
Abstract
Background
Montelukast (MT) is a leukotriene D4 antagonist. It is an effective and safe medicine for the prophylaxis and treatment of chronic asthma. It is also used to prevent acute exercise-induced bronchoconstriction and as a symptomatic relief of seasonal allergic rhinitis and perennial allergic rhinitis.
Objective
The aim of this study was to evaluate the bioequivalence (BE) of two drug products: generic MT 5 mg chewable tablets versus the branded drug Singulair® pediatric 5 mg chewable tablets among Mediterranean volunteers.
Methods
An open-label, randomized two-period crossover BE design was conducted in 32 healthy male volunteers with a 9-day washout period between doses and under fasting conditions. The drug concentrations in plasma were quantified by using a newly developed and fully validated liquid chromatography tandem mass spectrometry method, and the pharmacokinetic parameters were calculated using a non-compartmental model. The ratio for generic/branded tablets using geometric least squares means was calculated for both the MT products.
Results
The relationship between concentration and peak area ratio was found to be linear within the range 6.098–365.855 ng/mL. The correlation coefficient (R2) was always greater than 0.99 during the course of the validation. Statistical comparison of the main pharmacokinetic parameters showed no significant difference between the generic and branded products. The point estimates (ratios of geometric means) were 101.2%, 101.6%, and 98.11% for area under the curve (AUC)0→last, AUC0→inf, and Cmax, respectively. The 90% confidence intervals were within the predefined limits of 80.00%–125.00% as specified by the US Food and Drug Administration and European Medicines Agency for BE studies.
Conclusion
Broncast® pediatric chewable tablets (5 mg/tablet) are bioequivalent to Singulair® pediatric chewable tablets (5 mg/tablet), with a similar safety profile. This suggests that these two formulations can be considered interchangeable in clinical practice.
Introduction
Montelukast (MT) is a monosodium salt; its IUPAC chemical name is (R,E)-2-(1-((1-(3-(2-(7-chloroquinolin-2-yl)vinyl)phenyl)-3-(2-(2-hydroxypropan-2-yl)phenyl) propylthio)methyl)cyclopropyl) acetic acid. The chemical structure of MT is shown in .
Figure 1 Chemical structure of montelukast.
MT is a leukotriene receptor antagonist; it specifically blocks the action of leukotriene D4 on the cysteinyl leukotriene receptor of the lungs and bronchial tubes and reduces the bronchoconstriction and also inflammation.Citation1,Citation2 Several clinical studies have demonstrated the efficacy of MT in the treatment and prophylaxis of chronic mild asthma in patients aged 2 years and older.Citation3–Citation7 It has also proved to be effective in the prevention of exercise-induced asthma in patients aged above 6 years.Citation8 Moreover, MT has been indicated in the symptomatic relief of seasonal and perennial allergic rhinitis.Citation9–Citation11 Bioavailability studies showed that the presence of food in the gastrointestinal tract did not affect bioavailability when the tablet was administered with a standard meal.Citation11 Moreover, instructions about the use of the brand granules report that they can be mixed with one teaspoonful of cold or room temperature baby formula or breast milk or with one spoonful of one of the following soft foods: applesauce, mashed carrots, rice, or ice cream.Citation12
The oral bioavailability of MS when administered as a 10 mg film-coated tablet in adults was found to be 58%–66%.Citation13 Singulair® was approved by the US Food and Drug Administration (FDA) for the treatment of asthma and allergic rhinitis in August 2012. It is usually administered once daily in a dose of 10 mg or 5 mg per tablet. Other dosage forms have been approved by the FDA; these include oral granules at a dose of 4 mg and chewable tablets at doses of 4 mg or 5 mg.Citation14,Citation15 In fact, the FDA has invited pharmaceutical manufacturers to develop and produce proper dosage forms, especially for pediatric patients. In this context, chewable tablets are known to be advantageous over liquid dosage forms in many aspects, such as higher palatability, better bioavailability, stability, dose precision, portability, and ease of administration, especially during traveling.Citation16–Citation18 However, many challenges may be encountered during the development of chewable tablets, such as the taste and the bioequivalence (BE) of the drug when compared with the original brand. In fact, unsuitable taste may result in poor patient compliance. Chewing the tablet may result in immediate disintegration and partial dissolution or degradation of the drug in the saliva, and this may affect the amount of drug available for absorption. In the current study, the generic MT chewable tablet was formulated to be tasty, bioavailable, and as safe as the original brand. Accordingly, the objective of this study was to investigate the BE of this formulation by comparing its pharmacokinetic (PK) parameters with the original brand by developing a liquid chromatography–mass spectrometry (LC-MS) method for the determination of MT. In addition, the safety of the two formulations, on the basis of clinical and laboratory observation and documentation of adverse events, was also investigated ().
Materials and methods
The study was a comparative, randomized, two-period, two-treatment, two-sequence, single-dose, open-label, crossover BE study of generic MT 5 mg chewable tablets versus the brand drug Singulair® pediatric 5 mg chewable tablets given to healthy subjects under fasting conditions.
Volunteers and clinical protocol
The study was conducted by Arab Pharmaceutical Industry Consulting Co. Ltd., Jordan in accordance with the requirements of the Declaration of HelsinkiCitation19 and under the current Good Clinical Practice guidelinesCitation20 and the International Conference for HarmonizationCitation21 guidelines. The study protocol and the informed consent forms were approved by the Institutional Review Board. Owing to methodological and ethical difficulties and in order to safeguard the pediatric population, FDA and European Medicines Agency (EMA) guidelines state that PK studies of drugs intended for use in a pediatric population can be carried out on healthy adults.Citation22,Citation23
Thirty-two adult male volunteers were recruited to participate in the study. The volunteers were aged between 18 years and 50 years, weighing between 57 kg and 93 kg with an average weight of 76 kg. The volunteers were subjected to a full medical and physical examination to confirm their health status and were not on any medication during the study period. A written informed consent form, which explained the nature of the study, was given to the volunteers. The volunteers were instructed to abstain from taking drugs for 1 month before the study initiation, to abstain from caffeine and alcohol-containing beverages for at least 16 hours prior to each study drug administration and throughout the study period, and to fast for at least 10 hours before drug administration.
The study used an open-label, randomized two-period crossover design with a 9-day washout period between doses in 32 healthy subjects under fasting conditions. The volunteers were randomly divided into two groups, each of 16 subjects. The first group was given the reference brand and the second group was given the test formulation with a crossover after the washout period. On the morning of the study, each volunteer gave a blood sample to serve as a blank for the drug assay. Each volunteer received an oral dose of the assigned formulation given with 240 mL of water in the sitting position. During each period, blood samples were taken from each volunteer for the calculation of the PK parameters at pre and up to 24 hours after drug dosing. Each sample volume was 8 mL, 1 hour before dosing, and 8 mL of samples were withdrawn at the following time points: 0.50 hour, 1.00 hour, 1.50 hours, 1.75 hours, 2.00 hours, 2.25 hours, 2.50 hours, 2.75 hours, 3.00 hours, 3.25 hours, 3.50 hours, 3.75 hours, 4.00 hours, 5.00 hours, 6.00 hours, 8.00 hours, 12.00 hours, 16.00 hours, and 24.00 hours after dosing. Blood samples were collected in tubes containing heparin and centrifuged to separate the plasma fraction of the blood. The resulting plasma was immediately stored at −70°C and analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS). Four hours after drug administration, a standard lunch meal containing soup (no carrots), a half chicken, rice with mixed vegetables (no carrots), yogurt, a loaf of bread, and salad (tomato and cucumber) was served, and subjects had free access to water 1 hour after drug administration.
Chemicals and reagents
MT working standard was supplied by Morepen Laboratories Ltd. (Himachal Pradesh, India), while the MT-d6 internal standard (ISTd) was supplied by TRC (North York, Canada). High-performance liquid chromatography-grade methanol and acetonitrile were purchased from ROMIL (Cambridge, UK), isopropanol was obtained from Carbon Group (Cork, Ireland), extra pure formic acid was obtained from Scharlau (Port Adelaide, Australia), diethylether was obtained from JHD (Guangzhou, People’s Republic of China), and high-performance liquid chromatography-grade water was supplied by Sartorius Purified Water (Goettingen, Germany). Control human plasma was harvested from donors.
Tested brand and formulated tablets
The generic tablets, Broncast® chewable tablets (5 mg/tablet), were obtained from Avalon Pharma (Middle East Pharmaceutical Industries Co. Ltd., Riyadh, KSA), batch number 303111. Singulair® pediatric chewable tablets (5 mg/tablet) were obtained from Merck Sharp & Dohme Ltd. (Northumberland, UK) and had a batch number 322559.
Instruments and chromatographic separations
An Agilent1200 LC system (Agilent Technologies, Santa Clara, CA, USA) interfaced with an API 4000 tandem MS system (AB Sciex, Framingham, MA, USA) was used in the analysis. The mass spectrometer was operated with a source temperature of 550°C. Electrospray in positive ionization mode with a needle voltage of 4 kV was used. Multiple reaction monitoring mode was used for the detection of the drug and its ISTd with monitoring of following transitions: 586.384/568.300 and 586.384/422.200 for MT and 592.288/574.300 and 592.288/427.300 for MT-d6.
An ACE 5 CN column was used with 5 cm length and 4.6 mm internal diameter (Hichrom Limited, Reading, UK). The mobile phase was composed of acetonitrile and 0.005 M ammonium acetate and formic acid in a ratio of 80:20:0.1 (v/v/v), respectively. The pH of the mobile phase was 4.30± 0.2. The injection volume was 10 µL, and the flow rate was 0.80 mL/min. The retention time for the drug and IS was 0.95 minutes.
Preparation of standard and working solutions
A drug solution was prepared by dissolving 0.08 g MT (99.4%) in methanol. The stock solution was diluted in a serial dilution using methanol to produce a final standard solution of 10.00 mL containing 76.22 µg/mL MT.
The stock solution for the internal solution was prepared by weighing 0.001 g, and the volume was made up to 100 mL with methanol. The IS stock solution was diluted in a serial dilution using methanol to produce a final standard solution of 10.00 mL containing 0.518 µg/mL.
The working solutions for calibration, quality control (QC), and IS were prepared by diluting the stock solutions 50 times. All the calibration and the QC samples were spiked with IS in which 0.1 mL of plasma was spiked by 30 µL of the stock internal solution to get final concentration of IS of 119.5 ng/mL; each sample was shaken for 30 seconds using a vortex shaker. Then 1 mL of acetonitrile was added to remove protein followed by vortex mixing and centrifuging at 14,000 rpm for 5 minutes. The supernatant was removed for analysis.
Validation procedures
The method was validated for its application to the analysis of MT in the biological fluids by spiking standards into blank plasma. Sampling lasted 24 hours after drug administration in order to finalize the BE study. The method was validated in accordance with the international guidelines, and all the validation parameters, including linearity, accuracy, precision, and limit of quantitation, were calculated for the developed method.
The linearity assessment was performed using a series of nine standard plasma solutions previously spiked with MT (calibrator), which were employed for constructing calibration curves covering a concentration ranging from 6.098 ng/mL to 365.855 ng/mL. The accuracy and precision were determined by using a minimum of six replicates per concentration level. The limit of quantitation was determined by injecting a series of diluted solutions with known concentrations.
In addition, stock solution stability in mobile phase was assessed using two standard mixtures that were equivalent to the lower limit of quantification and upper limit of quantification along with the IS.
Short- and long-term matrix-based stabilities were assessed using two QCL and QCH MT concentrations. Stability after freeze and thaw cycles was assessed using two sets of QC samples that were subjected to three freeze-thaw cycles (stability samples). The stability of the samples was compared to a freshly prepared calibration curve and was analyzed in a single run in comparison with the QC sample (comparison sample).
Whole blood stability was assessed by spiking whole blood samples with two different concentrations. The matrix effect was investigated for MT and the IS. The matrix factor (MF) should be calculated in each lot of matrix, by calculating the ratio of the peak area in the presence of matrix to the peak area in the absence of matrix (pure solution of the MT). The IS-normalized MF was also calculated by dividing the MF of the analyte by the MF of the IS. The QC samples were used to evaluate the performance of the assay. They were prepared by spiking blank plasma with MT. The QC samples were prepared to have low, medium, and high concentrations (MT: 18.293 ng/mL, 304.879 ng/mL, and 457.319 ng/mL). Four QC samples were incorporated with each analysis run as unknown samples. The concentration in each QC sample was determined from the calibration curve and was compared with the nominal concentration. The analysis run was accepted if at least three out of four QC samples were within 15% of the nominal concentration. No carryover or matrix effect was found.
Pharmacokinetic and statistical analysis
The PK parameters were estimated using standard non- compartmental methods. The peak plasma concentration (Cmax) and the corresponding time of peak plasma concentration (Tmax) were taken directly from the data. The elimination rate constant (ke) was estimated from the slope of the semi-logarithmic plot of the terminal elimination phase of the plasma concentration–time curve. The equation t1/2= ln2/ke was used to calculate the elimination half-life time (T1/2). The areas under the plasma concentration–time curves for the drug from (AUC0→last) and the area to infinity (AUC0→inf) were calculated by using the linear trapezoidal method. Extrapolation to the infinity was achieved by dividing the last measurable plasma concentration Clast by terminal rate constant ke. AUC0→inf is obtained from the sum of the estimated and extrapolated parts (AUC0→inf = AUC0→last + AUClast→inf). For the purpose of BE analysis, one-way analysis of variance was used to assess the effect of formulations, periods, sequences, and subjects on AUC0→last, AUC0→inf, and Cmax. A commercially available software package (Thermo Scientific Kinetica, version 5.1) was used for the calculations.
Results
Results of validation procedures
The newly developed and validated LC-MS/MS method was found to be rapid, sensitive, reproducible, and accurate for the analysis of MT in plasma. The relationship between concentration and peak area ratio of MT/IS was found to be linear within the range 6.098–365.855 ng/mL for MT. The linear equation was y=0.00780x−0.003 with a correlation (R2) of 0.9975 during the course of the validation. The method was found to be sensitive with lower limit of quantification of 6.098 ng/mL. Mean recovery was 109.45%, 104.55%, and 101.88% for QCL, QCM, and QCH, respectively. The method was found to be precise and accurate for samples up to 1,829 ng/mL. Short-term stability testing of MT in plasma proved that the drug was stable for up to 24 hours at room temperature (RT). Stock solution short-term stability proved that the drug was stable up to 10 hours at RT. A post-preparative stability study showed that the drug was stable for up to 95 hours at RT in the autosampler. The whole blood stability study proved that the drug was stable for 1 hour when left in ice.
Results of PK study
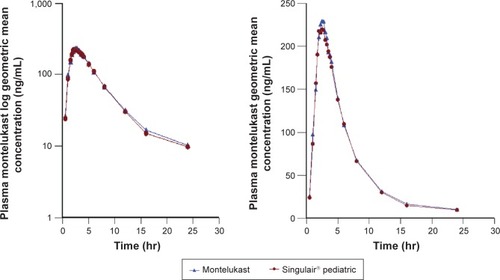
Both MT 5 mg chewable tablets, Broncast and Singulair®, were well tolerated by all the subjects, and they were discharged in good health. shows the plasma concentrations of both brands indicating that the two brands are superimposable.
Figure 2 Plasma montelukast geometric mean concentration (ng/mL) versus time (hr) curves and log plasma montelukast geometric mean concentration versus time (hr) curves following a single oral dose of 5 mg chewable tablets.
Abbreviation: hr, hour.
shows a summary of the PK parameters for the two products of MT 5 mg chewable tablets. The point estimates (ratios of geometric mean) were 101.258%, 101.556%, and 98.106% for AUC0→last, AUC0→inf, and Cmax, respectively. No statistically significant difference between the two formulations was found. These PK parameter values lie within the FDA and EMA specified BE limit (80%–125%).Citation21,Citation22 Our results in this part of the study showed equivalent clinical efficacy for the two brands. There were no serious or significant adverse events, with both formulations being well tolerated when administered as a single dose.
Table 1 Summary of calculated PK parameters
Discussion
According to FDA, a brand name drug is defined as a drug marketed under a proprietary, trademark-protected name and a generic drug is the same as a brand name drug with regard to active ingredient, dosage form, safety, strength, route of administration, quality, performance as assessed from the PK profile, and intended use and contains the same salt, ester, or chemical form. Generic versions of a drug can vary in shape, scoring configuration, packaging, and excipients. If all the previous criteria are met, then the two drugs are considered to be therapeutically equivalent.Citation23
During the development phase of an oral solid dosage form, several preformulation and formulation trials and tests are carried out to achieve a generic product that can be interchangeable with the original brand in terms of efficacy and safety. Accordingly, in vitro dissolution in different pH media is conducted on the generic product, and it must show a similar dissolution profile or overlap with the reference brand. In many cases, these tests cannot replace the in vivo tests that demonstrate the efficacy and safety of the generic product since it may contain different excipients that affect the rate of release in vivo.
Several previous studies have tested the BE of new generic MT tabletsCitation24 or other dosage forms as granulesCitation25 and disintegrating tablets.Citation26 The aim of this study was to test the bioavailability of MT 50 mg tablets produced by Avalon Pharma versus the reference MT 5 mg (Singulair® pediatric) produced by Merck Sharp & Dohme Ltd. The two dosage forms were administered to 32 fasting male volunteers in order to eliminate the influence of food on drug absorption.
This developed and validated analytical method has many advantages over the existing analytical methods of MT in the biological system. Our method used MS/MS detector compared to other studies that used only fluorescent and UV detectors in their analysis.Citation15,Citation27 Moreover, this method was validated, while other existing methods were not.Citation28 However, some analytical methods were validated for quantification of MT in combination with other drugs, such as gliclazide, nifedipine, and fexofenadine, in the human plasma but were not applied in any BE study.Citation29,Citation30
An LC-MS method validated as described earlier was utilized for quantification of MT 5 mg chewable tablets. Analysis was successfully applied without interference from the excipients used in the tablet formulation. The method provided the appropriate accuracy, sensitivity, linearity, precision, repeatability, and selectivity with high-sample throughput being economically convenient procedure for the support of PK studies. In fact, all validation parameters were conducted according to the international guidelines, and they were within the accepted limits as reported in .
Regarding the efficacy of our generic product, statistical comparison of the main PK parameters, AUC0→last, AUC0→inf, Cmax, and Tmax, clearly indicated no significant difference between test and reference tablets. The values obtained were compliant with the FDA and EMEA requirements for BE of generic drugs since the AUC0→inf, AUC0→last, and Cmax mean ratios are within the 80%–125% interval.Citation31,Citation32
The limitations of the study as per all BE studies were the relatively small sample size and administration of a single dose in healthy male volunteers. Also the inclusion of adults instead of pediatric volunteers could be reported as another limitation, but this was according to FDA guidelines and in order to safeguard the pediatric population from unnecessary risk. Several clinical studies have highlighted the efficacy of Singulair® pediatric in the treatment of asthma in both adults and childrenCitation3–Citation7 and for the symptomatic relief of allergic rhinitis.Citation9–Citation11 Given the BE demonstrated for Broncast 5 mg chewable tablets, this product is expected to be equally effi-cacious and well tolerated.
Safety is also important. In our study, the administered drugs were tolerated and all the participated volunteers completed the study without showing any signs of adverse effects and were released in good health. Accordingly, this product offers an efficacious treatment option for individuals with asthma or seasonal/perennial allergic rhinitis with relatively lower cost in comparison with the established brand.
Conclusion
The statistical analysis of the results was performed on AUC0→last, AUC0→inf, and Cmax using the analysis of variance method and showed that both test chewable and reference tablets (Singulair® pediatric 5 mg chewable tablet) were bioequivalent, since they deliver equivalent quantities of active ingredient to the systemic circulation at equivalent rates for both AUC0→last and Cmax ratios within the 80%–125% interval proposed by the FDA and EMA. These results showed that the formulation of this new generic tablet was good, which is important for achieving good therapeutic benefits and avoiding any potential problems that may arise from poor formulation.
Acknowledgments
The authors wish to express their sincere gratitude to one and all who directly or indirectly have contributed in any form to this work.
Disclosure
The authors report no conflicts of interest in this work.
References
- LabelleMBelleyMGareauYGauthierJYGuayDGordonRDiscovery of MK-0476, a potent and orally active leukotriene D4 receptor antagonist devoid of peroxisomal enzyme inductionBioorg Med Chem Lett199553283288
- De LepeleireIReissTFRochetteFMontelukast causes prolonged, potent leukotriene D4-receptor antagonism in the airways of patients with asthmaClin Pharmacol Ther199761183929024176
- ScaparrottaADi PilloSAttanasiMMontelukast versus inhaled corticosteroids in the management of pediatric mild persistent asthmaMultidiscip Respir Med2012711322958412
- BlakeKVMontelukast: data from clinical trials in the management of asthmaAnn Pharmacother199933121299131410630831
- BozekAWarkocka-SzoltysekBFilipowska-GronskaAJarzabJMon-telukast as an add-on therapy to inhaled corticosteroids in the treatment of severe asthma in elderly patientsJ Asthma201249553053422551116
- DucharmeFMNoyaFJAllen-RameyFCMaieseEMGingrasJBlaisLClinical effectiveness of inhaled corticosteroids versus mon-telukast in children with asthma: prescription patterns and patient adherence as key factorsCurr Med Res Opin201228111111922077107
- KeithPKKochCDjandjiMMontelukast as add-on therapy with inhaled corticosteroids alone or inhaled corticosteroids and long-acting beta-2-agonists in the management of patients diagnosed with asthma and concurrent allergic rhinitis (the RADAR trial)Can Respir J200916suppl A17A31A
- MerckPrescribing Information Singulair®;2013 Available from: http://www.merck.com/product/usa/pi_circulars/s/singulair/singulair_pi.pdf
- CingiCGunhanKGage-WhiteLUnluHEfficacy of leukotriene antagonists as concomitant therapy in allergic rhinitisLaryngoscope201012091718172320717951
- ModgillVBadyalDKVergheseAEfficacy and safety of montelukast add-on therapy in allergic rhinitisMethods Find Exp Clin Pharmacol201032966967421225018
- YamamotoHYamadaTSakashitaMEfficacy of prophylactic treatment with montelukast and montelukast plus add-on loratadine for seasonal allergic rhinitisAllergy Asthma Proc2012332e17e2222525385
- Physician Disc reference; PDR Network, LLC2015 Available from: http://www.pdrhealth.com/drugs/singulairAccessed: July 2015
- US Food Drug AdministrationFDA Approves First Generic Versions of Singulair to Treat Asthma, Allergies2012 Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm314436.htm
- Sandoz: SPCMontelukast Sodium 4 mg Oral Granules2013 Available from: http://www.medicines.org.uk/emc/medicine/27591
- ChengHLeffJAAminRPharmacokinetics, bioavailability, and safety of montelukast sodium (MK-0476) in healthy males and femalesPharm Res19961334454488692739
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH)Stability Testing of New Drug Substances and Products: Q1A(R2). ICH Harmonised Tripartite Guideline. Current Step 4 VersionGenevaICH2003 Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1A_R2/Step4/Q1A_R2_Guideline.pdfAccessed December 17, 2012
- ZaidANAbu GhoshAOSweilehWMAl-JabiSWJaradatNAChewable tablets: is this dosage form well evaluated by Palestinian health professionals?Islam Univ J (Series of Natural Studies and Engineering)20071528394
- World Medical Association“Declaration of Helsinki” as Amended by the 59th World Medical AssemblySeoul, South KoreaWorld Medical Association, Inc2008
- European Medicines AgencyNote for Guidance on Good Clinical Practice (CPMP/ICH/135/95)London, UKEuropean Medicines Agency1997
- International Conference of Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human UseICH Harmonization Tripartite Guideline Guidelines for Good Clinical PracticeGeneva, SwitzerlandICH1996
- Food and Drug AdministrationGuidance for Industry: Statistical Approaches to Establishing Bioequivalence2001 Available from: http://www.accessdata.fda.gov/scripts/cder/dissolution/dsp_SearchRe-sults_Dissolutions.cfm?PrintAll=1
- Guidance for Industry Exposure-Response Relationships – Study Design, Data Analysis, and Regulatory Applications Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatory-information/guidances/ucm072109.pdf
- Guideline on the role of pharmacokinetics in the development of medicinal products in the paediatric population Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003066.pdf
- KanjanawartSGaysonsiriDTangsucharitPComparative bioavailability of two montelukast tablet formulations after single-dose administration in healthy Thai male volunteersInt J Clin Pharmacol Ther20114952553021781653
- ZhengRKimBHPharmacokinetics and bioequivalence of montelukast granules and montelukast tablets in Korean healthy volunteersInt J Clin Pharmacol Ther20145253053624786017
- DamleBDuczynskiGJeffersBWCrownoverPCoupeALaBadieRRPharmacokinetics of a novel orodispersible tablet of montelukast in healthy subjectsClin Ther20143623624424447534
- MuñozEOcampoDHEspinalEEYépesNBioequivalence study of two 10 mg montelukast immediate-release tablets formulations: a randomized, single-dose, open-label, two periods, crossover studyJ Bioequivalence Bioavailability2014638690
- AbbasMKhanAMAminSRiffatSAshrafMWaheedNBioequivalence study of montelukast tablets in healthy Pakistani volunteersPak J Pharm Sci201326225525923455193
- EzzeldinEAbo-TalibNFTammamMHShahatAADevelopment and validation of LC/MS/MS method for the simultaneous determination of montelukast, gliclazide, and nifedipine and its application to a pharmacokinetic studyBiomed Chromatogr20142881048105624424850
- MuppavarapuRGuttikarSRajappanMKamarajanKMullangiRSensitive LC-MS/MS-ESI method for simultaneous determination of montelukast and fexofenadine in human plasma: application to a bioequivalence studyBiomed Chromatogr20142881048105624424850
- European Medicines AgencyNote for Guidance on the Investigation of Bioavailability and Bioequivalence (CPMP/EWP/QWP/1401/98)London, UKEuropean Medicines Agency2001
- Food and Drug AdministrationGlossary of Terms2012 Available from: http://www.fda.gov/drugs/informationondrugs/ucm079436.htm