
Abstract
In the era of personalized medicine, diagnostic approaches are helping pharmaceutical and biotechnology sponsors streamline the clinical trial process. Molecular assays and diagnostic imaging are routinely being used to stratify patients for treatment, monitor disease, and provide reliable early clinical phase assessments. The importance of diagnostic approaches in drug development is highlighted by the rapidly expanding global cancer diagnostics market and the emergent attention of regulatory agencies worldwide, who are beginning to offer more structured platforms and guidance for this area. In this paper, we highlight the key benefits of using companion diagnostics and diagnostic imaging with a focus on oncology clinical trials. Nuclear imaging using widely available radiopharmaceuticals in conjunction with molecular imaging of oncology targets has opened the door to more accurate disease assessment and the modernization of standard criteria for the evaluation, staging, and treatment responses of cancer patients. Furthermore, the introduction and validation of quantitative molecular imaging continues to drive and optimize the field of oncology diagnostics. Given their pivotal role in disease assessment and treatment, the validation and commercialization of diagnostic tools will continue to advance oncology clinical trials, support new oncology drugs, and promote better patient outcomes.
Clinical trial paradigm shift
Traditional approaches to drug development and clinical trials based on the premise of “one size fits all” are becoming less cost-effective and suboptimal clinically.Citation1 This is due mostly to the escalating costs required to develop a new drug, diminishing returns on drug investments, and a high rate of failure for Phase III clinical trials, particularly in oncology drug development.Citation2 As a result, the number of new drugs approved by the US Food and Drug Administration (FDA) has remained relatively flat and the number of new drug submissions has decreased significantly over the last decade.Citation3 The commitment of the FDA to drive innovation in drug development through its Critical Path Initiative appears to have contributed to a modest uptick in new drug approvals in the last several years.
In line with the FDA initiative to improve the efficiency of drug development and the success rates of late-phase studies, drug sponsors require methodologies that can shorten the length of clinical trial cycles, reduce the number of human subjects required, and provide more reliable early clinical phase assessments for go/no go trial decisions. Many sponsor companies are using companion diagnostic assays and diagnostic imaging studies to help streamline the clinical trial process.
Companion diagnostic assays (also referred to in the literature as pharmacodiagnostics or theranostics) provide a test that can identify the presence or absence of a biomarker that is predictive of a patient’s phenotype. This approach relies on a detailed understanding of the molecular basis of disease in an individual patient that can subsequently be used to follow-up with a tailored course of treatment based on the presence of specific disease biomarkers. Different classes of biomarkers include somatic mutations, polymorphisms, and gene/protein expression profiles that are associated with a particular disease state. In addition to identifying patients likely to respond to a personalized treatment approach, the incorporation of a diagnostic imaging technique or a diagnostic imaging study in clinical trials allows clinicians and scientists to non-invasively assess the presence, location, and extent of disease for objective, quantitative monitoring of disease progression and response to treatments.
Diagnostic approaches provide key benefits
Throughout the clinical trial process, the ability to detect and visualize patient biomarkers using companion diagnostic assays and diagnostic imaging tools provides clinicians and drug developers with tools that facilitate faster, safer, and more efficient clinical trials (). Early on, they can be used to determine and optimize trial eligibility and enrollment by confirming the presence and quantity of a drug target in an individual patient. During a clinical trial, companion diagnostic assays and diagnostic imaging can be used to monitor and improve treatment responses and patient outcomes by identifying and predicting patient sub-populations that are most likely to respond to a given treatment. Diagnostic approaches not only indicate the presence of a molecular target, but can also inform the off-target effects of a therapeutic, providing increased predictive power for toxicity and adverse effects associated with a drug. Finally, companion diagnostics and diagnostic imaging can inform whether a treatment is reaching its target, providing drug sponsors with an alternative to strict titration studies for determining optimal dosing. Taken together, these approaches are providing new avenues for identifying appropriate patient cohorts for inclusion in a study, monitoring disease, and assessing drug efficacy in individual patients, all of which contribute to potential economic benefits for drug sponsors.
Figure 1 Companion diagnostics-based treatment strategy for oncology clinical trials.

As an example, comparative data from drugs approved for the treatment of non-small cell lung cancer (Xalkori®, Pfizer, Inc., New York, NY, USA; and Tarceva®, OSI Pharmaceuticals, Inc., Melville, New York, USA and Genentech, Inc., South San Francisco, CA, USA) illustrate the financial benefit of incorporating a companion diagnostic early in the course of drug development. Xalkori, which was codeveloped with a companion diagnostic (Vysis ALK Break Apart fluorescence in situ hybridization probe, Abbott Laboratories, Abbott Park, IL, USA) required approximately threefold fewer patients in clinical trials (960 compared with 3,110), showed an approximately threefold reduction in time from Phase I to approval (1.8 years compared with 5.3 years), and had an overall reduced relative development cost per patient (100% compared with 154%).Citation4 Furthermore, the addition of epidermal growth factor receptor (EGFR) testing (Hoffman-La Roche Ltd., Basel, Switzerland) as a companion diagnostic for Tarceva in 2013 helped advance the drug to a first-line treatment in a select population and resulted in positive growth forecasts for a drug already on the market for over 8 years.Citation4
The importance of diagnostic approaches in drug development is highlighted by the growing global cancer diagnostics market which is expected to reach an estimated value of USD 168.6 billion by the year 2020.Citation5 Many newer drugs are being codeveloped with a companion diagnostic assay or imaging diagnostic, sparking the growth of more than 125 companies that provide companion diagnostic products and services.Citation6 Additionally, nearly two-thirds of breakthrough therapy designations recently granted by the FDA include a companion diagnostic.Citation7 At present, much of the activity in companion diagnostics development is focused in the area of oncology. There are currently 23 companion diagnostics approved by FDA, 22 of which are approved in oncology.Citation8 The first companion diagnostic, a HER2 immunohistochemistry assay, was developed in the late 1990s for use with trastuzumab (Herceptin®), a monoclonal antibody approved for the treatment of breast cancer. Since then, companion diagnostics have been developed for many targeted oncology therapeutics, including Tarceva, Iressa®, Erbitux®, and Vectibix® ().Citation9
Table 1 List of companion diagnostics approved by the US Food and Drug Administration
Advancing oncology trials with diagnostic imaging
Although companion diagnostic assays continue to improve personalized medicine, there are a number of significant limitations in current diagnostic assay approaches. Specifically, a positive signal generally informs the treating clinician or investigator that a target biomarker is present and, with quantitative assays, to what extent it is present in individual patients. However, the majority of approved diagnostic assays supply very little, if any, information regarding the location and distribution of a target biomarker. In oncology clinical trials, specific knowledge of a target lesion location can be essential, providing accurate biopsy localization and helping to design a treatment plan for tumors involving critical organs (eg, liver, lung, or bone marrow). Another limitation of using companion diagnostics is assay sensitivity (ie, the ability to detect true positives). Yet another limitation of companion diagnostic assays is the relatively narrow scope of biomarker evaluation. Research in the last several years has demonstrated that detection of a therapeutic target is not sufficient to predict drug efficacy and needs to be supplemented by additional data to assess for potential resistance. For example, the presence of KRAS mutations in colorectal cancers expressing EGFR often leads to resistance to anti-EGFR therapy.Citation10 Lastly, companion diagnostic assays may require large amounts of tissue samples for the evaluation of multiple biomarkers. This is especially challenging for certain solid tumors where tissue samples may be limited. In such instances, objective assessment by other diagnostic methods is essential for effective use of a companion diagnostic assay.
In clinical oncology studies, diagnostic imaging helps overcome these limitations by providing a reliable methodology to assess the presence, location, and extent of disease in response to treatment. For many years, computed tomography (CT) and magnetic resonance imaging (MRI) have been primary diagnostic imaging tools used for oncology disease assessments. As the use of diagnostic imaging techniques became widespread in clinical trials, a set of standardized imaging assessment criteria from the World Health Organization were established.Citation11 In the year 2000, a modified set of criteria called the Response Evaluation Criteria In Solid Tumors (RECIST 1.0) was introduced as part of collaborative efforts between the European Organization for Research and Treatment of Cancer, the National Cancer Institute in the USA, and the National Cancer Institute of Canada Clinical Trials Group.Citation12 RECIST refined an objective set of criteria that defined when tumor lesions in cancer patients improve (partial or complete response), remain unchanged (stable disease), or worsen (progressive disease) during treatment. Since its introduction, RECIST has been updated (RECIST 1.1) to introduce standards for the assessment of lymph nodes, redefine “measurable” lesions and assessment of disease progression, as well as establish recommendations for standardized image acquisition.Citation13 Today, a large number of oncology clinical trials employ RECIST to objectively assess cancer treatment response in solid tumors. Advances in imaging technologies and our understanding of disease have resulted in additional consortia guidelines for standardizing diagnostic imaging in oncology clinical trials. Most notably, the Cheson criteria (1999, 2007, and 2014) have established guidelines for the use of diagnostic imaging using CT, MRI, and fluorodeoxyglucose (FDG)-positron emission tomography (PET) as well as clinical findings for the assessment of lymphoma patients. In addition, the RANO criteria have been established for gliomas, and a number of other criteria have been introduced to specifically assess hepatocellular carcinoma, acute myeloid leukemia, prostate cancer, and the effects of immunotherapies on tumor responses.Citation14–Citation19
As these criteria have evolved, it has become clear that conventional anatomical imaging techniques, although very useful, have not supplied all of the objective assessments needed to make accurate early phase go/no go decisions. Initiatives by the Radiological Society of North America, including the Quantitative Imaging Biomarker Alliance, to advance volumetric assessments of tumor lesions continue to gain momentum, and researchers are showing increased interest in developing tools for the evaluation of metrics derived from CT and MRI studies. As an example, techniques such as dual energy CT and spectral CT imaging are being used to better differentiate and characterize certain cancers. These types of image analysis in conjunction with efforts to assess the relationship of CT and MRI to the molecular biology of various tumors, is helping to shape the new fields of radiomics and radiogenomics. Although these approaches hold great potential for oncology clinical trials, it is likely to be several years or more before they can be implemented in a clinical environment.
Molecular imaging diagnostics on the front line
The field of molecular imaging is rapidly evolving with many different technologies in various stages of development. At present, nuclear imaging techniques, including PLANAR, single photon emission computed tomography (SPECT), and PET remain the dominant approach for the diagnosis and treatment of cancers. PET and SPECT imaging requires the use of a radiotracer that is injected into a patient prior to interrogating its spatial distribution. PET relies on the detection of gamma photon pairs resulting from the annihilation of positrons (annihilation radiation) originating from a biologically active radiotracer. Using specialized detectors that encircle a patient, (ie, ring scanners) two-dimensional or three-dimensional images of radioactivity distribution within the body can be reconstructed. Similarly, SPECT requires a radiotracer, typically a heavy isotope, and relies on the detection of single gamma photons emitted directly from the radiotracer. SPECT tracers travel in the bloodstream and highlight areas of blood flow. Since SPECT tracers can be imaged at the time of injection, they can be used to detect changes in blood flow to various organs in a variety of disease states. SPECT tracers can also be linked to different biochemical analogs and antibodies to detect tissue specific distribution of cellular targets.
One of the first imaging diagnostic agents used in endocrinology and oncology studies was radiolabeled sodium iodide (131INaI). This compound has been used effectively to identify individuals with hyperthyroidism, monitor residual thyroid tissue post-surgery, and as a follow-up in treatment for thyroid cancer metastesis. Clinicians routinely rely on other nuclear medicine techniques to identify appropriate patient cohorts likely to respond to treatment and to monitor treatment responses for various cancers. For example, the use of 99mTc-labeled methylene diphosphonate and 18F-labeled NaF in bone scans to assay for bone metastasis in breast and prostate cancer patients, the use of 111In-labeled anti-CD20 antibodies for imaging lymphoid malignancies, and the investigational use of 123I, 99mTc, and 18F-labeled prostate-specific membrane antigen (PSMA) for monitoring prostate cancer patients ().
Table 2 Commonly used radiotracers in PET or SPECT studies
The ability to probe for molecular targets in cancer patients has opened the door to better, more accurate assessment of disease. Molecular imaging using various PET tracers provides enhanced visualization of tumors, their metabolic activity, and other biological phenotypes (eg, proliferation, hypoxia, expression of target receptors). Furthermore, widespread application of non-invasive imaging like PET/SPECT and gamma scintigraphy, enables the use of many additional tracers in oncology clinical trials (). For example, 18F-FDG and 18F-FLT are used to monitor glycolytic activity and proliferation of tumors, respectively, and technetium 99mTc-labeled antibody and peptide compounds are routinely used to label tumors and diagnose sites of cancer.
The elucidation and validation of novel oncology targets using high throughput screens is opening the door to development of potent and selective antibodies and other molecules capable of targeting tumor-specific or tumor-enriched receptors.Citation20 These agents can be linked to radionuclides, fluorophores, or other imaging probes (eg, 111In, 99mTc) to confirm the presence and anatomical location of cellular targets or developed as cytotoxic therapeutics (eg, 90Y, 131I). Several well studied proteins serve as oncology targets for imaging diagnostics including PSMA, the estrogen receptor (ER), and the folate receptor (). PSMA is a protein amplified on the surface of nearly all prostate cancer cells and is a validated target for the detection of primary and metastatic prostate cancer. Radiolabeled small molecules targeting PSMA are well tolerated tools for the detection of metastatic prostate cancer. A number of academic centers and pharmaceutical companies are developing and testing molecules labeled with 18F, 99mTc, and 123I that specifically target PSMA. Molecules capable of targeting ER are proving to be extremely valuable for improving breast cancer treatment. Several studies have shown that 16α-18F-fluoro-17β-estradiol (18F-FES), an ER-specific PET tracer, can reliably detect ER-positive tumor lesions and that its uptake correlates well with immunohistochemical scoring for the presence of ER.Citation21–Citation23 Currently, 18F-FES is being evaluated in breast cancer patients to determine patient ER status, to help differentiate between benign and malignant lesions, and to differentiate between metastases originating from different tumor types.Citation24 Similarly, the folate receptor is overexpressed in many cancer tissues and represents a target for selectively imaging and delivering therapeutics to cancer cells. A companion imaging diagnostic (99mTc-labeled folate-targeted molecule) has already been developed to identify tumors that overexpress the folate receptor, and clinical data have shown that patients with metastases that are positive for the folate receptor benefit from treatment with the corresponding folate-targeted small molecule drug conjugate.Citation25,Citation26 In addition to showcasing the importance of targeted imaging agents for oncology treatment, this approach provides a paradigm for the codevelopment of an imaging diagnostic and a therapeutic agent and may contribute to the design of more efficient drug development workflows in the future.
Table 3 Key oncology targets for which there are molecular imaging diagnostics
The emergence and development of new SPECT and PET radiopharmaceuticals, and the use of hybrid imaging modalities such as SPECT/CT, PET/CT, or PET/MRI (which combine anatomic imaging with physiological imaging in a single device and enable the acquisition of coregistered anatomical and physiological scans) provide a more complete picture of a patient’s disease state. In this respect, the use of molecular imaging is helping to modernize recommendations for the evaluation, staging, and response assessments of cancer patients. As an example, the Cheson criteria was recently revised to require 18F-FDG assessment as the dominant imaging technique for evaluation of FDG-avid lymphomas.Citation27
Significant improvements in hardware for PET and SPECT imaging, primarily driven by the oncology market, continue to advance widespread application of molecular imaging. Modern day PET and SPECT scanners increasingly are using newer crystal detector materials and solid state photon detectors that are smaller in size, provide increased sensitivity, and have better spatial resolution. New collimator designs and specialized gantries help reduce imaging time and radiation doses, thereby increasing patient safety and comfort. Additionally, newer image reconstruction techniques and software incorporate iterative reconstruction, time-of-flight data, and resolution recovery, which results in improved image contrast, image resolution, and reduce image noise.Citation28
The value of molecular imaging approaches for drug development has also led to the design and widespread integration of PET and SPECT techniques in preclinical animal models of disease. Small animal micro-PET and micro-SPECT imaging systems (as well as small-scale anatomical and hybrid imaging systems) are commercially available and are being used in the early drug discovery process to monitor drug toxicity and efficacy in efforts to advance the most promising oncology candidate drugs to human clinical trials.Citation29
Molecular imaging gets quantitative
Quantitative approaches using molecular imaging are extremely valuable as they provide insight as to what is occurring at the cellular level and are often predictive of a tumor response before anatomical changes can be observed. The introduction, validation, and use of quantitative molecular imaging continues to drive and optimize the field of imaging diagnostics. In addition to identifying the presence, location, and distribution of a specific tumor biomarker, radiopharmaceuticals can be used to objectively obtain quantitative measurements, including region of interest assessments of single or multiple areas. Most clinical trials that use molecular imaging rely on relative or semiquantitative approaches, since absolute quantitation methods using radionuclides are very complex and impractical for routine clinical studies. A common measurement used in molecular imaging for assessing treatment responses is the standardized uptake value (SUV). The SUV represents the ratio of the concentration of radioactivity in a selected region to the total injected dose of radioactivity distributed evenly throughout a patient’s body. SUV measurements can be calculated as a mean value (SUVmean) or as a maximum value (SUVmax), and can be further normalized to a patient’s lean body mass or whole body mass. Additional treatment response information can be gained by quantitative assessment of the changing pattern of uptake at multiple different time points ().
Figure 2 Assessing treatment response using PET and CT.
Abbreviations: CMR, complete metabolic response; CT, computed tomography; FDG, fluorodeoxyglucose; PET, positron emission tomography.
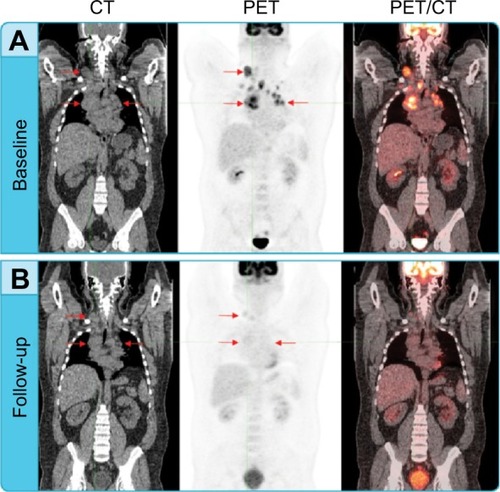
Other quantitative measurements used in clinical trials include glycolytic index determination, which is a measure of the total metabolic activity of a specific targeted area (eg, target tumor lesion) and the standardized uptake peak value (SUVpeak), currently used with the Positron Emission Tomography Response Criteria in Solid Tumors (PERCIST).Citation30 For longitudinal studies, dynamic measurements acquired over time can be used to generate time/activity curves. In particular, dynamic time/activity curve analysis is useful for assessing diagnostic probes, including their ability to localize in a specific tumor site and duration or residence time in a target site. This type of data can be very useful for determining optimal dosing using the therapeutic equivalent of an imaging companion diagnostic.
Although molecular imaging offers a wide spectrum of applications in drug development and clinical trials, there is only a single imaging companion diagnostic approved by the FDA, called FerriScan®. FerriScan uses MRI to select patients and manage therapy for non-transfusion-dependent thalassemia. The lack of additional FDA-approved imaging companion diagnostics highlights the opportunity and need for additional agents to be adapted, tested, and validated as diagnostic assays. In order for a molecular imaging test to become an integral part of any clinical trial investigation, the specific molecular imaging study has to be validated in prior investigations as an integrated component of a prospective analysis where it is not utilized to direct treatment decisions. Upon validation, a molecular imaging test can be used as an integral component of clinical trials and may even be required by regulatory agencies in clinical cases prior to and/or following drug administration.Citation31 As an example, 18FDG-PET imaging is being used in lymphoma clinical trials in conjunction with the Lugano criteria.Citation26 Given their pivotal role in clinical trials, it is likely that we will see an increase in the number of imaging companion diagnostics and integrated molecular imaging studies for oncology clinical trials. Even in cases where an imaging companion diagnostic is not incorporated into a clinical trial paradigm, it is important to recognize that the combination of a companion diagnostic assay with the appropriate imaging diagnostic can supply complementary information that cannot be ascertained from either methodology alone.
Companion diagnostics and regulatory requirements
The importance of companion diagnostics for current and future pharmacotherapy has attracted the attention of global regulatory agencies. For new therapies requiring the use of a diagnostic to qualify patient populations, companion diagnostics must meet typical design control and submission requirements to ensure safety and efficacy. As a result, regulatory agencies are increasing their visibility and offering more structured platforms for diagnostic companies to interact with them.
The FDA has taken significant steps in the last decade to define the companion diagnostic pathway. The FDA has published a drug-diagnostic codevelopment concept paper and created a personalized medicine group within the Office of In Vitro Diagnostics and Radiological Health. Last year, the FDA released a formal guidance called “In vitro companion diagnostic devices”, indicating regulatory pathways and requirements for companion diagnostic devices and therapeutic products.Citation32 The guidance defines in vitro companion diagnostic devices (IVDs), informs industry and FDA staff on premarket regulatory pathways and enforcement policies, and describes regulatory approval requirements relevant to therapeutic product labeling. In the case of clinical trials, where companion diagnostic assays are used to inform treatments, there are stringent requirements for submission of an investigational device exemption, usually as part of an investigational new drug application (IND). Given that the majority of companion diagnostic assays are considered high-risk devices (class III), there is also a requirement for a premarket approval application.
Although there are a number of therapeutic drugs that require companion diagnostic testing in the EU, the European Medicines Agency has been less transparent regarding companion diagnostics. The IVD Directive 98/79/EC regulates in vitro diagnostic medical devices in the EU and IVD devices require a CE mark to indicate compliance. Currently, any companion diagnostic entering the EU market is classified as a low-risk device based on CE marking by the manufacturer (self-certification). This results in a major divergence in the approval process between the USA and the EU. There are major changes underway in IVD legislation, including regulation that will provide a single regulatory framework for all EU member states. Under a new draft guidance which entered Parliament last year, companion diagnostics will be assigned as class C devices, requiring design examination certification by a Notified Body.
The process of achieving regulatory approval for new diagnostic imaging agents also remains extremely challenging, highlighted by the fact that only a handful of new radiotracers have received FDA approval in the last decade.Citation33 Although radiotracers are typically administered at doses that are orders of magnitude lower than therapeutics and are designed to measure molecular processes rather than modify them, they are regulated as though they carry the equivalent risk of a therapeutic. In fact, the commercial development of a new imaging agent shares many of the same challenges as therapeutic drug development, including target validation, lead selection, establishing high affinity and uptake, achieving adequate clearance, and demonstrating low toxicity.Citation2
The FDA has issued a three-part guidance in 2004 surrounding the regulatory pathway to the commercialization of new imaging agents, which covers safety assessments, clinical indications, and the design, analysis, and interpretation of clinical studies.Citation34–Citation36 In an effort to facilitate the regulatory process for imaging diagnostics, the FDA has established an exploratory IND for therapeutics and diagnostics, which provides an early look at the distribution and metabolism of new tracers in a small number of patients using early human screening and microdosing experiments. Imaging tracers that show promising results can proceed through traditional clinical trial phases and the filing of a formal IND. The exploratory IND process covers safety and efficacy for measuring a molecular process, but falls short in providing approval for larger clinical trials. To help overcome this, the Society of Nuclear Medicine has put forth a two-step approval process (safety and efficacy in measuring a molecular process and clinical utility and efficacy) specifically for diagnostic imaging agents.Citation37 More recently, the Society of Nuclear Medicine created the Molecular Imaging Clinical Trials Network with the use of centralized INDs for non-proprietary radiolabeled tracers to facilitate access to investigational molecular imaging radio-pharmaceuticals for clinical trials. The European Medicines Agency has also issued a formal guidance document that outlines the qualification process for biomarker development (EMEA/CHMP/SAWP/72894/2008).Citation38 This guidance, updated in 2014, outlines the scientific pathway leading to either a Committee for Medicinal Products for Human Use qualification opinion or qualification advice on innovative methods or drug development tools.
Closing thoughts
In vitro companion diagnostic assays and in vivo molecular diagnostic imaging continue to advance the field of personalized medicine and are changing the way in which clinicians are treating cancer and other human diseases. Assays and imaging agents are being developed alongside therapeutics to stratify patients and maximize the potential treatment benefit of new oncology therapeutics. These approaches are not only changing the landscape of clinical trials, but are also contributing to important changes in drug development and treatment. With the discovery of new oncology targets and imaging tracers comes increased capabilities to probe, monitor, and evaluate cancer on a molecular level. It is clear that more widespread implementation of imaging diagnostic tools will advance oncology clinical trials and help support new drug approvals in this rapidly expanding therapeutic area.
Disclosure
RVH, MO, RS, and EB are full-time employees of BioClinica and RF is a consultant for BioClinica. The authors report no other conflicts of interest in this work.
References
- DiMasiJAHansenRWGrabowskiHGThe price of innovation: new estimates of drug development costsJ Health Econ20032215118512606142
- KolaLLandisJCan the pharmaceutical industry reduce attrition rates?Nat Rev Drug Discov2004371171615286737
- HayMThomasDWCraigheadJLEconomidesCRosenthalJClinical development success rates for investigational drugsNat Biotechnol201432405124406927
- AgarwalAResslerDSnyderGThe current and future state of companion diagnosticsPharmgenomics Pers Med201589911025897259
- Transparency Market ResearchCancer diagnostics market (tumor biomarker tests, imaging, endoscopy and biopsy) – global industry analysis, size, share, growth, trends and forecast, 2014–2020 Available from: http://www.transparencymarketresearch.com/cancer-diagnostics-market.htmlAccessed July 20, 2015
- NaylorSColeTOverview of companion diagnostics in the pharmaceutical industryDrug Discovery World2010 Available from: http://www.ddw-online.com/personalised-medicine/p92845-overview-of-companion-diagnostics-in-the-pharmaceutical-industry.spring-10.htmlAccessed July 20, 2015
- VarondAJTrends in personalized medicineRegulatory FocusRegulatory Affairs Professional Society2013 Available from: http://www.raps.org/WorkArea/DownloadAsset.aspx?id=5275
- NicolaidesNCO’ShannessyDJAlboneEGrassoLCo-development of diagnostic vectors to support targeted therapies and theranostics: essential tools in personalized cancer therapyFront Oncol2014414124982846
- US Food and Drug AdministrationList of cleared or approved companion diagnostic devices (in vitro and imaging tools) Available from: http://fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm301431.htmAccessed March 27, 2015
- DouillardJYOlinerKSSienaSPanitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancerN Engl J Med20133691023103424024839
- MillerABHoogstratenBStaquetMWinklerAReporting results of cancer treatmentCancer1981472072147459811
- TherassePArbuckSGEisenhauerEANew guidelines to evaluate the response to treatment in solid tumors (RECIST guidelines)J Natl Cancer Inst20009220521610655437
- EisenhauerEATherassePBogaertsJNew response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1)Eur J Cancer20094522824719097774
- ChesonBDGreenbergPLBennettJMClinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasiaBlood200610841942516609072
- ScherHIHalabiSTannockIDesign and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working GroupJ Clin Oncol2008261148115918309951
- WolchokJDAxel HoosAO’DaySGuidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteriaClin Cancer Res2009157412742019934295
- ChesonBDBennettJMKopeckyKJRevised recommendations of the international working group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemiaJ Clin Oncol2003214642464914673054
- LencioniRLlovetJMModified RECIST (mRECIST) assessment for hepatocellular carcinomaSemin Liver Dis201030526020175033
- van den BentMJWefelJSSchiffDResponse assessment in neurooncology (a report of the RANO group): assessment of outcome in trials of diffuse low-grade gliomasLancet Oncol20111258359321474379
- HoelderSClarkePAWorkmanPDiscovery of small molecule cancer drugs: successes, challenges and opportunitiesMol Oncol2012615517622440008
- McGuireAHDehdashtiFSiegelBAPositron tomographic assessment of 16 alpha-[18F] fluoro-17 beta-estradiol uptake in metastatic breast carcinomaJ Nucl Med199132152615311869973
- MintunMAWelchMJSiegelBABreast cancer: PET imaging of estrogen receptorsRadiology198816945483262228
- PetersonLMMankoffDALawtonTQuantitative imaging of estrogen receptor expression in breast cancer with PET and 18F-fluoroestradiolJ Nucl Med20084936737418287268
- van KruchtenMGlaudemansAWde VriesEFPET imaging of estrogen receptors as a diagnostic tool for breast cancer patients presenting with a clinical dilemmaJ Nucl Med20125318219022241912
- DosioFMillaPCattelLEC-145, a folate-targeted vinca alkaloid conjugate for the potential treatment of folate receptor-expressing cancersCurr Opin Investig Drugs20101114241433
- LeamonCPFolate-targeted drug strategies for the treatment of cancerCurr Opin Investig Drugs2008912771286
- ChesonBDFisherRIBarringtonSFRecommendations for initial evaluation, staging, and response assessment of Hodgkin non-Hodgkin lymphoma: the Lugano classificationJ Clin Oncol2014323059306825113753
- SlomkaPJPanTBermanDSGermanoGAdvances in SPECT and PET hardwareProg Cardiovasc Dis20155756657825721706
- JangBMicroSPECT and microPET imaging of small animals for drug developmentToxicol Res2013291624278622
- WahlRLJaceneHKasamonYLodgeMAFrom RECIST to PER-CIST: evolving considerations for PET response criteria in solid tumorsJ Nucl Med200950122S150S19403881
- MankoffDAPrymaDAClarkASMolecular imaging biomarkers for oncology clinical trialsJ Nucl Med20145552552824614222
- US Food and Drug AdministrationIn vitro companion diagnostic devices2014 Available from: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM262327.pdfAccessed July 20, 2015
- AgdeppaEDSpilkerMEA review of imaging agent developmentAAPS J20091128629919415506
- US Food and Drug AdministrationGuidance for industry developing medical imaging drug and biological products. Part 1: Conducting Safety Assessments2004 Available from: http://www.fda.gov/downloads/Drugs/…/Guidances/ucm071600.pdfAccessed July 20, 2015
- US Food and Drug AdministrationGuidance for industry developing medical imaging drug and biological products. Part 2: Clinical indications2004 Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071603.pdfAccessed July 20, 2015
- US Food and Drug AdministrationGuidance for industry developing medical imaging drug and biological products. Part 3: Design, analysis, and interpretation of clinical studies2004 Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm071604.pdfAccessed July 20, 2015
- McEwanAVan BrocklinHDivgiCAction plan for emerging molecular imaging technologiesJ Nucl Med20084937N40N
- European Medicines AgencyQualification of novel methodologies for drug development: guidance to applicants2014 Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500004201.pdfAccessed July 20, 2015