citation29 The present data provide evidence that carbamazepine and phenytoin are substrates for P-glycoprotein at the blood–brain barrier. Thus, overexpression of such efflux transporters in focal epileptogenic cerebral tissue may impair the ability of an antiepileptic drug to pass through the blood–brain barrier. P-glycoprotein can pump antiepileptic drugs out of cerebral cells and/or tissue, decreasing drug concentrations in the extracellular cortical fluid, which could explain why when the first antiepileptic drug prescribed was not effective, switching to a combination of antiepileptic drugs or to another antiepileptic drug still yielded similarly negative results, despite the various pharmacologic mechanisms of antiepileptic drug action.
Verapamil has been considered to be able to antagonize P-glycoprotein function effectively.Citation30–Citation33 By inhibiting P-glycoprotein with verapamil, we found that the concentration of phenytoin was significantly increased in extracellular cortical fluid from animals with medically intractable epilepsy. Similarly, verapamil markedly increased carbamazepine concentrations in extracellular cortical fluid. However, it remains controversial whether carbamazepine is a substrate for P-glycoprotein. One view is that carbamazepine undergoes extensive metabolism, with the initial oxidative pathways catalyzed by cytochrome (CYP3)A4 and CYP2C8,Citation34 and there is overlap between substrates for CYP3A4 and P-glycoprotein.Citation35 Therefore, the effect of verapamil on carbamazepine is probably due to inhibition of CYP3A4 and not P-glycoprotein.Citation36 However, our investigations confirm that the decreased concentrations of carbamazepine and phenytoin in extracellular cortical fluid are associated with upregulation of P-glycoprotein in the epileptogenic cerebrum. Therefore, further studies are needed to clarify the relationship between P-glycoprotein and carbamazepine in drug resistance.
The mechanism by which P-glycoprotein comes to be highly expressed in cerebral tissue from rats with medically intractable epilepsy is unclear. However, two studies have shown that chemotherapeutic agents and chemical carcinogens, such as barbiturates, cisplatin, and the antibiotic rifampicin, can induce P-glycoprotein and increase its expression.Citation37,Citation38 Thus, antiepileptic drugs may induce an increase in P-glycoprotein expression in epileptogenic cerebral tissue. Other studies have shown that selection of antiepileptic drugs is crucial for new patients with epilepsy, especially in childhood. The initial response to antiepileptic drug treatment is highly predictive of the long-term outcome, and appropriate drug selection should avoid drug resisitance.Citation39–Citation41 In addition, seizures may upregulate the expression of P-glycoprotein.
In conclusion, our findings demonstrate that P-glycoprotein is upregulated in the epileptic cerebrum. Increased P-glycoprotein levels reduced the ability of antiepileptic drugs to penetrate the blood–brain barrier in rats with medically intractable epilepsy, whereas inhibition of P-glycoprotein improved antiepileptic drug concentrations in extracellular cortical fluid, suggesting that P-glycoprotein is involved in resistance to antiepileptic drugs in medically intractable epilepsy.
Acknowledgments
This investigation was supported by grants from the Natural Science Foundation of Shanghai (09ZR1405500) and the Shanghai Municipal Health Bureau (2008–08) to YC.
Disclosure
The authors report no conflicts of interest in this work.
References
- Elia M Amato C Bottitta M An atypical patient with Cowden syndrome and PTEN gene mutation presenting with cortical malformation and focal epilepsy Brain Dev 2012 34 10 873 876 22469695
- Elia M Falco M Ferri R CDKL5 mutations in boys with severe encephalopathy and early-onset intractable epilepsy Neurology 2008 23 71 13 997 999 18809835
- Capovilla G Beccaria F Montagnini A Short-term nonhormonal and nonsteroid treatment in West syndrome Epilepsia 2003 44 8 1085 1088 12887441
- Loscher W Potschka H Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases Prog Neurobiol 2005 76 1 22 76 16011870
- Potschka H Loscher W Multidrug resistance-associated protein is involved in the regulation of extracellular levels of phenytoin in the brain Neuroreport 2001 12 11 2387 2389 11496115
- Tishler DM Weinberg KI Hinton DR Barbaro N Annett GM Raffel C MDR1 gene expression in brain of patients with medically intractable epilepsy Epilepsia 1995 36 1 1 6 8001500
- Seegers U Potschka H Loscher W Transient increase of P-glycoprotein expression in endothelium and parenchyma of limbic brain regions in the kainate model of temporal lobe epilepsy Epilepsy Res 2002 51 3 257 268 12399076
- Morimoto K Sato H Sato K Sato S Yamada N BW1003C87, phenytoin and carbamazepine elevate seizure threshold in the rat amygdale-kindling model of epilepsy Eur J Pharmacol 1997 339 1 11 15 9450611
- Hung CC Chen CC Lin CJ Liou HH Functional evaluation of polymorphisms in the human ABCB1 gene and the impact on clinical responses of antiepileptic drugs Pharmacogenet Genomics 2008 18 5 390 402 18408562
- Potschka H Fedrowitz M Loscher W P-glycoprotein and multidrug resistance-associated protein are involved in the regulation of exrracellular levels of the major antiepileptic drug carmbamazepine in the brain Neuroreport 2001 12 6 3557 3560 11733711
- Baltes S Gastens AM Fedrowitz M Potschka H Kaever V Löscher W Differences in the transport of the antiepileptic drugs phenytoin, levetiracetam and carbamazepine by human and mouse P-glycoprotein Neuropharmacology 2007 52 2 333 346 17045309
- Morimoto K Goddard GV Seizure-triggering mechanism in the kindling model of epilepsy: I. EEG changes during stimulation from the site of stimulation Jpn J Psychiatry Neurol 1988 42 3 618 619 2853806
- Racine RJ Modification of seizure activity by electrical stimulation. II. Motor seizure Electroencephalogr Clin Neurophysiol 1972 32 3 281 294 4110397
- Löscher W Rundfeldt C Kindling as a model of drug-resistant partial epilepsy: selection of phenytoin-resistant and nonresistant rats J Pharmacol Exp Ther 1991 258 2 483 489 1650829
- Sharom FJ ABC multidrug transporters: structure, function and role in chemoresistance Pharmacogenomics 2008 9 1 105 127 18154452
- Polgar O Bates SE ABC transporters in the balance: is there a role in multidrug resistance? Biochem Soc Trans 2005 33 1 241 245 15667317
- Choudhuri S Klaassen CD Structure, function, expression, genomic organization, and single nucleotide polymorphisms of human ABCB1 (MDR1), ABCC (MRP), and ABCG2 (BCRP) efflux transporters Int J Toxicol 2006 25 4 231 259 16815813
- Lee G Dallas S Hong M Bendayan R Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations Pharmacol Rev 2001 53 4 569 596 11734619
- Shen S Zhang W ABC transporters and drug efflux at the blood-brain barrier Rev Neurosci 2010 21 1 29 53 20458886
- Ohtsuki S Terasaki T Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development Pharm Res 2007 24 9 1745 1758 17619998
- Loscher W Potschka H Blood-brain barrier active efflux transporters: ATP-binding cassette gene family NeuroRx 2005 2 1 86 98 15717060
- Elsinga PH Hendrikse NH Bart J Vaalburg W Waarde A PET studies on P-glycoprotein function in the blood-brain barrier: how it affects uptake and binding of drugs within the CNS Curr Pharm Des 2004 10 13 1493 1503 15134571
- Loscher W Potschka H Role of multidrug transporters in pharmaco-resistance to antiepileptic drugs J Pharmacol Exp Ther 2002 301 1 7 14 11907151
- Sisodiya SM Lin WR Harding BN Squier MV Thom M Drug resistance in epilepsy: expression of drug resistance proteins in common cause of refractory epilepsy Brain 2002 125 1 22 31 11834590
- Volk HA Potschka H Loscher W Increased expression of the multidrug transporter P-glycoprotein in limbic brain regions after amygdala-kindled seizures in rats Epilepsy Res 2004 58 1 67 79 15066676
- Jeub M Beck H Siep E Effect of phenytoin on sodium and calcium currents in hippocampal CA1 neurons of phenytoin-resistant kindled rats Neuropharmacology 2002 42 1 107 116 11750920
- Potschka H Loscher W In vivo evidence for P-glycoprotein-mediated transport of phenytoin at the blood-brain barrier of rats Epilepsia 2001 42 10 1231 1240 11737157
- Lazarowski A Czornyj L Lubienieki F Girardi E Vazquez S D’Giano C ABC transporters during epilepsy and mechanisms underlying multidrug resistance in refractory epilepsy Epilepsia 2007 48 5 140 149 17910594
- Rizzi M Caccia S Guiso Limbic seizures induce P-glycoprotein in rodent brain: functional implications for pharmacoresistance J Neurosci 2002 22 14 5833 5839 12122045
- Iannetti P Spalice A Parisi P Calcium-channel blocker verapamil administration in prolonged and refractory status epilepticus Epilepsia 2005 46 6 967 969 15946342
- Summers MA Moore JL McAuley JW Use of verapamil as a potential P-glycoprotein inhibitor in a patient with refractory epilepsy Ann Pharmacother 2004 38 10 1631 1634 15328394
- Jambroszyk M Tipold A Potschka H Add-on treatment with verapamil in pharmacoresistant canine epilepsy Epilepsia 2011 52 2 284 291 21219313
- Pirker S Baumgartner C Termination of refractory focal status epilepticus by the P-glycoprotein inhibitor verapamil Eur J Neurol 2011 18 12 e151 22097953
- Kerr BM Thummel KE Wurden CJ Human liver carbamazepine metabolism – role of CYP3A4 and CYP2C8 in 10,11-epoxide formation Biochem Pharmacol 1994 47 11 1969 1979 8010982
- Kim RB Wandel C Leake B Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein Pharm Res 1999 16 3 408 414 10213372
- Owen A Pirmohamed M Tettey JN Carbamazepine is not a substrate for P-glycoprotein Br J Clin Pharmacol 2001 51 4 345 349 11318771
- Lu Y Yan Y Wang XF Antiepileptic drug-induced multidrug resistance P-glycoprotein overexpression in astrocytes cultured from rat brains Chin Med J 2004 117 11 1682 1686 15569486
- Felix RA Barrand MA P-glycoprotein expression in rat brain endothelial cells: evidence for regulation by transient oxidative stress J Neurochem 2002 80 1 64 72 11796744
- Coppola G Auricchio G Federico R Carotenuto M Pascotto A Lamotrigine versus valproic acid as first-line monotherapy in newly diagnosed typical absence seizures: an open-label, randomized, parallel-group study Epilepsia 2004 45 9 1049 1053 15329068
- Coppola G Licciardi F Sciscio N Russo F Carotenuto M Pascotto A Lamotrigine as first-line drug in childhood absence epilepsy: a clinical and neurophysiological study Brain Dev 2004 26 1 26 29 14729411
- Löscher W How to explain multidrug resistance in epilepsy? Epilepsy Curr 2005 5 3 107 112 16145617