 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
Acyclovir has pharmacokinetic limitations, including poor oral bioavailability of 15%–30%, high variability, and short elimination half-life of 2.3 hours. These limitations necessitate frequent administration of acyclovir, up to five times daily, leading to poor patient compliance, which in turn leads to a reduction in therapeutic efficacy and development of resistance.
Methods
A gastroretentive sustained-release (GR) formulation of acyclovir, based on a combination of swelling and mucoadhesive mechanisms, has been developed. Composition has been optimized after evaluation of different polymers, carbomer, polyethylene oxide, and sodium alginate alone and/or in combination. GR formulations were characterized for in-process quality-control tests, drug release and release rate kinetics, similarity factor analysis, swelling index, and matrix erosion.
Results
A formulation containing a combination of carbomer and polyethylene oxide had the highest similarity of drug release compared with a target drug-release profile obtained by pharmacokinetic simulations. The measurement of mucoadhesive strength, carried out with a texture analyzer, showed that the mucoadhesive strength of the GR formulation was significantly higher than that of the immediate-release (IR) tablet. The optimized GR formulation was found to be retained in the upper part of the gastrointestinal tract for 480 minutes; the IR tablet was retained for only 90 minutes as measured using a gastrointestinal retention study in albino rabbits. The GR formulation was also found to maintain more sustained plasma concentrations than the IR tablet. Mean residence time of the GR formulation was 7 hours versus 3.3 hours for the IR formulation. The relative bioavailability of the GR formulation was 261% of the IR formulation.
Conclusion
The GR formulation of acyclovir, based on swelling and mucoadhesive mechanisms, has prolonged retention in the upper gastrointestinal tract, sustained in vitro drug release, prolonged in vivo absorption, and better bioavailability than the IR formulation. Such a formulation would improve patient compliance and increase the efficacy of therapy.
Introduction
Oral sustained-release (SR) dosage forms have retained prominence for the past 3 decades due to their clinical advantages in comparison with their immediate-release (IR) forms.Citation1 However, the conventional SR formulations are not suitable for drugs possessing a narrow absorption window in the upper part of the gastrointestinal tract (GIT). These formulations are rapidly cleared from the upper GIT, resulting in the release of a significant fraction of the drug in non-absorbing distal segments of the GIT. This leads to a short absorption phase and poor bioavailability of the drug.Citation2 Many drugs, such as ciprofloxacin, ofloxacin, levodopa, iron, and acyclovir, are preferentially absorbed from the upper GIT. It has been reported that, when drugs with a narrow absorption window are formulated as gastroretentive SR (GR) formulations, they have higher bioavailability due to an extended absorption phase.Citation3 The ciprofloxacin once-daily tablet and ofloxacin once daily are the well-known commercially available GR formulations. After oral administration, GR formulations are retained within the stomach and therein release the drug in a controlled manner, so the drug is supplied continuously to its absorption sites in the upper GIT. This would be the best mode of administration for these drugs to achieve the pharmacokinetic and pharmacodynamic advantages of SR dosage forms.Citation4
In our previous study, we prepared, optimized, and characterized mucoadhesive microspheres of acyclovir.Citation5 We found a significant increase in bioavailability and mean residence time (MRT) when acyclovir was administered as mucoadhesive microspheres in comparison with the plain drug. The simplicity and scalability of the manufacturing process is important for the preparation of any formulation at the commercial level. Methods employed in the manufacture of microsphere-based GR formulations suffer significant shortfalls and limitations (eg, the multistep process, use of organic solvents that must be removed from the final formulation, requirement for high shear conditions, and a lengthy post-processing time period) that potentially hinder commercial success. The preparation of SR tablets is well established at the commercial level due to the use of technology similar to that used to manufacture IR tablets. In the present study, we attempted to prepare, optimize, and characterize GR tablet formulations of acyclovir using a combination of swelling and mucoadhesion mechanisms. GR dosage forms using various approaches, such as high-density systems,Citation6 floating systems,Citation7 expandable systems,Citation8,Citation9 superporous hydrogels,Citation10 mucoadhesive/bioadhesive systems,Citation11 and magnetic systems,Citation12 have been reported in the literature. In the present study, a combination of mucoadhesion and swelling was used. While bioadhesion ensures adhesion of the dosage form onto gastric mucosa, rapid and high degrees of swelling help to delay clearance through the pyloric sphincter.
Acyclovir has pharmacokinetic limitations, such as poor oral bioavailability (15%–30%), high variability, and a short elimination half-life of 2.3 hours.Citation13,Citation14 These limitations necessitate the frequent administration of acyclovir, up to five times daily, leading to poor patient compliance, which in turn leads to a reduction in therapeutic efficacy and development of resistance. Acyclovir is soluble in acidic pH and is predominantly absorbed from the upper GIT. There are indications that the drug is absorbed only from the upper GIT.Citation15 In commercially available IR dosage forms, the fraction of dose absorbed is very low due to the short residence time of the dosage form at the absorption site. As a result, most of the drug is excreted in the feces (50%–60%), in an unabsorbed form.Citation16
The aim of the present study was to prepare and optimize a GR tablet formulation based on swelling and mucoadhesive mechanisms. Although some studies have already been conducted on GR tablet dosage forms of acyclovir,Citation17,Citation18 this study is the first to formulate a GR dosage form based on target dose and an in vitro drug-release profile obtained through pharmacokinetic simulations.Citation19 An attempt was also made to evaluate the mechanism of gastroretention by studying the swelling and matrix erosion kinetics and by quantitative evaluation of bioadhesion using a texture analyzer. In vivo radiographic and pharmacokinetic studies were performed in rabbits to evaluate gastroretention and associated pharmacokinetic advantages over conventional dosage forms.
Materials and methods
Materials
Acyclovir was obtained as a gift sample from Ranbaxy Laboratories Limited (Gurgaon, India). Carbomer (Carbopol 974P) was a gift sample from Lubrizol Advanced Materials India Private Limited (Mumbai, India). Polyethylene oxide (Polyox WSR 303) was purchased from Colorcon Asia Private Limited (Goa, India). Sodium alginate (Keltone® HVCR) and microcrystalline cellulose (Avicel PH 101) were received as gift samples from Signet Chemical Corporation Private Limited (Mumbai, India). Povidone K 30 (Plasdone K 29/32) was purchased from International Specialty Products (Hyderabad, India). Colloidal silicon dioxide (Aerosil 200) was purchased from Evonik Industries (Mumbai, India). Magnesium stearate (Hyqual®, vegetable source) was purchased from Mallinckrodt Baker India (Mumbai, India). Isopropyl alcohol was purchased from Avantor Performance Materials India Limited (Faridabad, India). Barium sulfate was procured from Merck (Mumbai, India).
Characterization of acyclovir
Angle of repose
Acyclovir was passed through a funnel kept at a height of 3 cm from the base. The powder was passed until it formed a heap and touched the tip of the funnel. The radius of the base of the conical pile and the height of the pile were measured, and the angle of repose was calculated using EquationEquation 1[1] ,
where θ is the angle of repose, h is the height of the pile, and r is the radius of the base of the pile.
Bulk density and tapped density
The weighed quantity of acyclovir (electronic weighing balance: GP 8201, Sartorius AG, Germany) was transferred into a 100 mL measuring cylinder without significant mechanical stresses during transfer. The volume occupied by the drug was measured initially and after subjecting to 1,250 taps in a tap density tester (ETD1020, Electrolab, Mumbai, India). Bulk and tapped density were calculated using EquationEquations 2[2] and Equation3
[3] , respectively,
where m is the mass of the drug (g), vi is the initial volume (mL), and vt is the tapped volume (mL).
Compressibility index
The compressibility index (CI) was expressed as a percentage and calculated using EquationEquation 4[4] or Equation5
[5] ,
Hausner’s ratio
Hausner’s ratio was determined by the ratio of tapped density and bulk density based on EquationEquation 6[6] ,
Drug–excipient compatibility study
The suitability of excipients with respect to their influence on the stability of acyclovir was evaluated through drug–excipient compatibility studies. Mixtures of acyclovir with individual excipients were prepared, and these mixtures were packed in glass vials (type I) as well as low-density polyethylene (LDPE) bags. These samples were stored in stability chambers (NEC2355; Newtronic, Mumbai, India). Samples packed in glass vials were stored at 60°C and 40°C, 75% relative humidity (RH), and those packed in LDPE bags were stored in 40°C, 75% RH. Samples were withdrawn after predefined time intervals and analyzed for guanine content, the major impurity of acyclovir, using high-performance liquid chromatography (HPLC).Citation20 Samples were also observed for physical/morphological changes in terms of color or lump formation.
Preparation of GR tablets
Compositions of various batches of acyclovir GR formulations are summarized in . Hydrophilic, mucoadhesive, polymer matrix tablets were prepared via the wet granulation method. Acyclovir, polymer(s), and microcrystalline cellulose were weighed (electronic weighing balance: AG204; Mettler-Toledo International Inc, Columbus, OH, USA/GP8201; Sartorius AG, Gottingen, Germany) and sifted together through a #40 ASTM sieve and blended in a polybag for 5 minutes. This blend was then granulated manually using a binder solution prepared by dissolving povidone K 30 in isopropyl alcohol. The wet mass was dried in a tray dryer for 60 minutes at 50°C to obtain loss on drying of <2% w/w (IR moisture analyzer-MA 100, Sartorius AG, Germany). The dried mass was passed through a #20 ASTM sieve to form granules. The resultant granules were lubricated by blending with magnesium stearate (previously sifted through a #60 ASTM sieve) in a polybag. This lubricated blend was compressed manually (tablet press model CMD3-16, Cadmach, India) using a 19 mm × 9 mm, modified capsule-shaped, concave, plain punch with beveled edges.
Table 1 Composition of gastroretentive formulations of acyclovir
In-process characterization
Properties of the acyclovir GR tablets, such as hardness, friability, thickness, and weight variation, were determined as in-process characterization. Hardness was determined by using a tablet hardness tester (VK200, Varian Inc, Cary, NC, USA). Friability was determined using Roche friability testing apparatus (EF-1W, Electrolab, Mumbai, India) as per procedures described in the Indian Pharmacopoeia (IP).Citation21 Weight variation was also performed according to the IP procedure.Citation22 Thickness of tablets was determined using a digital vernier caliper (Mitutoyo Corporation, Kawasaki, Japan).
In vitro drug-release studies
In vitro drug-release studies were performed using USP type II dissolution apparatus (paddle) at 50 rpm (VK7010 with VK8000 Auto sampler, Varian Inc). The drug-release medium was 900 mL of 0.1 N HCl at a temperature of 37°C±0.5°C. An aliquot (10 mL) was withdrawn at specific time intervals (1, 2, 4, 6, 8, 10, and 12 hours), and drug content was determined by ultraviolet-visible spectrophotometer (Cary 50, Varian Inc) at 255 nm. Immediately after withdrawal of the sample, 10 mL of 0.1 N HCl maintained at 37°C±0.5°C was replaced to maintain the volume of dissolution medium constant. We ensured that none of the ingredients used in the tablet formulations interfered with the analysis. When the formulations were subjected to in vitro drug-release studies, their swelling behavior and other physical changes were also observed.
Similarity factor analysis
Similarity factor (f2) is a simple and model-independent approach to comparing drug-release profiles.Citation23 It is a logarithmic reciprocal square root transformation of the sum of the squared error and is a measurement of the similarity in the percent dissolution (drug release) between the two curves. Two drug-release curves are said to be similar if f2 is between 50 and 100; f2 was determined using EquationEquation 7[7] ,
where Rt is the percentage drug release of reference at time t, and Tt is the percentage drug release of test at time t.
We determined f2 for all formulations. The target drug-release profile obtained in pharmacokinetic simulations was used as a reference, and respective drug-release profiles of different batches of GR tablets were used as a test.
Release kinetics
Drug-release data of selected batches were fitted into zero-order, first-order, Higuchi, Korsmeyer–Peppas, and Hixson–Crowell equations.Citation24 To study the mechanism of drug release, the well-known exponential equation (Korsmeyer equation), often used to describe drug-release behavior from polymeric systems, was used.Citation25
where Mt is the amount of drug released at time t; Mf is the amount of drug released after infinite time; k is a release rate constant, incorporating structural and geometric characteristics of the tablet; and n is the diffusional exponent indicative of the mechanism of drug release. The n value is obtained from the regression line of a plot of log% drug released versus log time. A value of n=0.45 indicates Fickian (case I) release; >0.45 but <0.89 for anomalous (non-Fickian) diffusion, indicating a combination of diffusion and erosion-controlled drug release; and 0.89 or above indicates super case II type of release, referring to erosion of the polymeric chain.
Swelling index and matrix erosion
Swelling index and matrix erosion studies were performed by a method similar to that reported by Al-Taani and Tashtoush,Citation26 using USP type II dissolution test apparatus (VK7010 with VK8000 Auto sampler, Varian Inc). The tablets were accurately weighed and dropped into the dissolution vessel containing 900 mL of 0.1 N HCl (pH 1.2) maintained at 37°C±0.5°C; speed of rotation was 50 rpm (n=3). At 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 4, 6, and 8 hours, the residual matrix was carefully removed from the dissolution vessel and weighed. The residual matrix was then dried in a hot air oven at 60°C for 12 hours and reweighed. The percentage swelling index (ie, the degree of swelling due to absorbed medium) was calculated using EquationEquation 9[9] ,
Percentage erosion was calculated from EquationEquation 10[10] ,
Measurement of mucoadhesive strength
The mucoadhesive strength of acyclovir GR tablets on goat gastric mucosa was determined using a texture analyzer (TA.XT.Plus, Stable Microsystems Ltd, Godalming, UK). A commercially available IR formulation, Zovirax® (GlaxoSmithKline, Mumbai, India), was used for comparison purposes. Use of goat gastric mucosa has been widely reported for evaluation of mucoadhesive strength of mucoadhesive formulationsCitation27,Citation28 and it is easily available. Immediately after slaughter, the stomach of the goat was removed and cleaned using ice-cold Krebs ringer solution (pH 6.8). This solution contained 115 mM sodium chloride; 5.9 mM potassium chloride; 1.2 mM each of magnesium chloride, sodium dihydrogen phosphate, and sodium sulfate; 2.5 mM calcium chloride; 25 mM sodium bicarbonate; and 10 mM glucose per liter of solution.
A piece of gastric mucosa measuring about 2 cm × 2 cm was mounted securely in the tissue holder of the texture analyzer, with mucosa facing upwards. The tablet was fixed to the probe with double-sided adhesive tape. The surface of the tablet was immersed in 0.1 N HCl for 30 seconds and then allowed to equilibrate for 90 seconds. The probe was lowered at a speed of 0.5 mm/second and allowed to be in contact with the mucosa with a force of 10 g for 300 seconds. The probe was then withdrawn at a speed of 0.5 mm/second with a trigger force of 5 g. The force required to detach the formulation from the tissue surface was determined as the peak value in resultant force-time plot. This experiment was carried out in triplicate and a fresh piece of gastric mucosa was used in each replicate. Three formulations containing a combination of carbomer and polyethylene oxide were included in this experiment (AGR-6, AGR-7, and AGR-8).
Gastroretention study
The gastroretention study was conducted on albino rabbits and was based on X-ray radiography. Albino rabbits were chosen as the experimental animals as previously reported in the literature for evaluation of gastroretention and bioavailability of gastroretentive formulations.Citation27,Citation29–Citation31 The study was conducted on six healthy rabbits weighing 1.8 kg–2.4 kg. Animals were grouped into two groups of three animals each. A small (6.0 mm) conventional tablet formulation and the optimized small GR tablet (6.0 mm) were administered, along with water, to the first and second groups, respectively. Both the formulations contained 72.5 mg of acyclovir per tablet. The optimized GR tablet contained a combination of AGR-7, ie, carbomer and polyethylene oxide (75:25). To make the tablet X-ray opaque, barium sulfate in 10% w/w concentration was included. The small tablets were also characterized for shape, size, thickness, hardness, friability, drug content, and in vitro drug release. The f2 of the drug-release profile of small tablets is more than 50 when compared with AGR-6. During the study, the animals were not allowed to eat, but water was available ad libitum. X-ray photography of the abdominal region was taken at 0, 1, 1.5, 2, 4, 6, 8, and 12 hours after administration of the tablets.Citation32 All animal investigations were performed after approval by the Institutional Animal Ethics Committee of the Department of Pharmaceutical Sciences and Drug Research, Punjabi University, Patiala, India, and in accordance with the disciplinary principles and guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals, Ministry of Environment and Forests, Government of India.
Pharmacokinetic study
Protocols similar to those used for the gastrointestinal retention study were used for the pharmacokinetic study. At 0, 0.5, 0.75, 1, 1.5, 2, 4, 5, 6, 8, 10, 12, 14, 18, and 24 hours, blood was collected from the ear vein in tubes coated with anti-coagulant, and centrifuged at 4,000 rpm for 10 minutes (Remi equipment, Mumbai, India). Acetonitrile was added to the supernatant to precipitate the proteins. The precipitated proteins were settled by centrifugation at 4,000 rpm for 15 minutes. The supernatant was filtered through a 0.45 μm filter, and the drug concentration was determined via the HPLC method.Citation33 A mixture of glacial acetic acid in water was used as the mobile phase. The injected fluid (20 μL) was eluted in C-18 column 4.6 mm × 250 mm, HyperSil, at room temperature, and acyclovir content was analyzed at 254 nm using a diode array ultraviolet detector (1260 Series, Agilent, Santa Clara, CA, USA). Pharmacokinetic parameters were calculated using non-compartmental analysis (WinNonlin 5.1.3, Certara, LP, St Louis, MO, USA).
Stability study
A stability study of the optimized formulation was conducted at accelerated stability test conditions as per International Conference on Harmonisation (ICH) guidelines.Citation34 Since the storage conditions of the accelerated test condition are harsher than intermediate- or long-term conditions, the former was used to evaluate the stability of the formulation within a shorter time period. Tablets were packed in aluminum blisters and stored at 40°C, 75% RH, in a stability chamber (NEC2355, Newtronic, India). Samples were withdrawn after 1, 2, and 3 months, and tested for assay, water content, guanine content, and drug release.
Results and discussion
Preparation and characterization of GR formulations
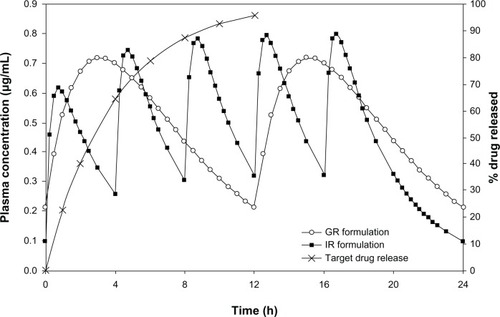
The strength of the acyclovir (725 mg) and target drug-release profiles were determined via pharmacokinetic simulations. Simulations were carried out using reported pharmacokinetic parameters of IR tablets of acyclovir.Citation14 Detailed methodology used for the simulations has been described in our previous publication.Citation19 shows the simulated steady-state plasma concentration profiles of the 200 mg IR formulation (administered five times daily) and the 723 mg SR formulation (administered two times daily), along with the target in vitro drug-release profile for the SR formulation.
Figure 1 Comparison of simulated steady state plasma concentration profiles of the IR and GR formulations of acyclovir along with target drug release profile.
Carbomer, polyethylene oxide, and sodium alginate were used as mucoadhesive release-controlling polymers, either individually or in combination. These polymers have good gel-forming abilities and high degrees of swelling,Citation35–Citation37 which will assist with retention of tablets in the stomach, without allowing them to pass through the pyloric sphincter. Moreover, their chemical structures allow them to form numerous hydrogen bonds, which is crucial for strong mucoadhesion.Citation38–Citation40 They are widely used in drug products and cosmetics as release-controlling polymers and viscosity modifiers.
shows the results of the drug–excipient compatibility study. All excipients used in the formulation of acyclovir GR tablets were included in this study. Compatibility of a drug with excipients is an important criterion when including a particular excipient in a formulation. It is predictive of possible stability failures of the drug product, particularly regarding impurities. There was no significant increase in impurity levels (guanine) with all excipients at 60°C in glass vials. In the case of the drug–carbomer mixture, there was a significant increase in impurity at 40°C, 75% RH, in samples packed in LDPE bags, but a similar increase was not observed in samples packed in glass vials. Since the samples packed in glass vials are completely protected from moisture, but those packed in LDPE bags are not, it can be concluded that the increase in impurities in LDPE bags is due to the absorption of moisture. This conclusion was also supported by soft lumps observed at 40°C, 75% RH, in the drug–carbomer mixture packed in LDPE bags. Hence, carbomer was not excluded from the formulation, as the final formulation can be protected from moisture by packaging in aluminum blister packs, which are completely impermeable to moisture. We observed no other chemical incompatibilities in our studies. Shahi et alCitation41 have also reported that there was no drug–excipient interaction between acyclovir and polyethylene oxide or povidone using differential scanning calorimetry (DSC) and Fourier transform infrared spectroscopy (FTIR) studies. Neither the impurity levels nor the physical appearance of mixtures with other excipients changed in any packs and conditions.
Table 2 Results of drug–excipient compatibility study of acyclovir
Results of various characterization tests performed on acyclovir are shown in . Acyclovir possesses an angle of repose value of greater than 40°, indicating a very poor flow property. The compressibility of acyclovir is also extremely poor, as the CI was found to be 39.47%.Citation42
Table 3 Characterization of acyclovir
Due to the poor flow properties of acyclovir, we selected a granulation process to prepare GR tablets. As the calculated dose of acyclovir was high (725 mg), the quantity of excipients has to be less, to avoid increasing the size of the tablet, which would be very difficult to swallow. Hence, a wet granulation process was selected, which provides intimate contact between the drug and polymer and better control of drug release at low concentrations of polymer. All the polymers used in the preparation were hydrophilic and swollen in the presence of water. This property makes the drying process of the wet mass after granulation very difficult, if water is used as a granulating solvent. Therefore, wet granulation was performed using isopropyl alcohol as the solvent. Isopropyl alcohol is a widely used granulating solvent in the pharmaceutical industry. It is a very safe solvent to use in oral dosage forms, and has been classified as a solvent with low toxic potential by ICH.Citation43
shows the results of in-process characterization of GR formulations. GR tablets of acyclovir were very compact. Their surface was very smooth and shiny. The weight variation and friability were found to comply with official limits. Tablets were of high mechanical strength, as inferred from the values of friability testing (0.05%–0.11%). There was no capping or lamination observed in any of the formulations.
Table 4 In-process characterization of different batches of acyclovir gastroretentive tablets
In vitro drug-release studies
Drug release of about 90% at 12 hours was considered to be complete release. shows the drug-release profiles in comparison with the target drug-release profile. It was observed that batch AGR-4 showed the fastest drug-release profile of all batches. Drug release decreased when the concentration of carbomer was increased from 7.5% to 10% (). Further increases in carbomer concentration to 15% resulted in drug release much faster than 10% carbomer and closer to 7.5% carbomer. Initial swelling of the batch containing 15% carbomer (AGR-3) was high, but the tablets split into two portions in longitudinal axis. This resulted in an increased surface area due to newly exposed surfaces and subsequently resulted in a faster drug-release profile.
Figure 2 In vitro drug release profiles of gastroretentive tablets of acyclovir.
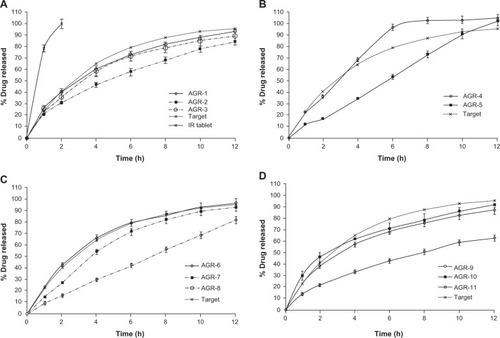
Batches containing polyethylene oxide alone did not swell much and followed an erosion pattern throughout the duration of release. No residual tablets remained after completion of drug release. Drug release was inversely proportional to the concentration of polymer (). Polyethylene oxide had a better release-controlling effect than carbomer up to 6 hours at the same polymer concentration of 10% (AGR-2 vs AGR-5). Since the target release profile is by first-order mechanism, further investigations were not carried out on batches containing polyethylene oxide alone. Batches AGR-6, AGR-7, and AGR-8 contained a combination of carbomer and polyethylene oxide. Drug release decreased with increasing proportions of polyethylene oxide (). This is in accordance with the observation that polyethylene oxide controls drug release better than does carbomer. Of these three batches, the drug-release profile of AGR-6, containing 7.5% carbomer and 2.5% polyethylene oxide, was closest to the target.
Batches containing a combination of carbomer and sodium alginate at equal concentrations split during drug release, although their drug-release profiles were closer to the target (). The drug-release profile of the batch containing 10% carbomer and 3.5% sodium alginate was much slower than the target.
The f2 of drug-release profiles of all batches against the target drug-release profile as reference are summarized in . Six of 11 batches had acceptable f2 values (between 50 and 100). Of the six batches with acceptable f2, AGR-3, AGR-9, and AGR-10 split during drug-release studies. In the absence of splitting, drug-release profiles of these batches must have been much slower. These three batches are not suitable for gastroretention due to the smaller size of split portions compared with the whole tablet.
Table 5 Similarity factor and splitting behavior of acyclovir gas-troretentive tablets
AGR-1, AGR-6, and AGR-7 had acceptable f2 values without splitting of tablets during drug-release testing. Since AGR-6 had the highest f2 value among the three batches, it was considered the most optimal formulation. Apart from AGR-6, both AGR-7 and AGR-8 were also taken up for further in vitro characterization, since the formulation strategy of these two batches was similar to that of AGR-6 (combination of carbomer and polyethylene oxide).
The drug-release data were fitted to zero-order, first-order, Higuchi, Korsmeyer–Peppas, and Hixson–Crowell equations to ascertain the kinetic modeling of drug release. The release rate kinetic data of batches containing a combination of carbomer and polyethylene oxide are shown in . In terms of R2 value, AGR-6 and AGR-7 had the best fit with the first-order release model, whereas AGR-8 fitted well with the zero-order release model. The drug transport mechanism of all three batches was found to be anomalous (non-Fickian) diffusion as determined by an n value >0.45 and <0.89. As the quantity of polyethylene oxide increased, the n value increased towards case II transport, that is, towards erosion-controlled zero-order release. Visual observation of drug-release profiles of these batches also concur with this observation ().
Table 6 Mathematical modeling and drug-release kinetics of acyclovir gastroretentive formulations
Swelling index and matrix erosion
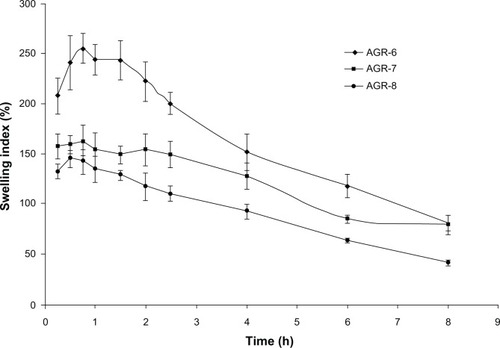
shows the swelling profiles of the acyclovir GR formulation. The swelling of all three batches was initially very rapid and then declined slowly in a sustained manner. The maximum swelling index obtained was 255.7% (0.75 hours), 163.5% (0.75 hours), and 145.3% (0.5 hours) for AGR-6, AGR-7, and AGR-8, respectively. The swelling index decreased with increasing proportions of polyethylene oxide. The swelling profile of all the batches also followed a similar trend. This behavior is similar to the drug-release profile. Swelling is an important characteristic of polymer that controls the drug release and increases the gastrointestinal retention of GR tablets. Swelling is also an important parameter for the mucoadhesion property of the formulation.Citation44 A number of studies showed the direct relationship between swelling and mucoadhesion.Citation45,Citation46 To develop maximum adhesion strength, an optimum concentration is needed for polymers to form a cohesive bond.
Figure 3 Swelling index of gastroretentive tablets of acyclovir.
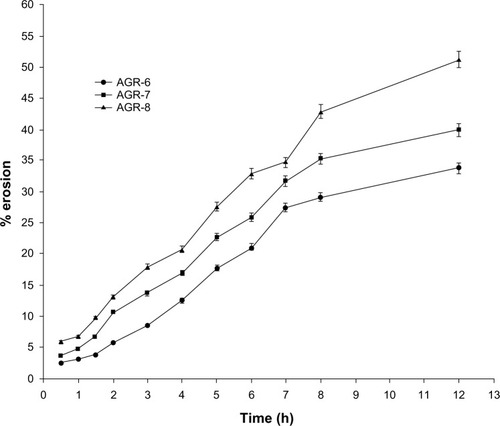
shows the results of the matrix erosion study. The results of matrix erosion are in accordance with the swelling index. Erosion was greater when the proportion of polyethylene oxide was higher. Erosion of AGR-6 and AGR-7 was much slower than the drug release, but was faster in AGR-8. This indicates that the drug release is more erosion-controlled as the proportion of polyethylene oxide increases. This is in agreement with the results of drug-release kinetics.
Figure 4 Matrix erosion of gastroretentive tablets of acyclovir.
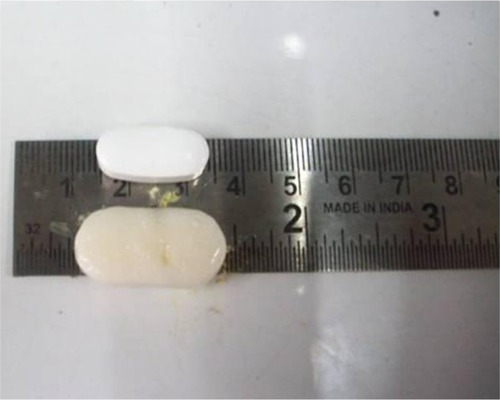
Rapid swelling and controlled erosion of the GR tablet are essential for better gastroretention. To avoid gastric emptying, the dosage form should be larger than about 13 mm.Citation47 shows the relative size of the optimized acyclovir GR tablet after 8 hours of dissolution. Results showed that the optimized GR tablet maintained a size > 13 mm up to 8 hours.
Figure 5 Relative size of optimized GR formulation.
Mucoadhesive strength
Mucoadhesive strength (measured in terms of detachment force using a texture analyzer) of the most optimal formulation (AGR-6) was much higher than the marketed IR formulation, Zovirax® (19.3g±4.7g vs 9.3g±0.8g) and was the highest of all three batches tested (). Detachment force decreased with increasing concentrations of polyethylene oxide.
Figure 6 Detachment forces of different batches of gastroretentive and conventional acyclovir tablets in mucoadhesion study.
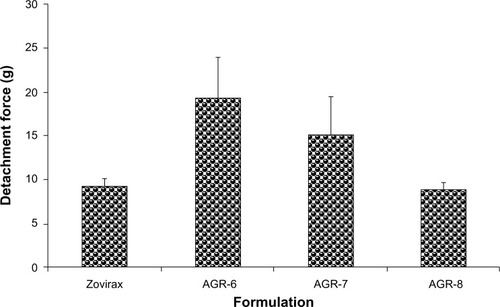
Although polyethylene oxide is highly hydrophilic and has a high degree of functional groups with hydrogen-bonding ability, AGR-8, containing a major proportion of polyethylene oxide, showed the lowest detachment force. It was similar to that of Zovirax®, which is a conventional dosage form. This could be because AGR-8 has the lowest water uptake of the three batches. Sufficient hydration of the polymer network is necessary for the complete opening of the inter-polymeric pores within the polymer matrix in addition to the mobilization of the polymer chains.Citation48
Gastroretention study
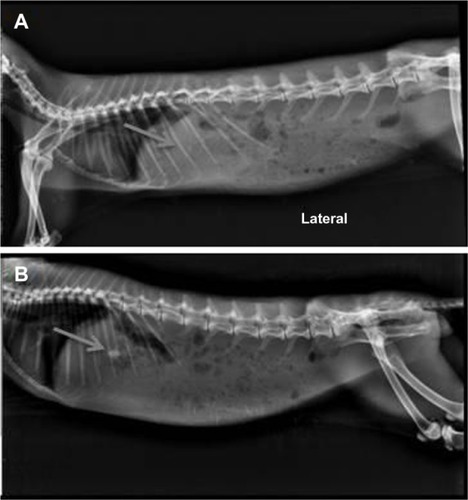
An in vivo radiographic study was conducted on healthy albino rabbits to determine the gastric retention time of the tablet. For this study, a GR tablet of 6.0 mm diameter and 110 mg weight was prepared for optimized batch AGR-6 (containing a combination of carbomer and polyethylene oxide in the ratio of 75:25). Batch AGR-6 was selected as the optimized batch due to its high bioadhesive strength and swelling index and acceptable drug-release characteristics. Images were taken at different time points to find the location of the tablet; gastric residence time was calculated based on this. The gastric residence time of the GR formulation was found to be 480 minutes in comparison with 90 minutes for the conventional IR tablet (). From this study, it is clear that the conventional IR tablet was quickly emptied from stomach, but the retention of the GR formulation was prolonged, up to 8 hours. The increased gastric retention time of the GR formulation is due to the synergistic mechanism of swelling and bioadhesion. These results are in accordance with the in vitro studies.
Figure 7 X-ray radiography photographs of rabbit administered with IR hours (A) and 8 hours (B) with optimized GR formulation (AGR-6).
Pharmacokinetic study
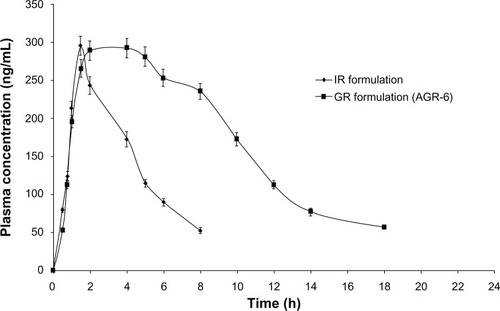
shows the comparative pharmacokinetic parameters of the optimized GR formulation (AGR-6, containing a combination of carbomer and polyethylene oxide in the ratio of 75:25) and the IR formulation. The GR formulation had a similar maximum plasma drug concentration (Cmax) and longer time to Cmax (tmax) compared with the IR formulation. The bioavailability of the GR formulation was significantly higher (2.6-fold) than that of the IR formulation. The plasma-concentration profile of the GR formulation indicated sustained absorption compared with the IR formulation (). The results are in strong agreement with those of the gastroretention study. Poor oral bioavailability (15%–30%) of acyclovir from conventional formulations is due to its narrow absorption window in the upper parts of the GIT. As the gastric residence time of the GR formulation is prolonged, small amounts of the drug are continuously released in the absorbable regions of the GIT, thereby increasing its bioavailability. Since the Cmax of the GR formulation is similar to that of the IR formulation, similar anti-viral activity can be expected. At the same time, prolonged absorption of the GR formulation will help in reducing the dosing frequency of the currently marketed IR formulation.
Table 7 Pharmacokinetic parameters of acyclovir after administration of GR and IR formulations
Figure 8 Mean plasma concentration profiles of IR and optimized GR formulation (AGR-6).
Stability study
shows the assay, water content, guanine content, and drug release of a batch with the same composition as the optimized batch, AGR-6, under accelerated stability-test conditions at 1, 2, and 3 months. There was no significant change in assay, water content, or guanine content. The drug-release profile was similar to the initial profile. It can be inferred from the data that the formulation is stable up to 3 months. The stability study is being continued up to 6 months.
Table 8 Stability data of optimized batch of acyclovir gastro-retentive sustained-release formulation (AGR-6)
Conclusion
A GR formulation of acyclovir has been developed with an in vitro drug-release profile similar to that of the target profile obtained through pharmacokinetic simulations. The formulation has been found to have a high degree of swelling and mucoadhesion properties. The gastric residence time of the optimized GR formulation was much higher than that of the IR formulation. The optimized GR formulation also had prolonged absorption and 2.6-fold higher bioavailability than the conventional formulation. An in vivo pharmacokinetic study showed a constant maintenance of plasma concentration for a prolonged period of time, essential for chronic treatment of viral disease. The results demonstrated that the developed GR formulation will increase the anti-viral activity of acyclovir and improve patient compliance by reducing the dosing frequency of conventional formulations. The GR formulation might represent a better alternative for sustained and efficacious delivery of acyclovir.
Acknowledgments
The authors are grateful to BioPlus Life Sciences Pvt Ltd, India, for providing financial assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
- Hoffman A Pharmacodynamic aspects of sustained release preparations Adv Drug Deliv Rev 1998 33 185 199 10837659
- Klausner EA Lavy E Friedman M Hoffman A Expandable gastroretentive dosage forms J Control Release 2003 90 143 162 12810298
- Qassim SMAinventor Tabuk Pharmaceutical Manufacturing Co, assignee Pharmaceutical compositions of ciprofloxacin European patent EP1880722B1 3 17 2010
- Hoffman A Stepensky D Pharmacodynamic aspects of modes of drug administration for optimization of drug therapy Crit Rev Ther Drug Carrier Syst 1999 16 571 639 10677802
- Dhaliwal S Jain S Singh HP Tiwary AK Mucoadhesive microspheres for gastroretentive delivery of acyclovir: in vitro and in vivo evaluation AAPS J 2008 10 322 330 18523891
- Riner JL Byford RL Stratton LG Hair JA Influence of density and location on degradation of sustained-release boluses given to cattle Am J Vet Res 1982 43 11 2028 2030 7181203
- Reddy LH Murthy RS Floating dosage systems in drug delivery Crit Rev Ther Drug Carrier Syst 2002 19 6 553 585 12822735
- Klausner EA Eyal S Lavy E Friedman M Hoffman A Novel levodopa gastroretentive dosage form: in vivo evaluation in dogs J Control Release 2003 88 1 117 126 12586509
- Kedzierewicz F Thouvenot P Lemut J Etienne A Hoffman M Maincent P Evaluation of peroral silicone dosage forms in humans by gamma-scintigraphy J Control Release 1999 58 2 195 205 10053192
- Chen J Blevins WE Park H Park K Gastric retention properties of superporous hydrogel composites J Control Release 2000 64 1–3 39 51 10640644
- Akiyama Y Nagahara N Nara E Evaluation of oral mucoadhesive microspheres in man on the basis of the pharmacokinetics of furosemide and riboflavin, compounds with limited gastrointestinal absorption sites J Pharm Pharmacol 1998 50 2 159 166 9530983
- Groning R Berntgen M Estimation of the gastric residence time of magnetic dosage forms using the Heidelberg capsule Pharmazie 1996 51 5 328 331 8710954
- Chiou WL Barve A Linear correlation of the fraction of oral dose absorbed of 64 drugs between humans and rats Pharm Res 1998 15 11 1792 1795 9834005
- Lewis LD Fowle ASE Bittiner SB Bye A Isaacs PET Human gastrointestinal absorption of acyclovir from tablet duodenal infusion and sipped solution Br J Clin Pharmacol 1986 21 4 459 462 3707815
- Gröning R Berntgen M Georgarakis M Acyclovir serum concentrations following peroral administration of magnetic depot tablets and the influence of extracorporal magnets to control gastrointestinal transit Eur J Pharm Biopharm 1998 46 3 285 291 9885300
- O’Brien JJ Campoli-Richards DM Acyclovir: an updated review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy Drugs 1989 37 3 233 309 2653790
- Fuertes I Miranda A Millán M Caraballo I Estimation of the percolation thersholds in acyclovir hydrophilic matrix tablets Eur J Pharm Biopharm 2006 64 3 336 342 16876392
- Garg R Gupta GD Preparation and evaluation of gastroretentive floating tablets of acyclovir Curr Drug Deliv 2009 6 5 437 443 19751200
- Sankar R Jain SK Determination of target in vitro drug release profile for extended release formulation of acyclovir through pharmacokinetic simulations Anti-Infect Agents 2013 11 2 204 211
- United States Pharmacopeia and National Formulary (USP 36-NF 31) Acyclovir tablets Rockville, MD The United States Pharmacopeial Convention 2013 2342 2343
- Indian Pharmacopoeia Uniformity of weight of single dose preparations Ghaziabad Indian Pharmacopoeia Commission 2010 1 192
- Indian Pharmacopoeia Friability of uncoated tablets Ghaziabad Indian Pharmacopoeia Commission 2010 1 193
- Moore JW Flanner HH Mathematical comparison of dissolution profiles Pharm Tech 1996 20 64 74
- Costa P Lobo JMS Modeling and comparison of dissolution profiles Eur J Pharm Sci 2001 13 2 123 133 11297896
- Siepmann J Peppas NA Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC) Adv Drug Deliv Rev 2001 48 2–3 139 157 11369079
- Al-Taani BM Tashtoush BM Effect of microenvironment pH of swellable and erodable buffered matrices on the release characteristics of diclofenac sodium AAPS PharmSciTech 2003 4 3 E43 14621975
- Ravindran VK Vasa S Subadhra S Banji D Banji O Rao YM Comparative study of mucoadhesive polymers carbopol 974P and sodium carboxymethyl cellulose for single unit dosage of imatinib mesylate Malaysian J Pharm Sci 2012 10 1 61 77
- Badhan AC Mashru RC Shah PP Thakkar AR Dobaria NB Development and evaluation of sustained release gastroretentive minimatrices for effective treatment of H. pylori infection AAPS PharmSciTech 2009 10 2 459 467 19381827
- Baskar GV Narayanan N Gaikwad R Abdul S Formulation and evaluation of gastro-retentive floating multi-particulate system of metoprolol tartarate Trop J Pharm Res 2010 9 2 181 186
- Nagarwal RC Ridhurkar DN Pandit JK In vitro release kinetics and bioavailability of gastroretentive cinnarizine hydrochloride tablet AAPS PharmSciTech 2010 11 1 294 303 20182827
- Thakar K Joshi G Sawant KK Bioavailability enhancement of baclofen by gastroretentive floating formulation: statistical optimization, in vitro and in vivo pharmacokinetic studies Drug Dev Ind Pharm 2013 39 6 880 888 22901056
- Chary RBR Vani G Rao YM In vitro and in vivo adhesion testing of mucoadhesive testing of mucoadhesive drug delivery systems Drug Dev Ind Pharm 1999 25 5 685 690 10219540
- United States Pharmacopeia and National Formulary (USP 36-NF 31) Acyclovir Rockville, MD The United States Pharmacopeial Convention 2013 2338 2339
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use [webpage on the Internet] ICH Q1C. Stability testing for new dosage forms 1997 Available from: http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html Accessed January 15, 2013
- Colorcon Polyox™, Water-Soluble Polymers for Pharmaceutical Applications Harleysville (PA) BPSI Holdings, LLC 2009 Available from: http://www.colorcon.com/literature/marketing/mr/Extended%20Release/POLYOX/English/Colorcon_PIB_POLYOX.pdf Accessed April 12, 2013
- Lubrizol Pharmaceutical Polymers for Oral Solid Dosage Forms Cleve-land (OH) Lubrizol 2011 Available from: http://www.lubrizol.com/Pharmaceutical-Ingredients/Documents/Brochures/Pharmaceutical-Polymers-for-Oral-Solid-Dosage-Forms.pdf Accessed April 12, 2013
- FMC Corporation Alginates for Pharmaceutical and Medical Applications: Performance Enhancing Products Philadelphia (PA) FMC Corporation 2008 Available from: http://www.fmcbio-polymer.com/Portals/ISP/Content/Docs/Alginates%20for%20Pharmaceutical%20and%20Medical%20Applications.pdf Accessed April 12, 2013
- Chen JL Cyr GN Compositions producing adhesion through hydration Manly RS Adhesion in Biological Systems New York Academic Press 1970 163 181
- Duchêne D Ponchel G Principle and investigation of the bioadhesion mechanism of solid dosage forms Biomaterials 1992 13 10 709 715 1420717
- Park H Robinson JR Physico-chemical properties of water insoluble polymers important to mucin/epithelial adhesion J Control Release 1985 2 47 57
- Shahi S Sonawane A Vanamore S Zadbuke N Formulation and in vitro characterization of acyclovir floating matrix tablets: a factorial design study J Applied Pharm Sci 2013 3 5 65 74
- Wells JI Aulton ME Preformulation Aulton ME Pharmaceutics: The Science of Dosage Form Design London Churchill Livingstone 1988 247 248
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use [webpage on the Internet] ICH Q3C(R5). Impurities: guideline for residual solvents 2011 Available from: http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html Accessed April 12, 2013
- Gu JM Robinson JR Leung SH Binding of acrylic polymers to mucin/epithelial surfaces: structure-property relationships Crit Rev Ther Drug Carrier Syst 1988 5 1 21 67 3293807
- Shaikh R Raj Singh TR Garland MJ Woolfson AD Donnelly RF Mucoadhesive drug delivery systems J Pharm Bioallied Sci 2011 3 1 89 100 21430958
- Roldo M Hornof M Coliceti P Bernkop-Schnürch A Mucoadhesive thiolated chitosans as platforms for oral controlled drug delivery: synthesis and in vitro evaluation Eur J Pharm Biopharm 2004 57 1 115 121 14729087
- Streubel A Siepmann J Bodmeier R Drug delivery to the upper small intestine window using gastroretentive technologies Curr Opin Phar-macol 2006 6 5 501 508
- Middleton DL Leung SS Robinson JR Ocular bioadhesive delivery systems Lenaerts V Gurny R Bioadhesive Drug Delivery Systems Boca Raton (FL) CRC Press Inc 1990 179 202