
Abstract
Celecoxib, a selective cyclooxygenase-2 inhibitor, is potentially useful for the treatment of colonic diseases such as colorectal cancer and colitis. However, the cardiovascular toxicity of celecoxib limits its routine use in the clinic. Generally, colon-specific delivery of a drug both increases the therapeutic availability in the large intestine and decreases the systemic absorption of the drug, most likely resulting in enhanced therapeutic effects against colonic diseases such as colitis and reduced systemic side effects. To develop a colon-specific prodrug of celecoxib that could reduce its cardiovascular toxicity and improve its therapeutic activity, dextran–glutamic acid–celecoxib conjugate (glutam-1-yl celecoxib-dextran ester [G1CD]) was prepared and evaluated. While stable in pH 1.2 and 6.8 buffer solutions and small-intestinal contents, G1CD efficiently released celecoxib in cecal contents. Oral administration of G1CD to rats delivered a larger amount of celecoxib to the large intestine than free celecoxib. G1CD prevented the systemic absorption of celecoxib and did not decrease the serum level of 6-ketoprostaglandin F1α, an inverse indicator of cardiovascular toxicity of celecoxib. Collectively, G1CD may be a polymeric colon-specific celecoxib prodrug with therapeutic and toxicological advantages.
Introduction
Nonsteroidal anti-inflammatory drugs have been studied as chemopreventive agents for colorectal cancer (CRC).Citation1,Citation2 Since chemoprevention involves the long-term use of oral agents, the numerous side effects of nonsteroidal anti-inflammatory drugs have led to the search for safer drugs.Citation3 Selective cyclooxygenase (COX)-2 inhibitors including celecoxib have been developed as safer alternatives to conventional COX inhibitors.Citation4 A number of in vitro and in vivo experiments have demonstrated the effectiveness of coxibs in the chemoprevention of CRC and for the treatment of familial adenomatous polyposis.Citation5–Citation7 In addition, selective COX-2 inhibitors have beneficial effects against experimental colitis.Citation8–Citation10 However, clinical studies have revealed that long-term use of coxibs increases the risk of serious dose-associated cardiovascular events.Citation11–Citation14 Considering that chemoprevention or treatment of colonic diseases such as CRC and colitis requires long-term pharmacotherapy, celecoxib cannot be routinely recommended for this indication owing to its potential to cause cardiovascular events.
In general, colon-targeted delivery of an orally administered drug is a pharmaceutical strategy that is used to achieve efficient treatment of colonic diseases such as CRC and colitis, and reduce systemic side effects. These beneficial effects result from increased therapeutic concentration at the target site (large intestine) while restricting systemic absorption.Citation15
A colon-specific prodrug needs to reach the colon after oral administration and activation in the gut lumen. Hydrophilic small molecules or polymers are used as colon-specific promoieties to limit the systemic absorption in the upper intestine. The linkage between a drug and the promoieties should be chemically and enzymatically stable during the transit through the stomach and small intestine.Citation15 After delivery to the colon, the prodrug is presumed to be activated by enzymes originating from the microbes that are abundant in that portion of the alimentary canal.Citation16
Dextran, a nonstarch polysaccharide with a linear α-1,6-glucopyranose chain and α-1,3-glucopyranose branching, is not digestible by host glycosidases but is degraded readily by dextranases derived from microbes residing in the large intestine.Citation15 For this reason, dextran has been used as a colon-specific polymeric carrier. There are many papers demonstrating that dextran–drug conjugates can deliver drugs specifically to the large intestine.Citation15,Citation17 During transit through the gastrointestinal tract, a dextran–drug conjugate does not release the drug in the upper intestine, presumably because the steric hindrance of the polymer matrix prevents enzymatic action at the linkage between dextran and the drug. Release of the drug takes place only after depolymerization of the dextran matrix by endodextranase, which is found in the colon as a result of the high bacteria count.Citation18
Dextran–succinic acid–celecoxib was reported as a dextran-based polymeric prodrug of celecoxib.Citation19 However, we found that succinyl celecoxib (SC) was not cleaved effectively enough to liberate celecoxib in the cecal contents, which may lead to inefficient release of celecoxib after colonic delivery. In this study, dextran–glutamic acid–celecoxib, where glutamic acid with a dicarboxylic acid was used as a linker, was prepared and evaluated as a polymeric colon-specific prodrug of celecoxib. Toxicological and therapeutic advantages were suggested by providing data on disposition and serum 6-ketoprostaglandin F1α (6-keto-PGF1α) after oral administration of the polymeric prodrug.
Materials and methods
Materials
1,1′-Carbonyldiimidazole (CDI), dextranase (Penicillium sp.), and 3,5-dinitrosalicylic acid (DNS) were purchased from Sigma Chemical Co. (St Louis, MO, USA). Succinic anhydride and 5-benzyl N-(Boc)-glutamate were purchased from Tokyo Chemical Industry (Tokyo, Japan). Dextran (molecular weight: 15–20 kDa) was purchased from Fluka (Sigma Chemical Co.). Celecoxib was ether-extracted from Celebrex capsules (Pfizer, Inc., New York, NY, USA). All other chemicals were reagent grade, commercially available products. Buffer solutions (pH 1.2 and 6.8) were prepared as described in USP XXIII. Thin layer chromatographys (TLCs) were performed on Merck Kieselgel 60 F254. The high-performance liquid chromatography (HPLC) system consisted of a Model 306 pump, a Model 117 UV detector, a Model 234 autoinjector, and a Model 805 manometric module from Gilson (Middleton, WI, USA). A symmetry column C18 (Waters, Milford, MA, USA) (250×4.6 mm) with a guard column (Waters, 3.9×20 mm) was used. Six-week-old male Sprague Dawley rats (Samtako Bio Korea, Kyeong-gi-do, South Korea) were housed in the university animal facility with controlled temperature, humidity, and dark/light cycle. The animal protocol used in this study has been reviewed and approved by the Pusan National University-Institutional Animal Care and Use Committee (PNU-IACUC) on their ethical procedures and scientific care.
HPLC analysis
Standard solutions of celecoxib in various biological specimens were prepared as described previously.Citation20 Standard or blank solution (1 mL) was mixed on a vortex mixer for 5 minutes, centrifuged at 10,000× g for 10 minutes at 4°C and filtered through a membrane filter (0.45 μm). The filtrate (20 μL) was injected on a symmetry C18 column, which was eluted with the mobile phase at a flow rate of 1 mL/min. The mobile phase consisted of 60% acetonitrile (ACN) in 0.067 M phosphate buffer (pH 4.0) containing 0.1% trifluoroacetic acid, which was filtered through 0.45 μm membrane filter before use. The eluate was monitored at 273 nm by a UV detector measuring the absorption with a sensitivity of absorbance units full scale (AUFS) 0.01. Gilson 712 software was used for data analysis. The retention time of celecoxib, glutam-1-yl celecoxib (G1C), and SC was 9.13, 3.20, and 9.33 minutes, respectively.
Preparation of SC
Succinic anhydride (2, 0.33 mg, 3.30 mmol) was added to a solution of celecoxib (1, 0.50 g, 1.31 mmol) in ACN (15 mL) in the presence of triethylamine (TEA, 4.3 mL). The reaction mixture was stirred at 55°C for 4 hours. The mixture was concentrated by evaporation and washed with 1 M hydrochloric acid (HCl) to afford the final product 1-succinylaminosulfonyl- 4-(5-[4-methylphenyl]-3-[trifluoromethyl] pyrazol-1-yl) benzene (3) as white powder. Synthetic scheme is shown in . Melting point (mp): 119°C–124°C; infrared (IR) (Nujol, cm−1): 1,718 (C=O SO2NHCO), 1,658 (C=O, carboxylic, shoulder); proton nuclear magnetic resonance (1H-NMR) (dimethylsulfoxide [DMSO]-d6, ppm): 2.30 (s, 3H), 2.35 (t, 2H, J=7.5 Hz), 2.45 (t, 2H, J=6.5 Hz), 7.17 (s, 1H), 7.16–7.21 (m, 4H), 7.56 (d, 2H, J=8.5 Hz), 7.95 (d, 2H, J=8.5 Hz); Elemental analysis: C21H18F3N3O5S (481.09) Calcd: C, 52.39; H, 3.77; N, 8.73; Found: C, 52.44; H, 3.82; N, 8.75.
Figure 1 Synthetic scheme of celecoxib derivatives.
Abbreviations: TEA, triethylamine; CDI, 1,1′-carbonyldiimidazole; ACN, acetonitrile; DMSO, dimethylsulfoxide; DMF, Dimethylformamide; TFA, trifluoroacetic acid.
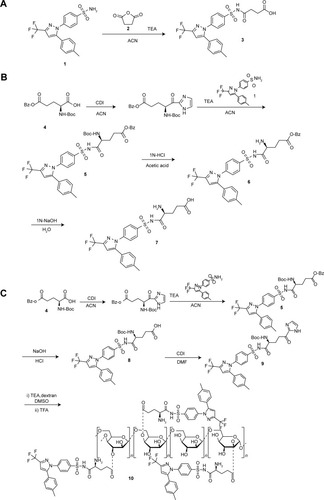
Preparation of G1C
5-Benzyl N-(Boc)-glutamic acid (4, 1.336 g, 3.96 mmol) was dissolved in 15 mL of ACN followed by the addition of CDI (0.76 g, 4.69 mmol), which was stirred at room temperature for 10 minutes. The mixture was added to celecoxib (1, 0.400 g, 1.05 mmol) and dissolved in ACN containing TEA (3.44 mL) and reacted at 55°C for 4 hours. After removal of solvents by evaporation, the residue was dissolved in ethylacetate/ether (1/1, 60 mL), washed with 5% NaHCO3, dried over anhydrous Na2SO4, and subjected to flash evaporation to obtain an oily residue. The oily residue was treated with 1 M NaOH (15 mL) at room temperature for 3 hours and subsequently with 1 M HCl (16 mL) at room temperature for 2 hours to obtain N-(Boc)-glutam-1-ylcelecoxib (5) as white precipitate. The precipitate was placed in small volume of 1 M HCl acetic acid for approximately 1 hour to remove the protection group (6), which was flash-evaporated to obtain G1C (7). Synthetic scheme is shown in . mp: 110°C–115°C; IR (Nujol, cm−1): 1,719 (C=O, SO2NHCO), 1,595 (C=O, zwitterionic carboxylate salt); 1H-NMR (DMSO-d6, ppm): 1.7 (m, 1H), 2.25 (m, 1H), 2.02 (m, 2H), 2.30 (s, 3H), 3.29 (m, 1H), 7.14 (s, 1H), 7.16–7.21 (m, 4H), 7.59 (d, 2H, J=8.5 Hz), 7.95 (d, 2H, J=8.5 Hz); Elemental analysis: C22H21F3N4O5S (510.12) Calcd: C, 51.76; H, 4.15; N, 10.98; Found: C, 51.82; H, 4.11; N, 11.20.
Preparation of glutam-1-ylcelecoxib-dextran ester (dextran–glutamic acid–celecoxib conjugate)
CDI (84.59 mg, 0.52 mmol) was added to N-(Boc)-glutam-1-yl celecoxib (8, 300 mg, 0.492 mmol) dissolved in dimethylformamide (280 μL), which was stirred at room temperature for 1 hour. The reaction mixture (9) was treated with dextran (250 mg) dissolved in DMSO (500 μL) and TEA (60.9 μL, 0.437 mmol), and reacted at 55°C for 4 hours. Excess ethanol was added to the mixture resulting in the formation of white precipitate, which was isolated by centrifugation. The isolated precipitate was treated with trifluoroacetic acid (0.2 mL) at room temperature for 40 minutes. Excess ethanol was added to precipitate the final product glutam-1-yl celecoxib-dextran ester [G1CD] (10, dextran–glutamic acid–celecoxib conjugate) as white powder. Synthetic scheme is shown in . Degree of substitution (DS) was defined as milligrams of celecoxib in 100 mg G1CD and was measured by HPLC analysis of G1C after hydrolysis of G1CD in 1 M NaOH for 1 hour. In this study, G1CD (DS: 9.5–10.5) was used for experiments.
Chemical stability
Each celecoxib derivative was incubated in pH 1.2 HCl buffer or pH 6.8 phosphate buffer at 37°C for 10 hours. At a predetermined time interval, a 20 μL portion of each solution was removed and the concentrations of celecoxib and G1C were analyzed by HPLC as described.
DNS method
DNS reagent solution was prepared by dissolving DNS (5 g) in 2 M NaOH (100 mL) and distilled water (250 mL). To this solution, sodium potassium tartratetetrahydrate (150 g) was added and dissolved and the volume was adjusted to 500 mL with distilled water. Maltose dissolved in 0.1 M acetate buffer (pH 5.4) at the concentration range of 0.093–0.75 mg/mL (200 μL) was mixed with DNS reagent solution (600 μL), boiled for 5 minutes, cooled for 10 minutes, and the absorbance measured at 540 nm by a UV spectrophotometer (Shimadzu, Tokyo, Japan). A calibration curve was generated using the results. G1CD (DS: 10, equivalent to 2.52 mg dextran/1 mL) dissolved in 0.1 M pH 5.4 acetate buffer was incubated with dextranase (15 DU/mL) at 37°C. At an appropriate time interval, a portion of the incubated sample was treated with DNS reagent solution according to the same procedure, and the amount of terminal reducing sugar was deduced from the calibration curve. Dextran (2.52 mg) was incubated with dextranase (15 DU/mL) at 37°C for 1.5 hours and this sample was used to calculate the percent degradation of G1CD.
Incubation of celecoxib derivatives in suspensions of gastrointestinal tract contents
Male Sprague Dawley rats (250–255 g) were sacrificed by CO2 and a midline incision was made. The contents of the small intestine and the cecal contents were separately collected and were suspended in isotonic phosphate buffer (pH 6.8). In each microtube, each celecoxib derivative in buffer (0.5 mL, 1 mM) was added to the suspension (0.5 mL) and incubated at 37°C under nitrogen (for incubation in the cecal contents). G1CD dissolved in the buffer (corresponding to 1 mM G1C) was used for this experiment. At an appropriate time interval, the samples were extracted with ethylacetate (0.5 mL) followed by centrifugation at 5,000× g for 5 minutes. Methanol (1.0 mL) was added to the residue obtained from evaporation of the organic layer (0.1 mL), vortexed, and centrifuged at 10,000× g for 10 minutes. The concentration of celecoxib in a 20 μL portion of the supernatant was determined by HPLC. SC and G1C were analyzed in samples without extraction.
Oral administration of G1CD and preparation of samples for blood and contents of rat intestinal tract
Male Sprague Dawley rats (250–255 g) were maintained on a stock diet and water ad libitum. These animals were fasted overnight (16 hours) prior to and during the experiments, while allowed free access to water. Water bottles were removed from the cages at least 30 minutes before drug administration. Celecoxib suspension (2.5 mg/0.3 mL/rat) in 0.5% sodium carboxymethylcellulose or G1CD solution (equivalent to 2.5 mg celecoxib/1 mL/rat) in distilled water was administered orally to rats by gavage.
At an appropriate time interval, the rats were anesthetized with isoflurane and 1 mL of blood was collected by intracardiac puncture through a heparinized syringe. Heparinized blood samples were immediately centrifuged at 6,000× g for 5 minutes, and the plasma was separated and transferred to microcentrifuge tubes. After blood collection, the rats were sacrificed using CO2 gas asphyxiation and the intestinal tract (proximal small intestine, distal small intestine, and cecum) were obtained by midline incision from the rat. The contents in the cecum was separated, transferred to falcon tubes, and mixed with pH 6.8 isotonic phosphate buffer to afford 20% (w/v) suspensions. Cecal samples were extracted with an equivalent volume of ethyl acetate for 5 minutes. For extraction of blood samples, fivefold volume of ethyl acetate was used. Each sample was centrifuged at 6,000× g for 5 minutes for effective phase separation. For analysis of celecoxib, a 1 mL (for cecal and small intestinal contents) or 0.1 mL volume (for blood) of pH 6.8 isotonic phosphate buffer was added to the residues obtained from evaporation of an aliquot of the organic layer (1 mL) in the microcentrifuge tubes, vortexed, and centrifuged at 10,000× g at 4°C for 10 minutes. The concentration of celecoxib in a 20 μL portion of the supernatant was determined by HPLC.
6-Keto-PGF1α immunoassay
The serum level of 6-keto-PGF1α was assessed using a commercially available immunoassay kit (enzyme-linked immunosorbent assay) according to the manufacturer’s instructions. Blood (1 mL) was drawn by intracardiac puncture. The clotted blood samples were centrifuged at 2,000× g for 30 minutes in the presence of indomethacin (10 μg/mL). The supernatants were collected and stored at −70°C until the 6-keto-PGF1α assay was performed.
Statistical analysis
The results are expressed as mean ± SEM. One-way ANOVA followed by Tukey’s (honest significant difference [HSD]) test was used for testing the difference between data. Differences with P<0.05 were considered significant. XLSTAT® Software (Addinsoft, Inc, Suite 503; New York, NY, USA) was used for statistical analysis.
Results
G1C but not SC releases celecoxib in cecal contents
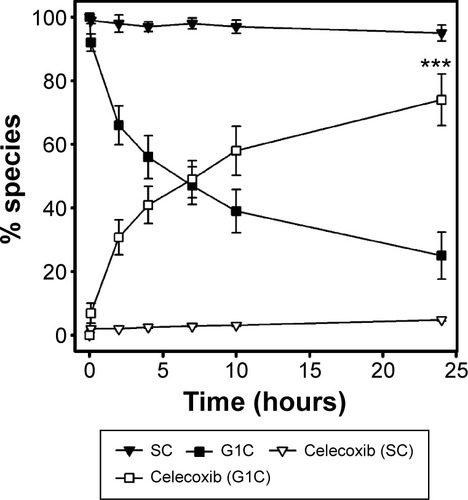
After colonic delivery of a dextran–glutamic acid–celecoxib conjugate, the celecoxib-linker is released from oligomerized dextran-linker–celecoxib and needs to be cleaved in order to release celecoxib.Citation15 To compare colonic celecoxib release from G1C with that from SC,Citation19 G1C and SC were incubated with the cecal contents of rats and the release of celecoxib was monitored. While SC released up to approximately 5% of the initial dose at 24 hours, G1C released up to approximately 55% and 75% of the initial dose at 10 and 24 hours, respectively (). These results indicate that glutamic acid is a more useful linker than succinic acid.
Figure 2 Release of celecoxib during incubation of succinyl celecoxib (SC) or glutam-1-yl celecoxib (G1C) with the cecal contents of rats.
Abbreviations: HPLC, high-performance liquid chromatography; SEM, standard error of mean.
G1CD releases celecoxib in cecal contents but not in small intestinal contents
Based on the earlier in vitro data, a dextran–glutamic acid–celecoxib conjugate, which has a glutamic acid linker between dextran and celecoxib, was prepared. To examine whether G1CD could be colon specific and deliver celecoxib to the large intestine without significant loss in the upper intestine, the polymeric conjugate was incubated for 6 hours in pH 1.2 or 6.8 buffer solutions, representing pH of the stomach and small intestine, respectively. In addition, G1CD was incubated with small intestine contents for 10 hours. Either celecoxib or G1C was not detected in the experiments.
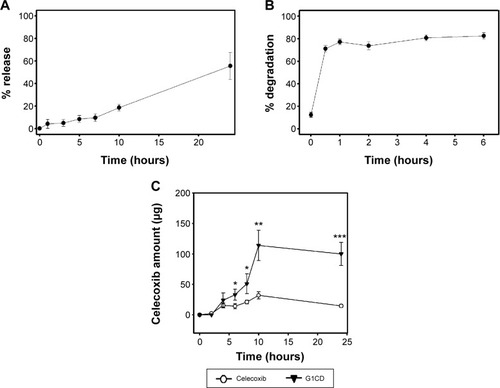
To examine whether G1CD delivered to the large intestine would liberate celecoxib, the conjugate was incubated with the cecal contents of rats and celecoxib release was monitored. As shown in , up to approximately 18% and 53% celecoxib were released at 8 and 24 hours, respectively. To test whether celecoxib release was dependent on microbial enzymes in the large intestine, the same experiment was performed with autoclaved cecal contents, which are used to test microbially triggered activation of a colon-specific prodrug.Citation21 In contrast, celecoxib was not detected until 12 hours after incubation of G1CD with the autoclaved cecal contents.
Figure 3 Glutam-1-yl celecoxib-dextran ester (G1CD) administered orally delivers and liberates celecoxib at the large intestine.
Abbreviations: HPLC, high-performance liquid chromatography; DNS, dinitrosalicylic acid; DS, degree of substitution; SEM, standard error of mean.
G1CD is a substrate of dextranase
The earlier data suggest that although esterases are abundant throughout the digestive tract, the ester bond between celecoxib-glutamic acid and dextran is cleaved (deesterified) only in the large intestine where microbial dextranases exist; thus the dextran backbone of G1CD can be degraded to produce oligomers, which reduce the steric hindrance imposed by dextran, most likely facilitating the cleavage of the ester link between dextran and G1C.Citation15 This rationale prompted us to examine whether the modified dextran, G1CD, was depolymerized by dextranase. Depolymerization was assessed using the DNS method following the incubation of G1CD with dextranase. Depolymerization appeared to be completed in 1 hour ().
G1CD delivers celecoxib to the large intestine
To examine the specificity of G1CD for the colon in vivo, G1CD was administered orally to rats and the amount of celecoxib in the contents of the small intestine and the cecum were measured at appropriate time intervals. For comparison, the same experiment was performed with free celecoxib. In the small intestine, celecoxib was detected at 52.4±12.4 μg in proximal small intestine (PSI) and 129±29.2 μg in DSI 4 hours after gavage of free celecoxib. In contrast, either celecoxib or G1C was not detected in the proximal and distal small intestine upon oral administration of G1CD. On the other hand, celecoxib began to appear in the cecum 4 hours after oral administration of G1CD. The peak concentrations of celecoxib were reached at 10 hours. The recovery of celecoxib was better after G1CD administration than that after celecoxib administration ().
G1CD does not affect the serum level of 6-keto-PGF1α
Long-term use of celecoxib for the prevention of CRC or the treatment of colitis may cause cardiovascular toxicity, which is correlated with the concentration of celecoxib in blood.Citation11 To examine whether G1CD could reduce the blood concentration of celecoxib, celecoxib or G1CD was administered orally to rats and the plasma concentrations of celecoxib were compared. As shown in , while the plasma concentration of celecoxib reached 2.8 μM at 2 hours after oral administration of celecoxib, celecoxib was not detectable in the plasma for 24 hours after oral administration of G1CD, indicating that G1CD limited the systemic absorption of celecoxib.
Figure 4 Glutam-1-yl celecoxib-dextran ester (G1CD) does not affect the serum level of 6-ketoprostaglandin F1α (6-keto-PGF1α).
Abbreviations: HPLC, high-performance liquid chromatography; ELISA, enzyme-linked immunosorbent assay.
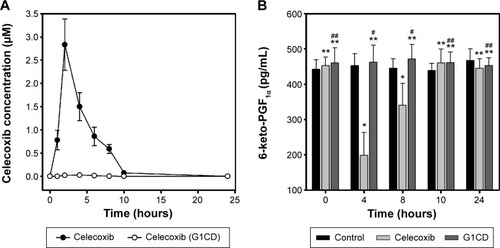
We next examined whether limiting the systemic absorption of celecoxib from G1CD could reduce the risk of cardiovascular toxicity of celecoxib. Celecoxib or G1CD was administered orally to rats and the serum levels of 6-keto-PGF1α, an inverse indicator of cardiovascular toxicity of celecoxib,Citation22 were monitored. As shown in , administration of free celecoxib decreased 6-keto-PGF1α levels by up to 40% of the initial level 4 hours later, and the level returned to the baseline level at approximately 10 hours later; on the other hand, G1CD did not affect the serum levels of 6-keto-PGF1α throughout the experimental period. These results were in line with the blood concentration data.
Discussion
In this study, G1CD was depolymerized and released celecoxib in the large intestine while remaining stable in the upper intestine. Oral administration of G1CD delivered a greater amount of celecoxib to the large intestine than that delivered using free celecoxib. Moreover, G1CD prevented the systemic absorption of celecoxib and did not decrease the serum level of 6-keto-PGF1α, an inverse indicator of cardiovascular side effects of celecoxib.
Consistent with previous papers, G1CD, a dextran-based colon-specific prodrug, shows specificity for the colon. Our in vitro data show that, while G1CD did not release celecoxib in simulated stomach and small intestine conditions, a significant release of celecoxib was observed in cecal contents. In parallel with this observation, oral administration of G1CD delivered greater amount of celecoxib to the large intestine without premature release of celecoxib in the small intestine. The colon specificity of G1CD should lead to a greater concentration at the target site and be therapeutically advantageous for the treatment of colonic diseases. In fact, a colon-specific celecoxib prodrug using a small-molecule carrier is reported to have enhanced anti-colitic effects in a trinitrobenzene sulfonic acid (TNBS)-induced rat colitis model.Citation8 The authors suggest that the enhanced anti-colitic effects are attributable to the increased colonic celecoxib concentration, which exerts these effects via pharmacologic mechanisms that may not be available when free celecoxib is used.Citation8
Despite greater accumulation of celecoxib at the target site (large intestine) probably leading to therapeutic enhancement, G1CD would prevent the systemic absorption of celecoxib, thus attenuating the cardiovascular side effects of celecoxib. Our data show that celecoxib was not detectable in the blood after oral administration of G1CD and in agreement with this observation, G1CD did not decrease the serum level of 6-keto-PGF1α associated with cardiovascular risk of celecoxib.Citation22 In contrast, oral administration of free celecoxib afforded a celecoxib concentration of up to 2.8 μM in blood and substantially decreased the prostaglandin levels.
In this study, we focused on whether G1CD can elicit colon targetability and its toxicological and therapeutic advantages over free celecoxib. Considering that polymer–drug conjugates generally exhibit sustained release of the drug,Citation23 G1CD could also control the release of celecoxib in the large intestine, thus resulting in desirable colonic celecoxib distribution. Since pathological lesions for CRC and inflammatory bowel disease exist in the distal region of the large intestine,Citation24 it would be beneficial to control colonic drug distribution even with colon-specific drugs. However, a more elaborate distribution study would be needed to identify whether G1CD offers such a benefit. Collectively, G1CD may be a polymeric colon-specific celecoxib prodrug with toxicological and therapeutic advantages.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (No 2009-0083538).
Disclosure
The authors report no conflicts of interest in this work.
References
- HerendeenJMLindleyCUse of NSAIDs for the chemoprevention of colorectal cancerAnn Pharmacother200337111664167414565811
- HilmiIGohKLChemoprevention of colorectal cancer with nonsteroidal anti-inflammatory drugsChin J Dig Dis2006711616412030
- BusPJVerspagetHWLamersCBGriffioenGChemoprevention of colorectal cancer by non-steroidal anti-inflammatory drugsScand J Gastroenterol Suppl200023210110411232485
- FortJCelecoxib, a COX-2 – specific inhibitor: the clinical dataAm J Orthop1999283 suppl131810193998
- ArberNEagleCJSpicakJPreSAP Trial InvestigatorsCelecoxib for the prevention of colorectal adenomatous polypsN Engl J Med2006355988589516943401
- DaviesNMGuddeTWde LeeuwMACelecoxib: a new option in the treatment of arthropathies and familial adenomatous polyposisExpert Opin Pharmacother20012113915211336575
- SteinbachGLynchPMPhillipsRKThe effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposisN Engl J Med2000342261946195210874062
- LeeYKimWHongSColon-targeted celecoxib ameliorates TNBS-induced rat colitis: a potential pharmacologic mechanism and therapeutic advantagesEur J Pharmacol2014726495624462351
- CuzzocreaSMazzonESerrainoICelecoxib, a selective cyclo-oxygenase-2 inhibitor reduces the severity of experimental colitis induced by dinitrobenzene sulfonic acid in ratsEur J Pharmacol200143119110211716847
- El-MedanyAMahgoubAMustafaAArafaMMorsiMThe effects of selective cyclooxygenase-2 inhibitors, celecoxib and rofecoxib, on experimental colitis induced by acetic acid in ratsEur J Pharmacol20055071–329129915659320
- BrophyJMCardiovascular risk associated with celecoxibN Engl J Med20053522526482650 author reply 2648–265015972876
- CaldwellBAldingtonSWeatherallMShirtcliffePBeasleyRRisk of cardiovascular events and celecoxib: a systematic review and meta-analysisJ R Soc Med200699313214016508052
- OviedoJASchroyPC3rdDoes celecoxib use increase the risk of cardiovascular events?Gastroenterology200512941348135016230088
- SolomonSDMcMurrayJJPfefferMAAdenoma Prevention with Celecoxib (APC) Study InvestigatorsCardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma preventionN Engl J Med2005352111071108015713944
- JungYKimYMWhat should be considered on design of a colon-specific prodrug?Expert Opin Drug Deliv20107224525820095945
- RubinsteinAMicrobially controlled drug delivery to the colonBiopharm Drug Dispos19901164654752207298
- SinhaVRKumriaRColonic drug delivery: prodrug approachPharm Res200118555756411465408
- SinhaVRKumriaRPolysaccharides in colon-specific drug deliveryInt J Pharm20012241–2193811472812
- ShrivastavaPKShrivastavaSKDextran carrier macromolecule for colon specific delivery of celecoxibCurr Drug Deliv20107214415120158488
- LeeYKimJKimHN-succinylaspart-1-yl celecoxib is a potential colon-specific prodrug of celecoxib with improved therapeutic propertiesJ Pharm Sci201210151831184222334096
- KimHKongHChoiBMetabolic and pharmacological properties of rutin, a dietary quercetin glycoside, for treatment of inflammatory bowel diseasePharm Res20052291499150916132362
- BuerkleMALehrerSSohnHYConzenPPohlUKrotzFSelective inhibition of cyclooxygenase-2 enhances platelet adhesion in hamster arterioles in vivoCirculation2004110142053205915451781
- MehvarRDextrans for targeted and sustained delivery of therapeutic and imaging agentsJ Control Release200069112511018543
- FriendDRReview article: issues in oral administration of locally acting glucocorticosteroids for treatment of inflammatory bowel diseaseAliment Pharmacol Ther19981275916039701522