
Abstract
Idiopathic pulmonary fibrosis (IPF) is the most common type of idiopathic interstitial pneumonia and is characterized by a poor prognosis, with an estimated 5-year survival of approximately 20%. Progressive and irreversible lung functional impairment leads to chronic respiratory insufficiency with a severely impaired quality of life. In the last 2 decades, novel treatments for IPF have been developed as a consequence of an increasing understanding of disease pathogenesis and pathobiology. In IPF, injured dysfunctional alveolar epithelial cells promote fibroblast recruitment and proliferation, resulting in scarring of the lung tissue. Recently, pirfenidone and nintedanib have been approved for the treatment of IPF, having shown efficacy to slow functional decline and disease progression. This article focuses on the pharmacologic characteristics and clinical evidence supporting the use of nintedanib, a potent small-molecule tyrosine kinase inhibitor, as therapy for IPF. After introducing the mechanism of action and pharmacokinetics, an overview of the safety and efficacy results from the most recent clinical trials of nintedanib in IPF is presented.
Introduction: epidemiology, diagnosis, and pathogenesis of idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is the most common type of idiopathic interstitial pneumonias, a heterogeneous group of chronic progressive disorders involving primarily the lung parenchyma. IPF mainly occurs in older male adults, on average 60–70 years old, with a history of smoking. The predominant symptom is shortness of breath, initially only during activity but in advanced disease also at rest. Dry cough is also present, and can heavily affect patients’ quality of life. The prognosis of IPF is poor, with a median survival time of 3–5 years from the time of diagnosis.Citation1 Epidemiologic data were derived principally from national registries and reports from interstitial lung disease (ILD)/IPF referral centers. In a recent review of the epidemiology of IPF, Hutchinson et alCitation2 reported that the incidence of IPF is increasing worldwide and is similar to that of conditions such as stomach, liver, testicular, and cervical cancer. The overall range of incidence of IPF varies from 0.22 to 93.7 per 100,000 per year worldwide, but excluding old studies and different populations (Asia and South America) is likely to be 2.8–9.3 per 100,000 per year in Europe and North America.Citation2 The prevalence of IPF is estimated to be between 200 and 500 cases per 100,000 people.Citation3,Citation4
The diagnosis of IPF requires the exclusion of known causes of pulmonary fibrosis and the presence of a typical radiologic and pathologic pattern called usual interstitial pneumonia (UIP).Citation5 Radiologically, UIP pattern is characterized by subpleural, basal-predominant reticular abnormalities and honeycombing, with or without traction bronchiectasis; the histology shows patchy involvement of the lung parenchyma by fibrosis and honeycombing in a predominantly subpleural/paraseptal distribution and the presence of fibroblastic foci.Citation5 In the presence of a definite UIP pattern on high-resolution computed tomography (HRCT), a typical clinical setting, and after exclusion of systemic disease causing fibrosis, a diagnosis of IPF is usually possible without performing lung biopsy. Bronchoalveolar lavage can be a useful tool to exclude other fibrotic lung diseases presenting with UIP pattern, like hypersensitivity pneumonitis.Citation6 In recent years, a new technique for bronchoscopic lung biopsy has been developed using flexible cryoprobes. In patients with suspected diffuse parenchymal lung disease, bronchoscopic cryobiopsy is a promising and minimally invasive approach to obtain lung tissue with high diagnostic yield.Citation7,Citation8 In any case, the diagnosis of IPF should be confirmed by a specialist center. It has been demonstrated that the accuracy of the diagnosis of IPF increases with multidisciplinary discussion among pulmonologists, radiologists, and pathologists experienced in the diagnosis of ILD.Citation5
Looking at the last 2 decades in the history of IPF, it is clear that the development of new drugs has occurred simultaneously with a growing understanding of disease pathogenesis. IPF is a complex multipathway and multigene disease. The current model regarding the pathogenesis of IPF implies aberrant fibrosis as a consequence of recurrent injury to alveolar epithelial cells in a susceptible host ().Citation9,Citation10 The excessive deposition of extracellular matrix (ECM), with consequent irreversible lung remodeling and honeycombing, is likely to be the result of different processes. Gene polymorphisms and transcriptional changes provide the background, linked to the inability of epithelial cells to respond appropriately to repetitive microinjuries like infections, chronic aspiration, tobacco, or mechanical stress.Citation11 Abnormal telomere shortening, as well as epigenetic mechanisms involving DNA methylation, histone tails modification, and dysregulation of microRNA expression, take place in aging lungs and lead to loss of epithelial integrity and epithelial senescence.Citation9 Further susceptibility gene variants have been identified in the gene encoding the toll-interacting protein (TOLLIP), an important regulator of innate immunity, and in the Mucin 5B encoding gene (MUC5B), one of the major gel-forming proteins in human airway secretions.Citation11 Interestingly, individuals with the susceptibility MUC5B rs35705950_T allele or the major TOLLIP rs5743890_A allele who develop IPF seem to have decreased mortality.Citation12,Citation13
Figure 1 Schematic representation of the pathogenetic mechanisms underlying IPF.
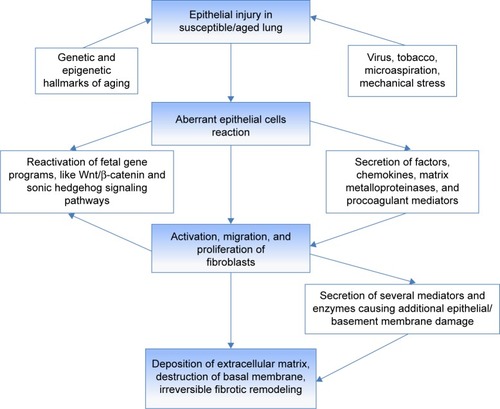
In an attempt to restore functional integrity, injured Type II alveolar epithelial cells aberrantly release pleonastic cytokines and growth factors, matrix metalloproteinases (MMPs), and procoagulant mediators, which promote the recruitment and activation of apoptosis-resistant fibroblasts, the key players in fibrotic tissue remodeling.Citation9,Citation10 Fibroblasts/myofibroblasts secrete several mediators and enzymes that provoke additional epithelial/basement membrane damage and perpetuate the aberrant wound healing response. Senescence of the immune system (production of autoantibodies, increased expression of toll-like-receptor,Citation9 downregulation of specific subtypes of T-cells) has been proposed as one of the most important factors inducing progression of IPF.Citation10 An increase in several microRNAs and noncoding RNAs influencing gene expression and a decrease in Argonaute (AGO)-1 and AGO2, which are indispensable for miRNA processing, have been revealed in lungs from patients with IPF with rapid progression in comparison to patients with stable IPF.Citation9,Citation10 Identifying signatures of disease progression, through circulating proteins alone or in combination, specific genotypes of single nucleotide polymorphisms, or epigenetic biomarkers, will be crucial to develop better stratification tools for patients and drive therapeutic decisions targeted to individual profiles. The knowledge deriving from translational biomarkers research will provide an important source of targets for drug development, because such proteins are seldom only an epiphenomenon.Citation14 However, one of the major problems in developing new treatments for IPF is the remarkable redundancy of the pathways involved in its pathogenesis. Similar to cancer, inhibiting single mediators or signaling pathways is largely ineffective in reducing the progression of IPF.Citation15
Management of IPF
The therapeutic approach for IPF integrates both nonpharmacologic and pharmacologic strategies.Citation5,Citation15 The goals of treatment in IPF are essentially to stop disease progression, reduce symptoms, prevent acute exacerbations – a life-threatening complication occurring in approximately 5%–15% of patientsCitation16,Citation17 – and finally prolong survival. Preventive care, rehabilitation, and symptom-based treatment should be started early in every patient to fight decline in quality of life.Citation18,Citation19 Inactivity due to shortness of breath leads to loss of muscular mass (sarcopenia) and chronic disabling fatigue in patients with IPF. Pulmonary rehabilitation may not only alleviate symptoms by reducing the threshold of dyspnea, but also improve functional status by stabilizing and/or reversing the extrapulmonary features of the disease.Citation15,Citation20 Patients should be referred to rehabilitation as standard of care as soon as possible after diagnosis, before the impact of symptoms on health-related quality of life becomes irreversible.Citation21,Citation22 Long-term oxygen treatment is essential for patients with resting or nocturnal hypoxemia.Citation5,Citation23 Although there are no data from randomized clinical trials on the utility of oxygen treatment in IPF, it has been demonstrated that oxygen therapy may improve quality of life by impacting physical and social performance.Citation23
Lung transplantation is an established therapeutic option for chronic lung diseases, but patients with IPF represent only 35% of transplant recipients.Citation24 In patients with IPF, lung transplant has been shown to reduce the risk of death by 75% compared with patients who remain on the waiting list.Citation25 Five-year survival rates range from 50% to 56%, with transplanted patients with IPF appearing to have more favorable long-term survival than those transplanted for other indications.Citation26 The efficacy of single- or double-lung transplantation in patients with IPF is still under debate. In a recently published exploratory analysis of transplant registry data from the USA, double-lung transplantation was associated with better graft survival (median: 65 months) than single-lung transplantation (50 months, P<0.001) in 4,134 patients with IPF.Citation27
In recent years, the landscape of pharmacologic treatment of IPF has completely changed. On the one hand, the definitive results of the PANTHER-IPF trial, a randomized, three-arm trial of prednisone, azathioprine, and N-acetylcysteine (NAC) in combination, NAC alone, or placebo, showed no effect of NAC monotherapy on the rate of change in forced vital capacity (FVC) in patients with IPF over a 60-week period.Citation28 On the other hand, the efficacy of the two antifibrotic molecules pirfenidone (Esbriet®, manufactured by F. Hoffmann-La Roche AG, Basel, Switzerland) and nintedanib (Ofev®, manufactured by Boehringer Ingelheim Pharma GmbH & Co. KG, Ingelheim, Germany) in slowing the progression of IPF was confirmed by the Phase III ASCEND trialCitation29 and the twin Phase III INPULSIS-1 and -2 trials,Citation30 respectively. On the basis of the results of these trials, in patients with IPF and mild or moderate impairment in FVC, both pirfenidone and nintedanib reduce decline in FVC by approximately 50% in 1 year, with acceptable safety profiles. This led to the approval of both medications for the treatment for IPF in the USA in 2014 and to the approval of nintedanib in Europe at the beginning of 2015 (pirfenidone had already been approved in Europe). Because of the slightly different study populations included in the trials of nintedanib and pirfenidone, it would be imprudent to compare these trials to contrast the efficacy of these agents. The recently updated ATS/ERS/JRS/ALAT clinical practice guideline gives conditional recommendations for the use of nintedanib and pirfenidone in the majority of patients with IPF.Citation31 No head-to-head studies comparing nintedanib and pirfenidone have been conducted, and the new recommendations did not recommend one treatment over the other.Citation31 Rather, the guideline emphasizes the need to consider individual patients’ values and preferences in making treatment decisions. Potential side effect profiles, comorbidities, and lifestyle should also be taken into account.
Pirfenidone is a small molecule with antifibrotic and anti-inflammatory properties.Citation32,Citation33 Its safety and efficacy have been evaluated in five randomized clinical trials in a total of 1,710 patients.Citation29,Citation34–Citation36 The ASCEND trial, in which 555 patients with IPF were randomized to receive either the maximum oral dose of pirfenidone (2,403 mg/d) or placebo, confirmed that pirfenidone significantly reduced FVC decline from baseline to week 52. Pirfenidone also significantly reduced the decline in 6-minute walk test (6MWT) distance and reduced disease progression, defined as an absolute decrease in FVC % predicted of ≥10% and/or a decrease in 6MWT distance of ≥50 m or death by 43% compared with placebo. The most frequent side effects associated with pirfenidone are gastrointestinal and skin related, including photosensitivity reactions and skin rash.Citation29,Citation35
Specific considerations for potential combination therapy in IPF
In various respiratory diseases such as asthma, chronic obstructive pulmonary disease, pulmonary hypertension, and lung cancer, combination regimens have been successfully established. With two antifibrotic drugs available for the treatment of IPF, it might be a logical next step to combine pirfenidone and nintedanib to obtain synergistic effects. In a randomized, double-blind, Phase II, dose escalation trial to assess the safety, tolerability, and pharmacokinetics of nintedanib in 50 Japanese patients with IPF stratified by preexisting pirfenidone treatment, a trend toward lower exposure of nintedanib was observed when nintedanib was added to pirfenidone compared to when it was given alone.Citation37 Although this study provides first data on concomitant treatment with nintedanib and pirfenidone, no conclusions on safety and efficacy can be drawn due to the low number of patients and the limited treatment duration (maximum of 28 days). Concomitant treatment with nintedanib and pirfenidone cannot be recommended until there are data to demonstrate its efficacy and tolerability/safety in patients with IPF.
Clinical pharmacokinetics of nintedanib
The clinical pharmacokinetics of nintedanib (formerly known as BIBF 1120) have been studied in healthy volunteers,Citation38 and in patients with cancer or IPF in a number of Phase ICitation39–Citation46 and IICitation37,Citation47,Citation48 clinical trials as monotherapy and in combination with other agents. After oral administration, nintedanib is rapidly absorbed. After 1–3 hours, maximum plasma concentration (Cmax) is achieved.Citation38,Citation40 The mean terminal half-life of nintedanib is 13–28 hours.Citation37,Citation38,Citation40 Overall drug exposure of nintedanib (area under the plasma concentration–time curve [AUC]) show dose-proportional increases.Citation40 No decrease in exposure was observed over a minimum 6-month treatment period.Citation40 Following multiple twice-daily administration of nintedanib, only slight accumulation of nintedanib exposure was observed, with a mean accumulation ratio of 1.66 based on AUC0–12 h and 1.33 based on Cmax.Citation49 The mean Cmax at steady state at the standard dosing in IPF of 150 mg twice daily is in the range of 35–40 ng/mL, which is approximately 64–74 nmol/L (FW =539.62 g/mol).Citation37,Citation40 Interpatient variability of parameters for nintedanib is moderate to high.Citation38,Citation40,Citation49 The apparent volume of distribution of nintedanib during the terminal phase is high, indicating a high tissue distribution of the drug and a high apparent total body clearance.Citation38 The major route of nintedanib elimination is through metabolism, with metabolites excreted via the biliary system into the feces. Urinary excretion for nintedanib is minor.Citation38 The metabolism of nintedanib has been explored in healthy volunteers. Nintedanib is cleaved at the methyl ester moiety, resulting in a carboxylate, which is then conjugated to glucuronic acid and in a further step to the 1-O-acylglucuronide. Hence, the metabolism of nintedanib is mainly CYP450 enzyme independent.Citation50
Mechanism of action in IPF
Nintedanib is a potent small-molecule receptor tyrosine kinase inhibitor targeting platelet-derived growth factor receptor (PDGFR)α and β (IC50 [half maximal inhibitory concentration] =59 and 65 nmol/L); fibroblast growth factor receptor (FGFR)-1, -2, -3, and -4 (IC50=69, 37, 108, and 610 nmol/L); and vascular endothelial growth factor receptor (VEGFR)-1, -2, and -3 (IC50=34, 21, and 13 nmol/L).Citation51 Nintedanib binds competitively to the ATP binding pocket of these receptors and so blocks intracellular signaling. Nintedanib also inhibits the proto-oncogene tyrosine-protein kinase Src (IC50=156 nmol/L), lymphocyte-specific tyrosine-protein kinase Lck (IC50=16 nmol/L), Lck/Yes novel tyrosine kinase Lyn (IC50=195 nmol/L), and Fms-like tyrosine kinase 3 Flt-3 (IC50=26 nmol/L).Citation51 Distinct functions that may impact the pathology of IPF have been described for specific tyrosine kinases. However, the exact contribution of inhibition of specific kinases to the activity of nintedanib in IPF has not been established.Citation52
Role of targeted growth factors in IPF
PDGF is a potent mitogen for fibroblastsCitation53 and is produced by alveolar macrophages and epithelial cells.Citation54,Citation55 PDGF appears to play an essential role in the expansion of myofibroblasts by stimulating proliferation, migration, and survival and acts as a stimulator of collagen synthesis.Citation54–Citation58 The result is a pathologic remodeled alveolar architecture with deteriorated gas exchange.Citation5,Citation16,Citation59 Inhibition of PDGFR with specific compounds reduces pulmonary fibrosis in a variety of animal models.Citation60–Citation64
FGFR-1 is expressed on epithelial cells, endothelial cells, smooth muscle cells, myofibroblast-like cells, and macrophages in the lungs of patients with IPF, and FGFR-2 on smooth muscle cells, myofibroblast-like cells, and neutrophils.Citation65 FGF-2 stimulates proliferation of lung fibroblasts from patients with IPF.Citation66 In vivo abrogation of FGF signaling reduces bleomycin-induced pulmonary fibrosis and improves survival in bleomycin-treated mice.Citation67 However, recent reports also describe a protective role of certain FGFs.Citation68–Citation70
The role of VEGF signaling in IPF is unclear. VEGF stimulates human lung fibroblast proliferation less potently than FGF or PDGF.Citation71 However, anti-VEGF gene therapy attenuates bleomycin-induced fibrosis in mice.Citation72 VEGF signaling is a prominent pathway in vascular remodeling, which is a pathologic feature in IPF, but it is controversial whether angiogenesis plays a key role in abnormal ECM remodeling and fibrosis in the lung,Citation73,Citation74 and extensive temporal and spatial heterogeneity in angiogenesis has been observed in patients with IPF.Citation75,Citation76 The balance between angiostatic and angiogenic mediators in patients with IPF remains controversial.Citation77
The roles of these growth factor receptors with respect to potential adverse events are unclear due to a lack of inhibitors specific for single kinases. Although certain so-called “class effects” like gastrointestinal disturbances or liver enzyme elevations have been proposed for VEGFR inhibitors,Citation78 such effects vary considerably in frequency and intensity between compounds and are not observed for all VEGFR inhibitors. No consistent pattern of “class effects” can be deduced for the available multispecific PDGFR inhibitors, but their most common side effects appear to be nausea and vomiting.Citation79 For FGFR inhibition, hyperphosphatemia seems to be a potential adverse effect.Citation80
Nintedanib interferes with fundamental processes in lung fibrosis in a variety of in vitro assays
Nintedanib has been shown to inhibit growth factor-induced proliferation and migration of primary human lung fibroblasts from patients with IPF (IPF-HLF) or control donors (N-HLF). Depending on the cell type and growth factor used, the IC50 values vary from below 1 to 226 nmol/L (). Nintedanib inhibited transforming growth factor β-induced fibroblast to myofibroblast transformation of IPF-HLF, as determined by α smooth muscle actin mRNA expression ().
Table 1 Activity of nintedanib in in vitro assays of lung fibrosis
Few data are available on the direct activity of nintedanib on ECM secretion in vitro. At high concentrations (1 μmol/L), nintedanib reduced TGFβ-stimulated collagen secretion and deposition by IPF-HLF. Nintedanib also reduced secreted tissue inhibitor of metalloproteinase (TIMP)-2 levels.Citation71 However, the enzymatic activity and secretion of MMP-2 were significantly reduced at lower concentrations (approximately 1 and 100 nmol/L, respectively).Citation71 Additionally, nintedanib is expected to have indirect activity on ECM secretion and deposition by reducing the number of fibroblasts/myofibroblasts in fibrotic lungs. Nintedanib attenuated proliferation of cell types involved in neoangiogenesis, endothelial cells, vascular smooth muscle cells, and pericytes, with IC50 values ranging from below 7 to 290 nmol/L ().
Antifibrotic and anti-inflammatory activity of nintedanib in animal models of lung fibrosis
The in vivo efficacy of nintedanib has been explored in three animal models: bleomycin-induced lung fibrosis in mice and rats and silica-induced lung fibrosis in mice. Pharmacokinetic/pharmacodynamic correlation experiments confirmed the effective dose in mice to be in the range of 30–100 mg/kg.Citation81 Nintedanib was administered orally, once daily, in both a preventive regimen starting directly after bleomycin or silica stimulation and a therapeutic regimen starting after the onset of fibrotic lung alteration. In general, nintedanib reduced lung inflammation and lung fibrosis across the animal models. The parameters explored and the extent of inhibitory activity differed between models. A daily dose of 30–100 mg/kg nintedanib in mice and 10–50 mg/kg in rats was efficacious and tolerated when administered for 10–30 days. In summary, nintedanib reduced lung inflammation demonstrated by reduced lymphocyte counts in the bronchoalveolar lavage fluid, interleukin-1β, chemokine (C-X-C motif) ligand 1/keratinocyte chemoattractant detected in lung tissue; diminished percentage of myeloid dendritic cells in lung tissue; and semiquantitative histologic analysis of hematoxylin and eosin-stained micrographs of the lungs. Furthermore, nintedanib diminished lung fibrosis demonstrated by reduced TIMP-1 and total collagen levels in lung tissue, by semiquantitative histologic analysis of the lungs and by reduction of mRNA expression of fibrosis-related marker genes (TGF-β1; procollagen I).
Recently, nintedanib has been shown to reduce the extent of missing integrity of the vascular architecture and to reduce the increase in vessel density and the diameter of enlarged vessels in bleomycin-induced vascular remodeling in fibrotic mouse lungs;Citation82 however, whether this antiangiogenic efficacy adds to its antifibrotic activity in IPF still needs to be elucidated. Nintedanib might also exert acute vasodilatory activity on the pulmonary circulation. The effect of nintedanib on endothelin-1 preconstricted pulmonary vessels was recently explored in precision-cut lung slices and isolated perfused lungs from mice. Nintedanib relaxed the pulmonary arterial bed significantly.Citation83 Whether this holds true for human pulmonary vessels remains unknown.
Although substantial preclinical evidence demonstrates that nintedanib has antifibrotic, anti-inflammatory, and antiangiogenic activity, it has not been possible to map specific inhibition of single kinases by nintedanib to precise functional outcomes.
Clinical efficacy, safety, and tolerability of nintedanib
Data from Phase II and III clinical trials in IPF
Nintedanib has been investigated in three international Phase II (TOMORROW) and Phase III clinical trials (INPULSIS®-1 and INPULSIS®-2) including almost 1,500 patients with IPF.Citation30,Citation84 These have been followed by open-label extension trials (NCT01170065 and INPULSIS®-ON) to provide a treatment option to all patients who completed the TOMORROW or INPULSIS® trials and to assess long-term safety and tolerability. In addition, the Japanese Phase II study that assessed the safety, tolerability, and pharmacokinetic profile of nintedanib when given alone or in addition to preexisting pirfenidone treatmentCitation37 was followed by an open-label extension trial (NCT01417156; ). Period 1 of the dose-finding TOMORROW trial suggested that 52 weeks’ treatment with nintedanib 150 mg bid results in reduced decline in FVC, fewer acute exacerbations, and preservation of health-related quality of life versus placebo.Citation84 On the basis of these results and an acceptable safety profile, a dose of nintedanib 150 mg bid, with the option to reduce the dose to 100 mg bid and/or interrupt treatment for the management of adverse events, was chosen as the treatment regimen for the replicate Phase III INPULSIS® trials. In the INPULSIS® trials, 1,066 patients from 24 countries were randomized in a 3:2 ratio to 52 weeks of treatment with nintedanib or placebo.Citation30 As in the TOMORROW trial, the primary end point was the annual rate of decline in FVC as a measure of disease progression. Key secondary end points were change in total score on the St George’s Respiratory Questionnaire (SGRQ) over 52 weeks and time to first investigator-reported acute exacerbation. Acute exacerbations adjudicated as confirmed or suspected acute exacerbations in pooled data from both trials was a prespecified sensitivity analysis. On the basis of inclusion and exclusion criteria, a broad range of patients was eligible, including patients with radiologic emphysema,Citation85 patients with mild FVC impairment at baseline,Citation86 ie, FVC >90% predicted, and patients with features of possible UIP on HRCT scans without surgical biopsy.Citation87
Table 2 Clinical development program for nintedanib in IPF
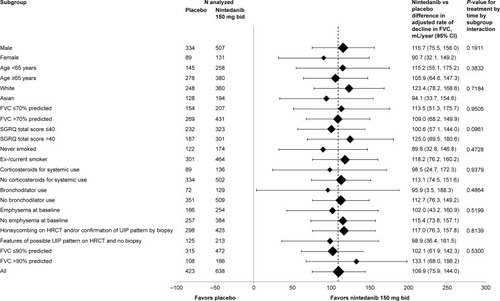
In both INPULSIS® trials, the primary outcome was achieved, with a reduction in the annual rate of FVC decline of approximately 50% in the nintedanib group compared to placebo. In a pooled analysis of data from the two trials, the adjusted annual rate of decline in FVC was 114 mL in the nintedanib group versus 224 mL in the placebo group (mean difference 110 mL/yr, P<0.001; ). The robustness and consistency of the results on the primary outcome were confirmed in a number of sensitivity analyses,Citation88 across outcomes for changes in FVC (change from baseline, categorical changes),Citation89 across various prespecifiedCitation90 and post hoc subgroup analyses, eg, defined by radiologic presence/absence of emphysema,Citation85 by baseline FVC % predicted (≤90% versus >90%)Citation86 and by HRCT diagnostic criteria (honeycombing on HRCT and/or confirmation of diagnosis by biopsy versus patients with features of possible UIP on HRCT and no biopsy; ).Citation87 Furthermore, the effect of nintedanib on slowing the decline in FVC in patients with IPF was maintained up to week 76 during period 2 of the TOMORROW trialCitation91 and supported by a numerical risk reduction in all-cause mortality of 30% (HR [hazard ratio] =0.70; P=0.095) in a pooled analysis of data from the TOMORROW and INPULSIS® trials.Citation92
Figure 2 Mean (SEM) observed change from baseline FVC (mL) over time in pooled data from the INPULSIS® trials.
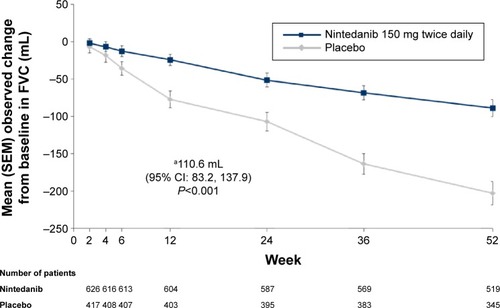
Figure 3 Forest plot for the annual rate of decline in FVC (mL/year) over 52 weeks by subgroups analyzed in pooled data from the INPULSIS® trials.
Abbreviations: FVC, forced vital capacity; CI, confidence interval; SGRQ, St George’s Respiratory Questionnaire; HRCT, high-resolution computed tomography; UIP, usual interstitial pneumonia.
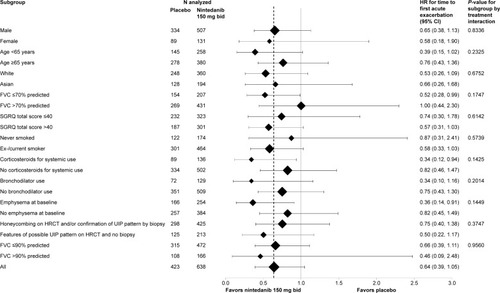
Nintedanib significantly reduced the risk of a first investigator-reported acute exacerbation versus placebo in INPULSIS®-2 (HR =0.38; P<0.01), but there was no difference in INPULSIS®-1 (HR =1.15; P=0.67). In the pre-specified sensitivity analysis based on adjudicated confirmed or suspected acute exacerbations, there was a statistically significant difference in favor of nintedanib, with a reduction in risk of 68% (HR =0.32; P<0.01).Citation30 In various prespecified and post hoc subgroup analyses based on pooled data from both trials, the treatment effect of nintedanib on time to first investigator-reported acute exacerbation was generally similar.Citation85–Citation87,Citation90 A more pronounced treatment effect was seen in patients with more advanced restrictive lung disease (FVC ≤70% predicted at baseline) in whom the majority of events were observed; investigator-reported acute exacerbations occurred in 7.7% and 14.9% of nintedanib- and placebo-treated patients with baseline FVC ≤70% predicted, and in 3.5% and 3.3% of nintedanib- and placebo-treated patients with baseline FVC >70% predicted, respectively ().Citation90 The mean difference in change from baseline in SGRQ total score at week 52 was −2.69 points in INPULSIS®-2 (P,0.02) and −0.05 points in INPULSIS®-1 (P=0.97), in favor of nintedanib, resulting in a numerical difference of −1.43 points in the pooled data set (P=0.09), which was consistent across subgroups.Citation85–Citation87,Citation90
Figure 4 Forest plot for time to first investigator-reported acute exacerbation (days) over 52 weeks by subgroups analyzed in pooled data from the INPULSIS® trials.
Abbreviations: HR, hazard ratio; CI, confidence interval; FVC, forced vital capacity; SGRQ, St George’s Respiratory Questionnaire; HRCT, high-resolution computed tomography; UIP, usual interstitial pneumonia.
The most frequently reported adverse event in the INPULSIS® trials was diarrhea, which was reported in 62% of patients treated with nintedanib compared to 18% in the placebo group. Diarrhea adverse events were mild to moderate in intensity in the vast majority of patients and led to premature treatment discontinuation in fewer than 5% of nintedanib-treated patients.Citation30 More than 90% of eligible patients (734/807) chose to receive open-label nintedanib in the ongoing INPULSIS®-ON extension trial, which recently confirmed the long-term safety and tolerability profile of nintedanib in an interim analysis.Citation93
Conclusion
In the last decade, growing knowledge of pathogenetic mechanisms underlying IPF has led to new treatments for this fatal disease. Nintedanib is a potent tyrosine kinase inhibitor with a distinct specificity targeting growth factors involved in fibrotic changes in the lungs of patients with IPF. The clinical efficacy of nintedanib in slowing disease progression has been shown in three clinical trials, resulting in the approval of this medication as a treatment for IPF independent of the extent of lung functional impairment.
Acknowledgments
Editorial assistance (edit by native speaker) was provided by Wendy Morris of Fleishman-Hillard Group, London, UK. The authors are responsible for all content decisions.
Disclosure
Francesco Bonella reports receipt of travel grants, research grants, speaker honoraria, and fees for serving on advisory boards from Boehringer Ingelheim, InterMune, Roche, Gilead, Centocor, and Serendex. Susanne Stowasser and Lutz Wollin are employees of Boehringer Ingelheim. The authors report no other conflicts of interest in this work.
References
- BjorakerJARyuJHEdwinMKPrognostic significance of histopathologic subsets in idiopathic pulmonary fibrosisAm J Respir Crit Care Med199815711992039445300
- HutchinsonJFogartyAHubbardRMcKeeverTGlobal incidence and mortality of idiopathic pulmonary fibrosis: a systematic reviewEur Respir J201546379580625976683
- RaghuGChenSYYehWSIdiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11Lancet Respir Med20142756657224875841
- NavaratnamVFlemingKMWestJThe rising incidence of idiopathic pulmonary fibrosis in the U.KThorax201166646246721525528
- RaghuGCollardHREganJJAn official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and managementAm J Respir Crit Care Med2011183678882421471066
- OhshimoSBonellaFCuiASignificance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosisAm J Respir Crit Care Med2009179111043104719246718
- CasoniGLTomassettiSCavazzaATransbronchial lung cryobiopsy in the diagnosis of fibrotic interstitial lung diseasesPLoS One201492e8671624586252
- KropskiJAPritchettJMMasonWRBronchoscopic cryobiopsy for the diagnosis of diffuse parenchymal lung diseasePLoS One2013811e7867424265706
- SelmanMPardoAStochastic age-related epigenetic drift in the pathogenesis of idiopathic pulmonary fibrosisAm J Respir Crit Care Med2014190121328133025496096
- SelmanMPardoARevealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral modelAm J Respir Crit Care Med2014189101161117224641682
- SpagnoloPGrunewaldJdu BoisRMGenetic determinants of pulmonary fibrosis: evolving conceptsLancet Respir Med20142541642824815806
- PeljtoALZhangYFingerlinTEAssociation between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosisJAMA2013309212232223923695349
- NothIZhangYMaSFGenetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association studyLancet Respir Med20131430931724429156
- CampoIZorzettoMBonellaFFacts and promises on lung biomarkers in interstitial lung diseasesExpert Rev Respir Med20159443745726146183
- TzouvelekisABonellaFSpagnoloPUpdate on therapeutic management of idiopathic pulmonary fibrosisTher Clin Risk Manag20151135937025767391
- KimDSParkJHParkBKLeeJSNicholsonAGColbyTAcute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical featuresEur Respir J200627114315016387947
- SongJWHongSBLimCMKohYKimDSAcute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcomeEur Respir J201137235636320595144
- CerriSSpagnoloPLuppiFRicheldiLManagement of idiopathic pulmonary fibrosisClin Chest Med2012331859422365248
- LeeJSMcLaughlinSCollardHRComprehensive care of the patient with idiopathic pulmonary fibrosisCurr Opin Pulm Med201117534835421760508
- NishiyamaOKondohYKimuraTEffects of pulmonary rehabilitation in patients with idiopathic pulmonary fibrosisRespirology200813339439918399862
- VainshelboimBOliveiraJFoxBDSoreckYFruchterOKramerMRLong-term effects of a 12-week exercise training program on clinical outcomes in idiopathic pulmonary fibrosisLung2015193334535425731736
- VainshelboimBOliveiraJYehoshuaLExercise training-based pulmonary rehabilitation program is clinically beneficial for idiopathic pulmonary fibrosisRespiration201488537838825341682
- EganJJFollow-up and nonpharmacological management of the idiopathic pulmonary fibrosis patientEur Respir Rev20112012011411721632799
- YusenRDEdwardsLBKucheryavayaAYThe registry of the International Society for Heart and Lung Transplantation: thirty-first adult lung and heart-lung transplant report – 2014; focus theme: retransplantationJ Heart Lung Transplant201433101009102425242125
- RussoMJIribarneAHongKNHigh lung allocation score is associated with increased morbidity and mortality following transplantationChest2010137365165719820072
- GottliebJLung transplantation for interstitial lung diseasesCurr Opin Pulm Med201420545746225058363
- SchafferJMSinghSKReitzBAZamanianRTMallidiHRSingle- vs double-lung transplantation in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis since the implementation of lung allocation based on medical needJAMA2015313993694825734735
- Idiopathic Pulmonary Fibrosis Clinical Research NetworkMartinezFJde AndradeJAAnstromKJKingTEJrRaghuGRandomized trial of acetylcysteine in idiopathic pulmonary fibrosisN Engl J Med2014370222093210124836309
- KingTEJrBradfordWZCastro-BernardiniSA phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosisN Engl J Med2014370222083209224836312
- RicheldiLdu BoisRMRaghuGEfficacy and safety of nintedanib in idiopathic pulmonary fibrosisN Engl J Med2014370222071208224836310
- RaghuGRochwergBZhangYAn official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis: executive summary: an update of the 2011 clinical practice guidelinesAm J Respir Crit Care Med20151922238248
- SchaeferCJRuhrmundDWPanLSeiwertSDKossenKAntifibrotic activities of pirfenidone in animal modelsEur Respir Rev201120120859721632796
- HilbergOSimonsenUdu BoisRBendstrupEPirfenidone: significant treatment effects in idiopathic pulmonary fibrosisClin Respir J20126313114322697264
- AzumaANukiwaTTsuboiEDouble-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosisAm J Respir Crit Care Med200517191040104715665326
- NoblePWAlberaCBradfordWZPirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trialsLancet201137797791760176921571362
- TaniguchiHEbinaMKondohYPirfenidone in idiopathic pulmonary fibrosisEur Respir J201035482182919996196
- StopferPRathgenKBischoffDPharmacokinetics and metabolism of BIBF 1120 after oral dosing to healthy male volunteersXenobiotica201141429731121204634
- StopferPRothWMrossKPharmacokinetic characterization of BIBF1120, an orally active triple angiokinase inhibitor (VEGFR, PDGFR, FGFR) in advanced cancer patientsEur J Cancer20154Suppl 26A73
- MrossKStefanicMGmehlingDPhase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumorsClin Cancer Res201016131131920028771
- OkamotoIMiyazakiMTakedaMTolerability of nintedanib (BIBF 1120) in combination with docetaxel: a phase 1 study in Japanese patients with previously treated non-small-cell lung cancerJ Thorac Oncol201510234635225299232
- KropffMKienastJBispingGAn open-label dose-escalation study of BIBF 1120 in patients with relapsed or refractory multiple myelomaAnticancer Res200929104233423819846979
- DoebeleRCConklingPTraynorAMA phase I, open-label dose-escalation study of continuous treatment with BIBF 1120 in combination with paclitaxel and carboplatin as first-line treatment in patients with advanced non-small-cell lung cancerAnn Oncol20122382094210222345119
- EllisPMKaiserRZhaoYStopferPGyorffySHannaNPhase I open-label study of continuous treatment with BIBF 1120, a triple angiokinase inhibitor, and pemetrexed in pretreated non-small cell lung cancer patientsClin Cancer Res201016102881288920460487
- BousquetGAlexandreJLe TourneauCPhase I study of BIBF 1120 with docetaxel and prednisone in metastatic chemo-naive hormone-refractory prostate cancer patientsBr J Cancer2011105111640164522027711
- du BoisAHuoberJStopferPA phase I open-label dose-escalation study of oral BIBF 1120 combined with standard paclitaxel and carboplatin in patients with advanced gynecological malignanciesAnn Oncol201021237037519889612
- OguraTTaniguchiHAzumaASafety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosisEur Respir J20154551382139225504994
- ReckMKaiserREschbachCA phase II double-blind study to investigate efficacy and safety of two doses of the triple angiokinase inhibitor BIBF 1120 in patients with relapsed advanced non-small-cell lung cancerAnn Oncol20112261374138121212157
- Van CutsenEPrenenHGuillen-PonceCA phase I/II, open-label, randomized study of BIBF 1120 Plus mFOLFOX6 compared to bevacizumab plus mFOLFOX6 in patients with metastatic colorectal cancerEur J Cancer201147Suppl 214LBA
- EisenTShparykYMacleodNEffect of small angiokinase inhibitor nintedanib (BIBF 1120) on QT interval in patients with previously untreated, advanced renal cell cancer in an open-label, phase II studyInvest New Drugs20133151283129323625328
- RothGJBinderRColbatzkyFNintedanib: from discovery to the clinicJ Med Chem20155831053106325474320
- HilbergFRothGJKrssakMBIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacyCancer Res200868124774478218559524
- WollinLWexEPautschAMode of action of nintedanib in the treatment of idiopathic pulmonary fibrosisEur Respir J2015451434144525745043
- ClarkJGMadtesDKRaghuGEffects of platelet-derived growth factor isoforms on human lung fibroblast proliferation and procollagen gene expressionExp Lung Res19931933273448319603
- AntoniadesHNBravoMAAvilaREPlatelet-derived growth factor in idiopathic pulmonary fibrosisJ Clin Invest1990864105510642170444
- BonnerJCRegulation of PDGF and its receptors in fibrotic diseasesCytokine Growth Factor Rev200415425527315207816
- PanLHYamauchiKUzukiMType II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPFEur Respir J20011761220122711491168
- KhalilNO’ConnorRNUnruhHWIncreased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosisAm J Respir Cell Mol Biol1991521551621892646
- PiguetPFRibauxCKarpuzVGrauGEKapanciYExpression and localization of tumor necrosis factor-alpha and its mRNA in idiopathic pulmonary fibrosisAm J Pathol199314336516558362967
- KatzensteinALMyersJLIdiopathic pulmonary fibrosis: clinical relevance of pathologic classificationAm J Respir Crit Care Med19981574 Pt 1130113159563754
- AbdollahiALiMPingGInhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosisJ Exp Med2005201692593515781583
- AonoYNishiokaYInayamaMImatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in miceAm J Respir Crit Care Med2005171111279128515735062
- LiMAbdollahiAGröneHJLipsonKEBelkaCHuberPELate treatment with imatinib mesylate ameliorates radiation-induced lung fibrosis in a mouse modelRadiat Oncol200946620025728
- RiceABMoomawCRMorganDLBonnerJCSpecific inhibitors of platelet-derived growth factor or epidermal growth factor receptor tyrosine kinase reduce pulmonary fibrosis in ratsAm J Pathol1999155121322110393853
- VuorinenKGaoFOuryTDKinnulaVLMyllärniemiMImatinib mesylate inhibits fibrogenesis in asbestos-induced interstitial pneumoniaExp Lung Res200733735737317849262
- InoueYKingTEJrBarkerEDaniloffENewmanLSBasic fibroblast growth factor and its receptors in idiopathic pulmonary fibrosis and lymphangioleiomyomatosisAm J Respir Crit Care Med2002166576577312204879
- HetzelMBachemMAndersDTrischlerGFaehlingMDifferent effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblastsLung2005183422523716211459
- YuZHWangDDZhouZYHeSLChenAAWangJMutant soluble ectodomain of fibroblast growth factor receptor-2 IIIc attenuates bleomycin-induced pulmonary fibrosis in miceBiol Pharm Bull201235573173622687409
- GupteVVRamasamySKReddyROverexpression of fibroblast growth factor-10 during both inflammatory and fibrotic phases attenuates bleomycin-induced pulmonary fibrosis in miceAm J Respir Crit Care Med2009180542443619498056
- DeterdingRRHavillAMYanoTPrevention of bleomycin-induced lung injury in rats by keratinocyte growth factorProc Assoc Am Physicians199710932542689154642
- ShimboriCBellayePSGauldieJFibroblast growth factor-1 attenuates TGF-β1-induced lung fibrosis by regulating TGF-β1 signalingAm J Respir Crit Care Med2015191A2347
- HostettlerKEZhongJPapakonstantinouEAnti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosisRespir Res20141515725496490
- HamadaNKuwanoKYamadaMAnti-vascular endothelial growth factor gene therapy attenuates lung injury and fibrosis in miceJ Immunol200517521224123116002726
- KingTEJrPardoASelmanMIdiopathic pulmonary fibrosisLancet201137898071949196121719092
- RenzoniEANeovascularization in idiopathic pulmonary fibrosis: too much or too little?Am J Respir Crit Care Med2004169111179118015161610
- FarkasLKolbMPulmonary microcirculation in interstitial lung diseaseProc Am Thorac Soc20118651652122052930
- FarkasLGauldieJVoelkelNFKolbMPulmonary hypertension and idiopathic pulmonary fibrosis: a tale of angiogenesis, apoptosis, and growth factorsAm J Respir Cell Mol Biol201145111521057104
- CuiAAnhennOTheegartenDAngiogenic and angiostatic chemokines in idiopathic pulmonary fibrosis and granulomatous lung diseaseRespiration201080537237819816001
- RoodhartJMLangenbergMHWitteveenEVoestEEThe molecular basis of class side effects due to treatment with inhibitors of the VEGF/VEGFR pathwayCurr Clin Pharmacol20083213214318690886
- ArrondeauJHuillardOTlemsaniCInvestigational therapies up to Phase II which target PDGF receptors: potential anti-cancer therapeuticsExpert Opin Investig Drugs2015245673687
- WöhrleSBonnyOBeluchNFGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in boneJ Bone Miner Res201126102486249721812026
- WollinLMailletIQuesniauxVHolwegARyffelBAntifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosisJ Pharmacol Exp Ther2014349220922024556663
- AckermannMKimYOSchuppanDEffects of nintedanib on the microvascular architecture in a bleomycin-induced pulmonary fibrosis modelAm J Respir Crit Care Med2015191A4395
- RiegADWollinLKrabbeJUhligSMartinCVasorelaxant properties of nintedanib in the murine pulmonary circulationAm J Respir Crit Care Med2015191A1956
- RicheldiLCostabelUSelmanMEfficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosisN Engl J Med2011365121079108721992121
- CottinVTaniguchiHRicheldiLEffect of baseline emphysema on reduction in FVC decline with nintedanib in the INPULSIS trialsPaper presented at: ICLAF 2014, 18th International Colloquium on Lung and Airway Fibrosis (ICLAF)September 20–24, 2014Mont Tremblant, Canada Available from: http://www.iclaf.com/conference/index.php/2014/ICLAF/paper/view/151Accessed November 4, 2015
- KolbMRicheldiLKimuraTStowasserSHallmannCdu BoisRMEffect of baseline FVC on decline in lung function with nintedanib in patients with IPF: results from the INPULSIS trialsAm J Respir Crit Care Med2015191A1021
- RaghuGWellsANicholsonAGConsistent effect of nintedanib on decline in FVC in patients across subgroups based on HRCT diagnostic criteria: results from the INPULSIS trials in IPFAm J Respir Crit Care Med2015191A1022
- KolbMCollardHRStowasserSGirardMSchlenker-HercegRRicheldiLSensitivity analyses from the INPULSIS trials of nintedanibEur Respir J201444Suppl 58A1908
- CottinVTaniguchiHCollardHRReduction in disease progression with nintedanib in the INPULSIS trialsEur Respir J201444Suppl 58A1906
- CostabelUInoueYRicheldiLEfficacy of nintedanib in idiopathic pulmonary fibrosis across pre-specified subgroups in INPULSISAm J Respir Crit Care Med Epub9222015
- RicheldiLCostabelUSelmanMEfficacy and safety of nintedanib in patients with IPF beyond week 52: data from the Phase II TOMORROW trialAm J Respir Crit Care Med2015191A1019
- CottinVNintedanib: a new treatment for idiopathic pulmonary fibrosisClin Invest20155621632
- CrestaniBOguraTPellingKInterim analysis of nintedanib in an open-label extension of the INPULSIS trialsEur Respir J201546Suppl 59OA4495