
Abstract
Type 2 diabetes (T2D), which has currently become a global pandemic, is a metabolic disease largely characterised by impaired insulin secretion and action. Significant progress has been made in understanding T2D aetiology and pathogenesis, which is discussed in this review. Extrapancreatic pathology is also summarised, which demonstrates the highly multifactorial nature of T2D. Glucagon-like peptide (GLP)-1 is an incretin hormone responsible for augmenting insulin secretion from pancreatic beta-cells during the postprandial period. Given that native GLP-1 has a very short half-life, GLP-1 mimetics with a much longer half-life have been developed, which are currently an effective treatment option for T2D by enhancing insulin secretion in patients. Interestingly, there is continual emerging evidence that these therapies alleviate some of the post-diagnosis complications of T2D. Additionally, these therapies have been shown to induce weight loss in patients, suggesting they could be an alternative to bariatric surgery, a procedure associated with numerous complications. Current GLP-1-based therapies all act as orthosteric agonists for the GLP-1 receptor (GLP-1R). Interestingly, it has emerged that GLP-1R also has allosteric binding sites and agonists have been developed for these sites to test their therapeutic potential. Recent studies have also demonstrated the potential of bi- and tri-agonists, which target multiple hormonal receptors including GLP-1R, to more effectively treat T2D. Improved understanding of T2D aetiology/pathogenesis, coupled with the further elucidation of both GLP-1 activity/targets and GLP-1R mechanisms of activation via different agonists, will likely provide better insight into the therapeutic potential of GLP-1-based therapies to treat T2D.
Introduction
Type 2 diabetes (T2D) is the most common and clinically important metabolic disorder which has become a global pandemic in recent decades and a major healthcare burden worldwide. In 2013, there were an estimated 382 million patients with diabetes globally.Citation1 Concerningly, the T2D incidence continues to increase, and it is projected that there will be >590 million patients diagnosed with this condition by 2035.Citation1,Citation2 The World Health Organisation (WHO) defines diabetes as a “metabolic disorder of multiple aetiology characterised by chronic hyperglycaemia with disturbance of carbohydrate, fat, and protein metabolism resulting from defects in insulin secretion, insulin action, or both.”Citation3 The most prevalent form of diabetes is type 2, as an estimated 90% of diabetes patients are diagnosed with this form,Citation2 and the majority of the remaining 10% of patients have type 1 diabetes (T1D), although there are other rare types.Citation4 T2D is largely caused by impaired insulin production and secretion by pancreatic beta-cells, as well as peripheral tissue insulin resistance.Citation5,Citation6 Given that ~90% of patients are obese or overweight at T2D diagnosis, the aetiology of T2D is largely thought to be linked to diets involving excessive nutrient consumption combined with insufficient energy expenditure.Citation7 There are a range of effective treatments that reduce hyperglycaemia in T2D patients, which mediate their effects by improving insulin secretion or decreasing peripheral tissue insulin resistance.Citation8 Despite this, post-diagnosis complications, especially long-term complications, are prevalent globally. As a result, diabetes remains a leading cause of blindness, end-stage renal disease, lower limb amputation and cardiovascular disease.Citation3
T2D and obesity have such an interdependent relationship that the term “diabesity” has been coined.Citation9 In recent decades, the number of people with T2D has more than doubled, and the increased global burden of T2D is thought to be largely due to an increase in obesity.Citation9,Citation10 Obesity has become a global pandemic over recent decades. Weight loss is associated with an improved prognosis for overweight T2D patients and obese individuals. Better glycaemic control has been reported in T2D patients who have lost weight, and excess body weight is associated with the risk of cardiometabolic complications, which are major causes of morbidity and mortality in T2D and obese individuals.Citation11,Citation12 Bariatric surgery has proven to be an effective treatment for diabesity, but it is expensive and there are numerous post-surgery complications: for example, vomiting and dumping syndrome, iron and B12 deficiency, and secondary hyperparathyroidism.Citation13 GLP-1 analogues (eg liraglutide and exenatide) are used to treat T2D as they promote insulin secretion and induce weight loss.Citation3 Since the GLP-1 receptor (GLP-1R) agonists are effective in treating diabesity, they could be pharmacological alternatives to bariatric surgery but without the post-surgery complications.Citation3,Citation13,Citation14
The current literature on T2D epidemiology, aetiology, pathogenesis and treatment is discussed in this review.
Obesity
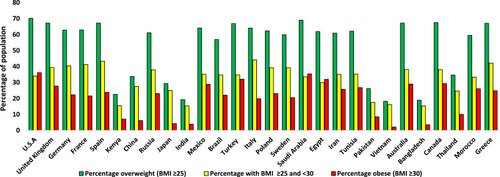
Obesity is caused by excessive energy intake and subsequent storage coupled with insufficient energy expenditure resulting in weight gain. It has become a huge healthcare concern over the last few decades, especially in developed countries. However, in some obese individuals, excessive dietary intake may have a genetic aetiology, such as leptin deficiency.Citation15–Citation17 Obese individuals have a BMI of ≥30 kg/m2, and individuals with a BMI of ≥25 and <30 are classified as overweight.Citation16 Obesity and overweight incidences have more than doubled since 1980, giving rise to >2.1 billion individuals with a BMI of >25 globally, as stated by the WHO.Citation15,Citation18 The WHO estimated that there were ~600 million obese individuals in 2014, and this number will continue to rise in the future.Citation18 Obesity is associated with an increased risk for T2D, hypertension, dyslipidaemia, cardiovascular diseases, musculoskeletal disorders (such as osteoarthritis), certain types of cancer, and premature mortality.Citation19,Citation20 It is well established that there is a generally directly proportional relationship between BMI and both fasting and postprandial insulin levels.Citation21,Citation22 A similar relationship also exists between BMI and the degree of insulin resistance.Citation22 The hyperinsulinemia associated with rising BMI is necessary to overcome insulin resistance and maintain normoglycemia.Citation22 Not all obese individuals develop insulin resistance though;Citation23 one study reported that 19, 34 and 60% of individuals with a BMI of <30, ≥30–35 and >35, respectively, were insulin resistant.Citation22 Insulin hypersecretion also becomes more frequent with increasing BMI;Citation24 28, 49 and 80% of individuals with a BMI of <30, >30–35 and >35, respectively, exhibited insulin hypersecretion.Citation22 Of the 2.1 billion individuals that are obese or overweight <25% have been diagnosed with T2D.Citation1,Citation18 The rise in T2D over the last few decades is generally thought to be attributed to an increase in the percentage of overweight individuals in the global population.Citation3,Citation25 shows how the percentage of overweight and obese individuals in the population vary between countries.
Figure 1 Variation in the percentage of obese and overweight individuals over 18 years of age in the population between different countries in 2016.Citation325 Values displayed are an average of the male and female percentage of the population for each country, which were taken from the WHO data.Citation325
Type 2 Diabetes
Diabetes mellitus was first reported over 3000 years ago in an Egyptian manuscript.Citation26 However, it was not until 1936 that the distinction between T1D and T2D was made.Citation27 T2D is by far the most common form of diabetes and accounts for ~90% of diabetes cases.Citation2,Citation7 T2D is a chronic metabolic complex multifactorial disease, involving many organs, that has become a global pandemic in recent decades.Citation1,Citation28 T2D is characterised by pancreatic beta-cell dysfunction, increased pancreatic alpha-cell function, and peripheral tissue insulin resistance. These alterations result in (1) hyperglycaemia due to impaired peripheral glucose uptake, (2) dyslipidaemia (hypertriglyceridemia and low high-density lipoprotein [HDL]-cholesterol) due to impaired peripheral fat uptake, (3) impaired amino acid uptake and ATP production into peripheral tissues, such as skeletal muscle, due to the impaired nutrient uptake, and (4) increased glucagon production, which further amplifies hyperglycaemia and hyperlipidaemia.Citation29–Citation32 T2D is thought to be largely provoked by diets involving excessive nutrient consumption, as ~90% of patients are obese or overweight in western countries.Citation3,Citation9,Citation16,Citation33 In contrast to that observed in western countries, a study in China found that 50.3% of T2D patients were not overweight,Citation34 and other Asian countries also have a high percentage of T2D patients with a BMI of <25.Citation35 It has become clear that genetic factors also play a role in conferring susceptibility, although no important risk genes have yet been identified.Citation36,Citation37 T2D is considered to be a heterogeneous disease as the severity of insulin deficiency, treatment requirements and prognosis varies between patients.Citation38,Citation39
Although treatments are available, diabetes remains a leading cause of blindness, end-stage renal disease, lower limb amputation and cardiovascular disease.Citation40 One study in the UK found that 48.9% of amputations from 2007 to 2010 were carried out on people with diabetes and diabetes conferred a 23.3x risk for amputations.Citation41 Additionally, diabetic patients also usually exhibit impaired wound healing, susceptibility to infection, neuropathology and impotence.Citation3 T2D has also been identified as a risk factor for Alzheimer’s and Parkinson’s diseases.Citation42,Citation43 Cardiovascular disease (CVD) accounts for 75% of all related deaths in diabetic patients and on average individuals with T2D have an increased risk of shorter life expectancy.Citation44,Citation45 There are a range of treatments available which improve insulin secretion and/or decrease peripheral tissue insulin resistance, which reduces hyperglycaemia.Citation46,Citation47 However, maintaining glycaemic control in patients is no easy task, as hypo- or hyperglycaemia can result from administration of the currently available treatments resulting in either too much or not enough, respectively, of an anti-hyperglycaemic effect.Citation3,Citation48 Regardless of the treatment regime, longitudinal studies in humans have demonstrated that beta-cell function generally deteriorates over time.Citation49,Citation50 It has been postulated that chronic hyperglycaemia and dyslipidaemia lead to a progressive decline in beta-cell function post-diagnosis.Citation50
Epidemiology of T2D
It has been estimated that there were ~382 million T2D patients globally in 2013 and that number has more than doubled in the last few decades.Citation1 The incidence and prevalence of T2D continue to increase. By 2035, it is estimated that there will be >590 million T2D people diagnosed.Citation1,Citation2 Although the prevalence and incidence of T2D vary between countries, T2D is still considered to be a global disease.Citation28 T2D used to be considered as a disease induced by ‘western lifestyles’ (high-calorie diets and sedentary lifestyles).Citation3 Interestingly, the rise in prevalence of T2D is estimated to be almost 4x as high in developing as in developed countries.Citation1,Citation25,Citation51 This is thought to be due to developing countries adopting ‘western lifestyles’ and the increase in obesity and the number of people being overweight in their populations.Citation1,Citation28,Citation52 In general, the age group with the highest risk of developing T2D is 40–60 years in developed countries and 60+ years in developing countries.Citation53 Although T2D is considered to be a disease associated with adulthood and the incidence of T2D increases with age, it is becoming more common for children to be affected.Citation54,Citation55 It is, however, likely that the number of cases of individuals diagnosed with T1D in adulthood is underestimated: it has been speculated that 5–15% of adult patients are misdiagnosed as having T2D when they may actually have T1D, which is currently an area of controversy in the literature.Citation56,Citation57
Diabetes in young people was previously thought to be T1D.Citation58 Until the early 1990s, it was rare for paediatric centres to have T2D patients. However, this has drastically changed since the late 1990s onwards with paediatric T2D accounting for up to 45% of newly diagnosed paediatric diabetes cases in the USA. The ”youth T2D” is not just a phenomenon limited to the USA.Citation59 Staggeringly, 80% of all new diabetes cases in Japan in children and adolescents were reported as T2D and other countries have also reported an increase in youth T2D.Citation60 However, many studies from Europe have reported that youth T2D is much rarer, accounting for only 1–2% of all diabetes mellitus cases.Citation55,Citation60 China has the highest number of T2D patients (~98 million) and India has the second-highest (around 65 million).Citation1 The USA was estimated to have around 24.4 million T2D patients in 2013.Citation61 Tokelau has been reported in 2013 as the country with the greatest prevalence of T2D (37.5%) in the national population. The Federated States of Micronesia, the Marshall Islands, and Kiribati all have T2D prevalence rates in their national populations of 35, 34.9 and 28.8%, respectively.Citation1 All other countries had T2D national population prevalence rates of under 26% in 2013.Citation1 Globally, the number of people with diabetes was estimated to be 285, 366, 382, 415 and 425 million in the years 2009, 2011, 2013, 2015 and 2017, respectively.Citation62 shows how T2D prevalence varied between countries in 2013 and 2019, and predictions for future data on T2D prevalence are also shown.Citation1,Citation62
Figure 2 (A) The varying estimated prevalence of T2D in 2013 and projections for 2035, between ages 20–79 years.Citation1 (B) The varying estimated prevalence of T2D in 2019 and projections for 2030 and 2045, between ages 20–79 years.Citation62 The numbers above are the values for each country, indicating the percentage increase of diabetes patients from 2013 to 2035 (A) or from 2019 to 2045 (B) for any given country, rounded to the nearest whole number. Data from these studies.Citation1,Citation62
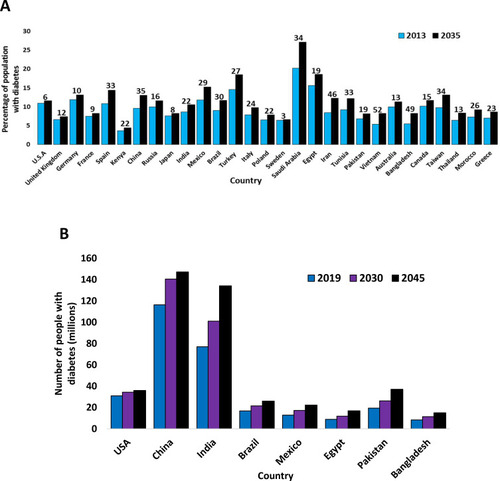
Diabetes was considered to be rare in the first half of the twentieth century in the USA; less than 1% of the population was diagnosed with diabetes in 1958.Citation63,Citation64 However, both the incidence and prevalence of T2D increased throughout the second half of the 20th century in developed countries, becoming an epidemic towards the end of the century and remaining as such into the 21st century.Citation1,Citation63 Over recent decades, T2D incidence and prevalence have also increased in developing countries, becoming a comparable health burden in these countries. In 1980, <1% of China’s population had diabetes but this increased to almost 10% by 2008.Citation25 In urban areas of South India, almost 20% of the population was estimated to be affected by diabetes in 2008.Citation65 In 2014, a study revealed that 46% of newly diagnosed T2D patients in India were under 40 years of age,Citation66 which is a contrast with the notion that patients in developing countries are usually over 60.Citation53 T2D incidence and prevalence have continued to increase globally in the 21st century as well. By 2045, it has been estimated that there will be over 700 million T2D patients worldwide.Citation1,Citation62 However, as highlighted in and , the future increase in the number of T2D patients will be partly due to the rise in the global population and not just increasing incidence.Citation1
Table 1 Variation of T2D Prevalence Between Global Regions in 2013 and Projections for 2035
Table 2 Variation of T2D Prevalence Between Global Regions in 2019 and Projections for 2030 and 2045
Studies have demonstrated that the incidence of T2D potently increased globally in the latter half of the 20th century (especially paediatric cases), and one study even reported the incidence of diabetes rose from 2.6 to 9.4% between 1980 and 1988 in the USA.Citation59,Citation67 Although life expectancy has generally increased globally over the last few decades, this does not fully explain the rise in T2D incidence and prevalence given the drastic increase in childhood cases, but it likely accounts for at least part of the rise in T2D in developing countries where patients are generally aged 60+ years.Citation47 Globally, it was estimated that diabetes accounted for ~12% of health expenditures in 2010 (at least $376 billion), and the healthcare cost will continue to rise to ~$490 billion in 2030.Citation28 Many developing countries do not have adequate infrastructure to treat this pandemic, meaning that diabetes is a serious concern for the future.Citation28 Additionally, there is continuing emerging evidence that the prevalence and incidence of diabetes are likely higher in developing countries than currently documented due to undiagnosed diabetes. Studies conducted in Africa estimated that 18% of adult diabetes patients in Libya and 75% of adult diabetes patients in Tunisia were undiagnosed.Citation68 Generally, the incidence of T2D continues to increase globally but at a steadier rate than in previous decades - this statement could be incorrect due to the uncertain global prevalence and incidence of undiagnosed diabetes.Citation1 The rise in the prevalence and incidence of T2D throughout the 20th and 21st centuries globally currently classifies this disease as a global pandemic; the rise in this increase is generally thought to be due to an increase in the percentage of the global population being overweight accompanied by individuals adopting more sedentary lifestyles.Citation28
Aetiology
Many risk factors have been identified for T2D.Citation3,Citation69 The main risk factor is obesity as being obese can increase the risk of developing T2D by 90-fold, and the majority of patients are overweight or obese.Citation9,Citation15 T2D risk is positively correlated with increasing BMI, and the risk rises exponentially with increasing BMI above 30.Citation15 In Western countries, it has been estimated that ~50% of T2D patients have a BMI of >30 and 30–40% have a BMI of 25–30.Citation70 However, in some Asian countries, ~50% of patients are not overweight.Citation34,Citation35 Surprisingly, underweight T2D patients have even been reported.Citation71,Citation72 Increased deposition of fat in the ectopic regions of the body (particularly visceral fat) also increases T2D risk by more than double.Citation3,Citation73 Genetics are known to play an important role in T1D but this is also the case for T2D.Citation37,Citation74 The concordance rate between monozygotic twins with T2D is higher (around 70%) compared with T1D (between 30 and 50%), and additionally, the lifetime risk of individuals for developing T2D with one affected parent is 40% and almost 70% if both parents are affected - these observations suggest that genetics play an important role in T2D susceptibility.Citation75,Citation76 Concordance rates between dizygotic twins with T2D (between 20 and 30%) are also higher than T1D (around 10%).Citation7,Citation75,Citation76 However, the highest odds ratio reported for a risk locus for T2D is 1.57 – this implies that more currently untested variants underlie T2D susceptibility.Citation37 The increased risk of developing T2D for relatives of type 2 diabetic patients may be due to them sharing similar diets and lifestyles as well as genetics.Citation7,Citation77 One study demonstrated the importance of genetics in T2D susceptibility independently of the diet as there was a higher prevalence of the disease in the twin population compared to the singleton populationCitation77– both populations had similar average BMI scores (26.1–26.3) with comparable standard deviation values (3.9–4.7). A cohort study found that alcohol and smoking also increased the risk of T2D, even in individuals who were classified as having low-risk diet and physical activity profiles.Citation78
Interestingly, longitudinal studies have demonstrated that “psychological stress-related circumstances” (such as stressful working conditions) or mental health problems (such as depression) increase the risk of T2D.Citation79 Globally, more males are diagnosed with T2D than females, and 14 million more men were diagnosed with this disease than the number of women in 2013.Citation80 Evidence demonstrates that adult men are at higher risk of T2D than women, which is at least partly thought to be due to differential adiposity storage patterns in men.Citation81 Studies have shown that men with T2D are more likely to develop CVD, but women with T2D who do develop CVD are more likely to have a worse prognosis, which is thought to be at least partly due to men being more likely to achieve medical targets in T2D (such as desirable plasma glucose control and blood pressure).Citation80,Citation81 The seasonality of T2D onset is not well studied, but a study conducted in Hungary found that seasonality followed a sinusoidal pattern; the peak month was March, and the trough month was August.Citation82 A recent study also found that Chinese individuals born outside of summer had a 9% increased chance of developing adult T2D than individuals born in summer.Citation83 Environmental changes over the last few decades could also play a role in T2D aetiology due to the use of pesticides, drugs and food additives in food processing and packaging.Citation84 However, minimal evidence exists linking T2D aetiology to altered food processing/packaging in recent decades.Citation84 Some environmental pollutants have been shown to alter β-cell function, and the best example is bisphenol A (used in food container manufacturing), which can cause impaired beta-cell function in animals.Citation85 However, it has not yet been determined whether prevailing environmental concentrations of these types of compounds can be a risk factor for diabetes.Citation84 Associations have been made between certain pathogens and T2D risk: herpes simplex virus type 1 and hepatitis C virus are risk factors for T2D, although it is not clear how this can be mechanistically explained.Citation86,Citation87 However, it has been established that hepatitis C promotes insulin resistance in the liver, which is thought to increase T2D risk.Citation88,Citation89 If this is the case, then this implies that T2D manifestation can be initiated by insulin resistance in the liver alone, which therefore suggests that the liver may play a much more important role in T2D aetiology/pathogenesis than is currently thought.
Pathogenesis
The main focus here is the current and future therapeutic potential of modulation of GLP-1R activity in T2D, but the altered activity of hormones involved in metabolic homeostasis observed in T2D are also discussed, as well as other factors, such as the nervous system and uncoupling protein 2 (UCP2), to highlight the complexity of its pathogenesis.
Typically, T2D does not manifest acutely in individuals but is preceded by an insidious phase of prediabetes.Citation90 Prediabetes is characterised by raised blood glucose levels (fasting plasma glucose levels of 6.1–6.9 mmol/L and two hours post glucose ingestion levels between 7.8–11 mmol/L) due to declining islet beta-cell mass and function but not enough to warrant a diagnosis of T2D.Citation90,Citation91 Patients with prediabetes are asymptomatic butCitation90 ~5–10% progress to T2D each year.Citation90,Citation91 Studies have demonstrated that weight loss and exercise can usually delay progression to T2D, or even prevent T2D from manifesting; lifestyle interventions reduce the risk of T2D progression in 40–70% of adults with prediabetes.Citation91,Citation92 It has been estimated that ~70% of individuals with persistent prediabetes will eventually develop T2D, and that >470 million people will have prediabetes by 2030.Citation91 Progressing to T2D is characterised by blood glucose levels of >7 mmol/L during fasting and a two hours post glucose ingestion reading of >11 mmol/L.Citation90,Citation93 The consensus in the literature is that T2D clinical manifestation is provoked by peripheral tissue insulin resistance, which is in turn, usually induced by obesity.Citation6,Citation30,Citation94 Obesity is characterised by elevated levels of cytokines and fatty acids, and it is thought that elevated levels of both provoke insulin resistance.Citation94 However, it has still not been determined how this occurs mechanistically.Citation95 Following the induction of insulin resistance, islet beta-cells can maintain normoglycaemia and metabolic homeostasis by increasing their secretion of insulin and/or by increasing their number.Citation49,Citation96,Citation97 Obesity has been estimated to induce an ~50% increase in islet beta-cell volume due to increased neogenesis.Citation49 However, over time islet beta-cells are seemingly unable to compensate for the insulin resistance and their ability to secrete insulin decreases and many islet beta-cells undergo apoptosis, which is thought to be a result of a variety of stressors, such as increased insulin demand, oxidative, endoplasmic reticulum, dyslipidemic, amyloidal, and inflammatory stress.Citation98,Citation99 The characteristic consequences of beta-cell pathology during T2D include impaired first-phase insulin secretion, ongoing insufficient insulin secretion to promote normolipidaemia (normal triglyceride levels, normal LDL cholesterol, and normal HDL-cholesterol) and normoglycaemia, dysfunctional glucose-sensing, and an increased proportion of proinsulin secretion.Citation98
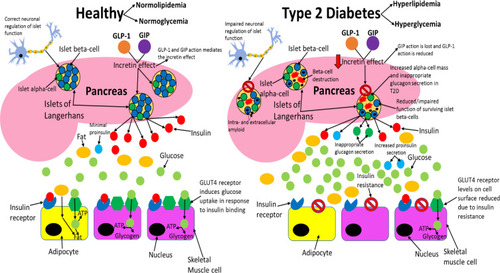
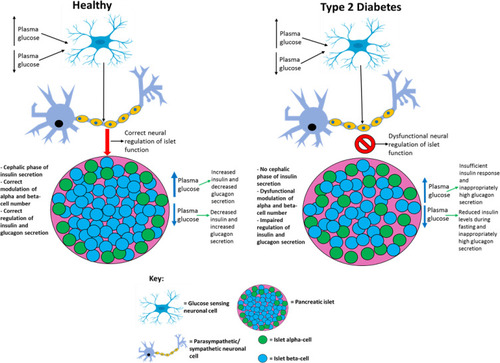
During prediabetes, pancreatic beta-cell number and function decline slowly, usually over a few years before T2D manifests.Citation91 It has been reported that the decline in islet beta-cell function can begin an average of 12 years before T2D diagnosis.Citation49 Interestingly, there have been reports of individuals who do not progress to T1D for >10 years despite persistent islet autoimmunity (slow progressors).Citation100 This demonstrates that the rate of islet beta-cell death can vary greatly between individuals before T1D or T2D diagnosis, enabling some individuals to remain disease-free for longer, possibly by similar mechanisms. The gradual decline in islet beta-cell number and function results in insulin levels becoming too low to promote metabolic homeostasis and T2D results.Citation49,Citation91 Hyperproinsulinemia has been reported in both T2D patients and individuals with prediabetes, suggesting that defective islet beta-cell insulin processing is integral to the early stages of disease pathology.Citation101,Citation102 In healthy subjects, proinsulin accounts for up to 20% of the insulin levels in circulation, but in T2D this value can reach up to 50% and total proinsulin levels have shown to be higher in T2D patients than healthy controls, which suggests that dysfunctional processing and secretion of insulin by the remaining islet beta-cell population further contributes to the decreased insulin levels observed in this disease.Citation101,Citation103 Symptoms of T2D manifest when insulin levels become too low to prevent hyperglycaemia, which includes dehydration, excessive thirst, increased susceptibility to infection, excessive urination, lethargy and blurred vision.Citation3,Citation47 Chronic superphysiological glucose concentrations also negatively affect the ability of islet beta-cells to secrete insulin, which further worsens hyperglycaemia and promotes T2D.Citation104 Peripheral tissues dependent on insulin to uptake nutrients from circulation can no longer do so to the same extent as before T2D.Citation47,Citation105 Hence, peripheral tissues adapt to rely on fat and catabolism of intracellular stored macromolecules, such as proteins, to generate the ATP they need.Citation3,Citation32 This results in weight loss (due to the breaking down of macromolecules), excessive eating and lethargy.Citation3,Citation32,Citation105,Citation106 compares healthy and type 2 diabetic phenotypes.
Figure 3 Comparing healthy and type 2 diabetic phenotypes. In healthy individuals, insulin is produced and secreted by beta-cells in the islets of Langerhans (found in the pancreas) when blood glucose levels are above 5mM. Beta-cells are the most abundant cell type in the islets (~70%) and alpha-cells (responsible for glucagon secretion) are the second most abundant (~20%). Insulin then binds to the insulin receptor (IR), which allows uptake of glucose into tissues by inducing translocation of GLUT4 receptors from intracellular vesicles to the plasma membrane. GLUT4 is primarily found in adipose tissue as well as skeletal and cardiac muscle. Glucose is then transported into the cell by GLUT4 from the bloodstream and catabolised in the cell for ATP production, which provides the fuel for intracellular processes, or glucose can be converted to either glycogen or fat for fuel storage after uptake. In individuals with T2D, many islet beta-cells have undergone apoptosis and the function of the surviving cells is impaired, which results in markedly reduced insulin levels in circulation. Additionally, peripheral tissue insulin resistance impairs the action of insulin, resulting in reduced uptake of glucose from circulation, as a result of decreased GLUT4 translocation to the membrane. Reduced insulin levels and action result in hyperglycaemia and hyperlipidaemia, and subsequent T2D associated symptoms manifest in the patient. Inappropriate glucagon secretion, diminished incretin hormone action, increased proinsulin secretion, impaired pancreatic islet neural regulation, and islet amyloid deposition are also characteristic of T2D. This figure and information in its legend are with data adapted from these studies.Citation105,Citation106,Citation111,Citation326–Citation328
It has been demonstrated that the insulin released during the early and late postprandial period by T2D patients is ~25 and 40%, respectively, of that produced by healthy individuals with a similar BMI.Citation107 Fasting insulin levels in T2D patients are ~50% of that observed in healthy subjects with a similar BMI,Citation107 demonstrating that T2D has a more significant effect on impairing the insulin response during the postprandial period in the early stages of the disease. Interestingly, insulin levels are higher in diabesity patients than in normal-weight T2D subjects,Citation21 implying that the islet beta-cell pathology is more severe in the latter group and that peripheral tissue insulin resistance contributes more to the disease phenotype in overweight T2D patients. Although it is known that the postprandial insulin response is similarly impaired in overweight and normal-weight T2D patients (determined by measuring the increase in insulin levels in comparison to fasting levels), absolute insulin levels are higher in overweight patients compared to disease-free normal-weight subjects, during both fasting and the postprandial period,Citation21 which strongly suggests that in overweight T2D subjects peripheral tissue insulin resistance is central to the clinical manifestation of this disease. T1D is more “severe” than T2D, as ~90% of beta-cells are destroyed by autoimmunity, which results in vastly reduced insulin levels.Citation108 Insulin treatment is essential for survival in T1D, as the absolute insulin deficiency results in the induction of ketoacidosis in an attempt to provide tissues dependent on insulin with adequate fuel.Citation106 Ketoacidosis is necessary during starvation to provide fuel to tissues when there is no adequate nutrient supply.Citation109 However, prolonged ketoacidosis is fatal due to the lowering of blood pH, which is why T1D is considered to be a deadlier form of diabetes than type 2, although a subset of T2D patients can develop ketoacidosis.Citation106,Citation110 Although ketoacidosis is largely considered to be a pathological response, fatality is inevitable under these circumstances, as impaired ATP production is fatal.Citation32,Citation106,Citation111
It has been firmly established that T2D is caused by the dysfunctional exocrine function of the pancreas, and peripheral insulin resistance is a requisite precursor for the development of this disease.Citation105,Citation112 Skeletal muscle insulin resistance has been associated with intracellular lipid accumulation.Citation94 However, endurance-trained insulin-sensitive athletes may have as much, if not more, lipid content in their skeletal muscles as insulin-resistant T2D patients.Citation113,Citation114 Thus, it seems that a high rate of ATP turnover in athletes prevents skeletal muscle insulin resistance, but a decreased demand for ATP in typically sedentary obese individuals with and without T2D induces insulin resistance.Citation94 It is reasonable to assume that insulin resistance in obese individuals without T2D may be desirable in skeletal muscle that has an abundance of stored nutrients and a low rate of ATP turnover, as the tissue’s ATP demands are met and hypertrophy would result from any further nutrient uptake. However, it is correct to state that peripheral tissue insulin resistance is pathological in T2D as it contributes to the disease phenotype.Citation30,Citation47 The insulin resistance in peripheral and hepatic tissues is present in obese and non-obese T2D patients, but not to the same extent.Citation115 Studies have shown that BMI is correlated to the degree of insulin resistance in Korean patients, even within the non-obese range.Citation116 Weight loss has been shown to decrease insulin resistance in both obese T2D patients and obese T2D-free individuals, suggesting that insulin resistance is related to increased nutrient storage.Citation117 Interestingly, insulin resistance is also observed in a proportion of T2D-free individuals within the normal weight range (BMI>18.5–25) and is reasonable to assume that this is pathological as, in theory, peripheral tissues are not at risk of hypertrophy due to insulin action and require continual nutrient influx to maintain ATP supply to meet their metabolic demands, as these individuals are not overweight and do not have an abundance of stored nutrients within their tissues.Citation23,Citation118 One study demonstrated that normalisation of both beta-cell function and hepatic insulin sensitivity in T2D patients was achieved by 8 weeks of dietary energy restriction alone (600 kcal a day), accompanied by decreased pancreatic and liver triacylglycerol stores.Citation119 After cessation of the low-calorie diet and returning to a normal diet for 12 weeks, 7 out of 10 participants remained in remission. Given that lipid accumulation both in and around the pancreas is associated with beta-cell pathology and confers T2D risk,Citation120 the observation that T2D remission was accompanied by a decrease in pancreatic fat content suggests that increased pancreatic fat storage is an important component of T2D aetiology/pathogenesis. In healthy individuals, insulin sensitivity varies naturally, which is thought to be beneficial for promoting metabolic homeostasis.Citation121–Citation123 The general consensus in the literature is that chronic insulin resistance, induced by obesity, is thought to be pathological overall in the metabolic syndrome, which is a syndrome characterised by a range of disorders that collectively predispose risk to T2D and cardiovascular pathology.Citation94
A study examined pancreatic tissue from 124 autopsies: 91 obese cases (BMI >27kg/m2; 41 with T2D, 15 with impaired fasting glucose [IFG], and 35 non-diabetic subjects) and 33 lean cases (BMI <25kg/m2; 16 types 2 diabetic and 17 non-diabetic subjects.Citation124 Obese humans with IFG and T2D had a 43 and 60%, respectively, decrease in islet beta-cell volume compared to obese nondiabetic individuals. This decrease in islet beta-cell volume in obese T2D patients was found to be due to a decreased number of islet beta-cells, and interestingly the volumes of these cells were found to be similar between non-diabetic obese and diabetic obese individuals. The frequency of islet beta-cell replication was found to be similar between obese nondiabetic and T2D patients. However, it was estimated that there was an ~3-fold increase in apoptosis in obese T2D individuals. The majority of diabesity cases were found to have islet amyloid in their islets (~80%), whereas only ~10% of obese individuals without T2D had amyloid deposits. Amyloid deposits had a similar prevalence in IFG obese individuals to that observed in T2D-free obese individuals. Islet beta-cell dysfunction in diabesity patients is therefore attributed to increased rates of apoptosis and amyloid deposition, and not to rates of replication and new islet formation as this is similar to non-diabetic controls.
Currently, why individuals of normal weight develop T2D is largely elusive, but pathogenesis appears to be similar to diabesity patients, although there are some striking differences according to the study conducted in 2003.Citation124 Islet beta-cell mass was shown to be decreased in lean T2D patients by ~41% in comparison to lean T2D-free controls, due to a decreased number of these cells. Islet amyloid was present in 88% of lean T2D patients but only 13% of lean healthy controls. New islet formation and beta-cell replication were similar between lean T2D patients and lean T2D-free individuals, but the frequency of beta-cell apoptosis was increased by 10-fold in lean T2D patients. Hence, lean T2D is also characterised by an increased rate of beta-cell apoptosis with a normal, but too slow a replication rate of these cells to restore their numbers to normal levels. It is not known if islet amyloid deposits are produced due to the chronic hyperglycaemia associated with this disease. It is also not clear if these deposits affect the prognosis of T2D.Citation124,Citation125
What has become clear is that islet amyloid deposits are very likely to be directly involved in the pathogenesis of T2D, although it has not been well established how mechanistically.Citation124,Citation126,Citation127 Islet amyloid polypeptide (IAPP) (more commonly known as amylin) is co-secreted with insulin from islet beta-cells in healthy individuals during the fed state and acts as an anti-hyperglycaemic agent by decreasing gastric motility, promoting satiety and inhibiting glucagon secretion.Citation128 The amylin analogue pramlintide is used for T1D and T2D treatment adjunct with insulin therapy in the USA if patients are not achieving adequate glucose control despite optimal insulin therapy.Citation129 Virtually all T2D patients have IAPP-containing plaques in their islets.Citation128 IAPP appearance was first noticed in the 1980s to correlate with hyperglycaemia in primates.Citation130 Several lines of transgenic mice have been generated which produce human IAPP, and they were shown to have islet amyloid accumulation, increased beta-cell apoptosis, decreased beta-cell mass, impaired glucose tolerance, and some developed diabetes.Citation128 The current consensus in the literature is that extracellular plaques of IAPP are not causative of beta-cell pathology, but rather intracellular oligomers of this protein observed in T2D patients (but not in T2D-free individuals) produce the cytotoxic effects.Citation128,Citation131 It has been elucidated that beta-cells are usually protected from IAPP oligomers by autophagy processes, so it seems that these processes are defective in prediabetes and T2D, which is likely pivotal for the pathogenesis of T2D.Citation128,Citation132 Both T1D and T2D manifest due to beta-cell apoptosis and the activated apoptotic pathways are thought to share similarities, but the aetiology of apoptosis is different between the two diseases.Citation133
Although obesity is a major risk factor for T2D, paradoxically overweight and obese patients have been reported to have had a lower mortality rate than normal-weight patients due to cardiovascular pathology associated with T2D and this phenomenon was termed “the obesity paradox”.Citation134,Citation135 Furthermore, weight loss has been reported to increase mortality and morbidity in T2D and cardiovascular co-morbidity patients.Citation136 Additionally, a study found that the risk of amputation in non-elderly diabetic men decreased with increasing body weight.Citation137 Peripheral and hepatic insulin resistance is present in obese and non-obese T2D patients, but not to the same extent in non-obese subjects.Citation115 Studies have shown that BMI is correlated to the degree of insulin resistance in Korean patients, even within the non-obese range.Citation116 Interestingly, a study from India found that fasting and postprandial plasma glucose levels were higher in T2D patients with a BMI <18.5.Citation71 The aetiology of T2D patients with a BMI of under 25 is thought to be largely attributed to beta-cell dysfunction, which is thought to be greater than that observed in obese patients.Citation71 A study conducted in Chicago found that glycaemic control was worse in T2D patients with a BMI of 18–25 than in individuals with a BMI of over 25, but these patients had lower insulin resistance.Citation138 The study found that smoking and alcoholism confer an increased risk for non-obese T2D than obese T2D, chronic alcoholism induces islet beta-cell dysfunction and apoptosis.Citation139 Strikingly, T2D has been shown to manifest in non-overweight individuals in the absence of insulin resistance, although this is thought to be rare.Citation140 This implies that pancreatic exocrine dysfunction can induce T2D independent of both insulin resistance and a BMI>25. Whether or not weight loss in T2D patients with a BMI of under 25 improves prognosis has not been determined.Citation71 It has been well established that Asians are at increased risk of non-overweight T2D. In Japan, more than 50% of patients have a BMI of under 25, but they have more abdominal fat at the equivalent BMI to Caucasians.Citation35,Citation71
T2D is not only attributed to beta-cell dysfunction but also alpha-cell dysfunction.Citation141,Citation142 Pathologically high plasma concentrations of glucagon are observed in T1D, advanced T2D, and diabetic ketoacidosis.Citation142 It is well established that islet alpha-cell populations increase in T1D, whereas in T2D, patients can have alpha-cell numbers increased or unaltered.Citation141 Paradoxically, T2D patients have increased levels of glucagon during the postprandial period, leading to augmentation of postprandial hyperglycaemia associated with impaired insulin secretion.Citation142,Citation143 Hence, it is correct to state that the behaviour of diabetic islet alpha-cells is different for healthy individuals, as an abundance of nutrients during the postprandial period prevents glucagon secretion in healthy individuals by raising ATP levels (as discussed earlier in this review).Citation144 Therefore, in T2D, the islet alpha-cells must have altered biochemical pathways inducing altered mechanisms of glucagon secretion. Studies have estimated that postprandial hyperglucagonemia may contribute to ~50% of the pathological increase in plasma glucose levels after glucose ingestion.Citation145–Citation147 Interestingly, fasting hyperglucagonemia has been reported in some T2D patients with moderate glycaemic control, and glucagon levels are ~50% higher in diabetic subjects.Citation146,Citation148 The dysfunctional glucagon secretion in T2D could be due to the decreased insulin levels, as insulin suppresses alpha-cell function.Citation105,Citation149 However, islet alpha-cells should have their exocytotic function suppressed during the fed state, as intracellular ATP levels should be high as a result.Citation144 However, studies have demonstrated that blocking insulin signalling in islet alpha-cells in mice results in defective suppression of the fed state on glucagon secretion, highlighting the pivotal role of insulin in the regulation of the alpha-cell.Citation105,Citation149
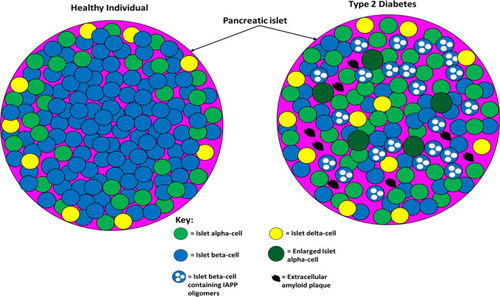
Interestingly though, dysregulated glucagon secretion is not significant in adolescents with T2D.Citation150 There is some evidence that the liver is more sensitive to glucagon in T2D patients, further exacerbating the hyperglycaemia, although there have been conflicting findings with this observation.Citation142 Both T1D and T2D induce major changes in the structure of animal and human pancreatic islets.Citation141 The changes in the islets of T2D patients have been postulated to induce altered signalling within the islets, which contributes to the phenotype of this disease.Citation141 The increase in alpha-cell mass likely results in increased alpha–alpha cell contacts at the expense of beta–beta cell contacts, which may alter intra-islet signalling.Citation151 The amyloid deposits in the islets have also been postulated to alter intra-islet signalling.Citation124 Islet alpha-cells are usually found in the periphery of the islets, but in diabetes, many of the alpha-cells are found in the centre as well; in rodent models, this can be reversed by restoring normoglycaemia.Citation141,Citation152 The proportion of delta–delta and delta–alpha cell contacts also increases, likely as a result of the beta-cell destruction in T2D.Citation151 In T2D rodent models, delta-cells were reported to migrate from the periphery of the islets to the centre.Citation152 How the morphology, migration and number of islet gamma- and epsilon-cells are altered in T2D is not well studied.Citation141 Although there have been reports suggesting that the delta-cell number is increased in diabetes, other studies suggested that the number of these cells was unaltered in patients’ islets.Citation153,Citation154 Interestingly, a recent study found that the delta-cell number and volume were decreased in baboons with impaired fasting glucose by ~41%, and this was due to apoptosis.Citation155 Evidence is emerging that a subset of islet beta-cells may undergo conversion into other endocrine islet cell types in T2D, which may account for the increased alpha-cell population observed in some T2D patients and their distorted insulin-to-glucagon ratios.Citation156 compares pancreatic islet architecture between healthy and type 2 diabetic individuals.
Figure 4 The difference between pancreatic islet architecture in healthy individuals and individuals with T2D. In healthy individuals, beta-cells are situated centrally and peripherally and are the most abundant cell type (~70%). Non-beta cells are found in the periphery of the islets and constitute ~30% of the cell population (20% alpha-cell and 10% other cell types). Islet architecture is altered in T2D with a greatly diminished population of beta-cells, more alpha-cells, more delta-cells, migration of alpha- and delta-cells into the centre, extracellular amyloid plaque deposits, intracellular IAPP oligomers in beta-cells, and enlarged alpha-cells. The altered architecture in T2D produces different intra-islet paracrine signalling which impairs metabolic homeostasis. This figure and information in its legend are with data adapted from Brereton et al.Citation141
The incretin effect is greatly diminished in T2D where GLP-1 and GIP account for <20% of the insulin release after glucose ingestion in patients.Citation157 The current consensus in the literature is that GLP-1 and GIP circulation levels are comparable between T2D patients and nondiabetic controls in response to nutrient challenges.Citation158 Hence, the diminished incretin effect is due to dysfunctional beta-cells.Citation159 GLP-1 infusions can normalise blood glucose levels, and GLP-1 analogues are used to treat T2D.Citation3 However, GIP action is lost in T2D and even infusions of GIP that result in supraphysiological concentrations fail to elicit a significant insulin secretory response in T2D patients.Citation160 Studies have demonstrated that intensive insulin administration treatment results in improved insulin secretion from islet beta-cells after GIP infusion.Citation161,Citation162 It has been established that hyperglycaemia induces down-regulation of GIPR expression in beta-cells, so it is plausible that in this study GIPR expression may have been upregulated in these cells due to reducing plasma glucose levels.Citation159 Therefore, although it is clear that GIP action is lost in T2D it could still have future therapeutic potential for treating T2D. GIP action in prediabetes has not been established to date, so it is currently not clear if impaired GIP action is involved in T2D pathogenesis or manifests post-diagnosis.
Additionally, pancreatic neuronal activity is also impaired in T2D.Citation163 The pancreas is richly innervated by the sympathetic and parasympathetic nervous system.Citation164 The cephalic phase of insulin secretion is lost in T2D and glucagon secretion is not suppressed during the fed state.Citation163 This implies that if the nervous system does suppress glucagon secretion in an insulin-independent manner during the fed state in healthy individuals then this is absent in T2D. The parasympathetic activity also plays a role in stimulating beta-cell proliferation in adult animals, which is at least partly regulated by acetylcholine binding to muscarinic receptors on beta-cells.Citation164,Citation165 The neuronal activity has also been postulated to modulate the alpha-cell number.Citation163 This demonstrates that the nervous system is needed for both islet function and islet-cell proliferation when appropriate. Parasympathetic and sympathetic nervous systems are known to influence endocrine pancreas postnatal development and plasticity in adult animals.Citation163 Dysfunctional regulation of the pancreatic islet by the nervous system contributes to the clinical phenotype of T2D, but whether or not this occurs before or after T2D diagnosis remains elusive, although it is reasonable to assume that it does, influencing the altered islet architecture observed in T2D.Citation141,Citation163 It has been confirmed that glucose-sensing cells in the central nervous system (CNS) are excited by either a rise (glucose excited neurons) or decrease (glucose inhibited neurons) in plasma glucose levels, and it has been postulated that these neurons regulate the sympathetic and parasympathetic branches of the autonomic nervous system, which are known to influence insulin and glucagon secretion.Citation163 It is likely that the pathology of these central neurons contributes to the T2D phenotype and may partly, or even exclusively, induce T2D pathogenesis by currently unknown mechanisms. One study clearly demonstrated the importance of the nervous system’s ability to regulate islet beta-cell number and function: GLUT2 neuronal knockout mice exhibited impaired glucose-stimulated insulin secretion (GSIS), hyperglucagonemia, and decreased beta-cell proliferation and mass in comparison to controls.Citation166 Importantly, treatment of the control mice with the ganglionic blocker chlorisondamine reduced the proliferation rate of beta-cells by 50%, but in GLUT2 neuronal knockout mice, chlorisondamine did not further reduce the proliferation rate. This study clearly shows that the activity of the neuronal glucose-sensing cells and islet alpha- and beta-cells function synergistically to promote appropriate plasma insulin and glucagon levels – this synergism is known to be dysfunctional in T2D.Citation163 summarises the differential neural regulation of the pancreas between healthy and type 2 diabetic individuals.
Figure 5 Comparing the differential neural regulation of pancreatic islets in healthy and type 2 diabetic individuals. T2D patients do not exhibit the cephalic phase of insulin secretion and based on the findings from studies it is possible that dysfunctional neuronal regulation of pancreatic islets contributes to the impaired GSIS, hyperglucagonemia, and decreased beta-cell proliferation and mass observed in this disease. This figure and information in its legend are with data adapted from these studies.Citation163,Citation166
The notion that glucolipotoxicity conditions in circulation (associated with obesity) induces islet beta-cell dysfunction has been an attractive area of research.Citation167 A number of in vitro studies, using insulin-secreting cells and isolated islets, have found that exposure of islet beta-cells to glucolipotoxicity conditions results in impaired GSIS, impaired insulin expression, and cell death by apoptosis.Citation167,Citation168 Importantly, several of these studies have demonstrated that fatty acids do not induce beta-cell dysfunction in the absence of elevated glucose, highlighting that fatty acids and glucose are not cytotoxic independently.Citation168,Citation169 The application of these experiments to in vivo situations is questionable as it has been demonstrated that non-diabetic obese subjects exhibit increased insulin secretion and an increase in beta-cell number and mass, and islet amyloid deposits are rare in these individuals as aforementioned in this review. Thus, it is reasonable to assume that glucolipotoxicity diets do not induce beta-cell pathology before the prediabetes phase and pre-T2D diagnosis. However, in vivo studies on healthy rats have demonstrated similar findings to that of in vitro when studying the effects of glucolipotoxicity on insulin secretion.Citation170 Given that weight loss and a controlled diet improve glycaemic control in overweight T2D patients, it seems likely that glucolipotoxicity diets worsen the disease phenotype post-diagnosis.Citation12 However, attributing T2D pathogenesis to glucolipotoxicity diets does not explain how individuals with a BMI of under 25 develop this disease. To further complicate the issue, data obtained from humans in lipid infusion studies have produced conflicting results as to whether or not glucolipotoxicity conditions impair or boost insulin secretion.Citation171
Studies conducted by Affourtit and colleagues attempted to explain mechanistically how glucolipotoxicity conditions might impair GSIS.Citation172,Citation173 Interestingly, they have found that the impaired GSIS observed during in vitro studies involving exposing pancreatic beta-cell line INS-1E to glucolipotoxicity conditions (palmitate + high glucose) was found not to be dependent on mitochondrial dysfunction.Citation173 From the observations of this study, it seems that whatever role glucolipotoxicity may have in impairing GSIS in T2D is disconnected from mitochondrial dysfunction, which is surprising given the role of the mitochondria in ATP and reactive oxygen species (ROS) generation needed for both phases of insulin secretion.Citation174,Citation175 The concept of “beta-cell rest” to treat T2D has emerged given the observations from both in vitro and in vivo studies.Citation98 Isolating islets from diabetic rodents and exposing them to euglycemic conditions for 8–12 h resulted in the return of both normal insulin secretory capacity and beta-cell function.Citation89 Furthermore, bariatric surgery (which results in reduced food intake post-surgery) and low-calorie diets can induce T2D remission, which can be viewed as in vivo evidence that reducing demand on the beta-cell (ie, “beta-cell rest”) improves endogenous insulin secretory capacity.Citation98 Administering diazoxide (which inhibits endogenous insulin secretion and therefore induces a degree of transient “beta-cell rest”) to T2D patients resulted in a 280 and 500% increase in insulin levels over baseline after stimulation tests in T2D low and high beta-cell reserve groups, respectively.Citation176 This study provides in vivo evidence that “beta-cell rest” can at least temporarily allow insulin secretory capacity recovery, and in vitro studies examining the effect of diazoxide on diabetic human islets produced similar findings.Citation177 Another in vivo study found comparable effects on improved beta-cell function following short-term continuous subcutaneous insulin infusion in T2D patients that were uncontrolled by conventional oral therapies.Citation88 This resulted in near euglycemia being achieved in the participants and glycaemic control was improved after cessation of insulin infusion. Islet beta-cell function tests conducted 2 days after cessation of insulin infusion revealed that the peak insulin response was notably improved. Reducing demand on the islet beta-cells by improving insulin sensitivity through weight loss and/or pharmacological means has been reported to improve T2D prognosis and delay/prevent pre-diabetic individuals progressing to T2D, suggesting that inducing “beta-cell rest” to even a degree alleviates the pathology that is associated with this disease.Citation92,Citation98 Despite the evidence generated by both in vitro and in vivo studies supporting that glucolipotoxicity impairs beta-cell function, these findings likely do not apply to non-obese/overweight diabetic individuals.
UCP2 is a protein present in the beta-cell mitochondria inner membrane, which was postulated to dampen GSIS by dissipating mitochondrial proton motive force as heat (due to its analogy with UCP1), thereby reducing the efficiency of ATP production. Approximately 20% of the proton leak in beta-cells is thought to occur due to the presence of UCP2.Citation178,Citation179 The detection of this protein in islet beta-cells is puzzling as sufficient ATP levels are vital for the correct function of these cells, and studies have shown that overexpression of UCP2 in isolated beta-cells impairs GSIS.Citation180–Citation182 It has been shown that a high proton motive force (PMF) results in the production of ROS, and as UCP2 activity can lower the PMF by increasing proton leak, it can then lower intracellular ROS, as well as increased production of ROS, is thought to be at least partly dependent on an increased mitochondrial PMF in islet beta-cells.Citation180,Citation183–Citation185 As ROS are largely thought to be cytotoxic molecules, it was assumed that the fact UCP2 attenuates ROS production could mean that this protein has a protective role in the cell.Citation32,Citation185,Citation186 This notion had led to a debate as to whether or not the presence of UCP2 in these cells is pathological given its ability to decrease ATP production or beneficial given its ability to regulate levels of ROS.Citation179 However, it has been demonstrated that UCP2 does not appear to regulate GSIS, as palmitate + high glucose impaired GSIS to the same extent in INS-1E cells with and without UCP2 (UCP2 was knocked down via RNAi).Citation187 UCP2 knockdown was shown to only minimally and not significantly increase the coupling efficiency (<10%) when INS-1E cells were exposed to 28mM glucose and palmitate. Oddly, it was found that palmitate with 28 mM glucose dampened the coupling efficiency in UCP2 depleted cells more than in UCP2 positive cells. Based on these findings, it appears that UCP2 is not involved in either GSIS or regulation of the efficiency of mitochondrial energy transduction in INS-1E cells in vitro.
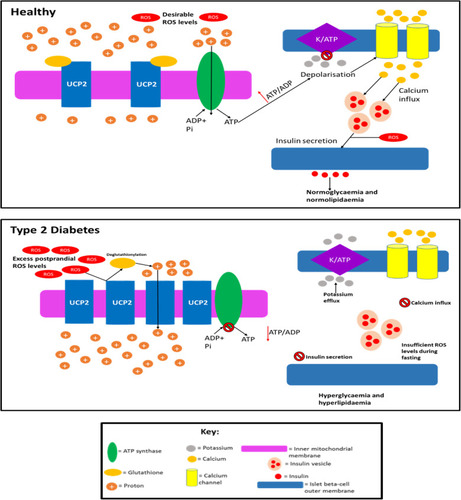
It has been postulated that UCP2 may not be an uncoupling protein despite its analogy with UCP1, but it likely functions as a carbon skeleton exporter from the mitochondrial matrix, and there is experimental evidence for this.Citation188 To further complicate the role of UCP2 in GSIS, in vivo studies have found that global UCP2 knockout mice exhibit either improved glucose tolerance and GSIS, or unaltered glucose tolerance and impaired GSIS depending on their genetic background.Citation179,Citation187 Beta-cell specific ablation of UCP2 produces glucose-intolerant mice that have increased proton gradients across their inner mitochondrial membranes (suggesting that this protein does mediate premature proton leak), but oddly, ATP levels and respiration rates are unaltered.Citation184 The enhancement of GSIS in the beta-cells of these mice was suggested to be due to the increased ROS levels associated with UCP2 knockout. The glucose intolerance observed in these mice was thought to be due to a greater alpha-cell area and higher glucagon content in the islets, and glucagon was secreted by alpha-cells even during high plasma glucose, suggesting that UCP2 presence in beta-cells is necessary to maintain appropriate alpha-cell behaviour during the fed state. Affourtit and colleagues have demonstrated that mitochondrial function plays an important role in cell viability, as palmitate-induced mitochondrial superoxide formation results in reduced viability; this can be prevented by co-exposure to palmitoleate (an unsaturated fatty acid).Citation172 Hence, mitochondrial activity during glucolipotoxicity may play an important role in beta-cell survival in vitro and in vivo. A critical gap in UCP2 research is that studies have not taken into account the glutathionylation state of this protein.Citation189 Glutathionylation and deglutathionylation are thought to deactivate and activate proton leak, respectively.Citation189,Citation190 There is evidence that this post-translational modification is regulated by intracellular ROS levels (increased ROS causes deglutathionylation and decreased ROS does the opposite); hence, it is now relatively well established mechanistically how ROS levels can regulate themselves in these cells.Citation189,Citation190 It is important that future studies examining UCP2 activity and levels also monitor glutathionylation status as well, which could explain any unexpected phenotypes. Currently, the role of UCP2 in healthy individuals and T2D patients is unclear, but if this protein behaves pathologically in T2D by inducing decreased ATP production, then this could explain the dysfunctional insulin secretion observed. proposes how UCP2 activity and quantity alter in T2D.
Figure 6 Speculation of how UCP2 activity and quantity is altered in T2D. In healthy individuals: UCP2 levels are basal, UCP2 is glutathionylated, ROS levels are low enough so that they do not cause cell damage but the levels are sufficient so that these molecules can amplify insulin secretion, and proton leak exists (~55% of islet beta cells’ metabolism is wasted due to proton leak which is not caused by UCP2) but is minimal, so enough of the protons can move down the gradient through ATP synthase to generate sufficient ATP levels. In T2D, UCP2 is deglutathionylated (due to increased ROS levels) and upregulated allowing for much greater proton leak. Consequently, the majority of the protons prematurely leak through the inner mitochondrial membrane and do not pass through ATP synthase, although there is likely a period before the UCP2 deglutathionylation where ATP production occurs, it is unlikely that enough ATP is generated during this time to promote adequate insulin secretion. Hence, overall ATP production is impaired in the cell resulting in the inadequate insulin secretion observed in T2D during both the postprandial period and fasting. ROS levels are likely too high (leading to cytotoxic damage) during the postprandial period as a result of severe nutrient oversupply to islet beta-cells, due to the enhanced hyperglycaemia and hyperlipidaemia in T2D during this period. The excess ROS levels result in UCP2 deglutathionylation and a subsequent dissipated PMF, meaning that ROS formation during fasting is impaired and insulin secretion is not amplified by ROS to alleviate the chronic hyperglycaemia and hyperlipidaemia in fasting T2D subjects. Although UCP2 may become glutathionylated upon ROS levels decreasing during fasting, this likely reverses during subsequent feeding as ROS levels once again become undesirably high, restarting this cycle. This figure and information in its legend are with data adapted from these studies.Citation32,Citation175,Citation178,Citation179,Citation189,Citation190,Citation329,Citation330
Glucolipotoxicity conditions induce beta-cell dysfunction in vitro but it is not clear if this is the case in vivo due to the reasons aforementioned. Additionally, GSIS defects in vitro do not appear to be attributable to the mitochondria, but this has not been confirmed in vivo, and the role of UCP2 and its glutathionylation state in GSIS in vivo is currently a source of confusion.
Extrapancreatic Tissues Involvement in T2D: Aetiology/Pathogenesis and Post-Diagnosis Complications
Whilst it is firmly established that pathological pancreatic function and peripheral tissue insulin resistance are central to T2D aetiology/pathogenesis, there is increasing evidence that other extrapancreatic pathologies may contribute.Citation30,Citation191,Citation192 The diabetic phenotype includes systemic pathology, such as in the CVS, skeletal muscle, nervous system (central and peripheral), skin, kidneys and gastrointestinal tract (GIT), which further exacerbates the disease phenotype and/or induces post-diagnosis complications.Citation30,Citation193–Citation195 However, research evidence over the last few decades suggests that some of these pathologies manifest during the prediabetic phase and is possibly therefore involved in both the disease pathogenesis and aetiology.Citation191,Citation192 For example, skin and liver pathology can manifest during the prediabetic stage, and even in individuals with metabolic syndrome before islet beta-cell dysfunction occurs.Citation191,Citation196
It is firmly established that chronic insulin resistance in skeletal muscle of people with T2D contributes to the diabetic phenotype and results in poor exercise tolerance.Citation3,Citation197 However, the altered metabolism of skeletal muscle in patients results in a reduced capacity to oxidise fat and promote fat storage, which further exacerbates the disease phenotype.Citation192 This behaviour of skeletal muscle is more pronounced in T2D as diabesity patients have a reduced ability to oxidise fat than obese-matched controls during exercise.Citation192,Citation198 Impaired energy utilisation results in decreased exercise tolerance and increased fat storage, which further promotes insulin resistance.Citation192 Interestingly, this pathological behaviour of skeletal muscle has also been detected in individuals with prediabetes, suggesting that these alterations may play a role in the development of both prediabetes and T2D.Citation192,Citation199 However, contradictory to this, some studies have shown that fat oxidation is increased in the muscle of T2D patients.Citation200 Additionally, metabolic inflexibility is also characteristic of T2D patients, whereby patients’ ability to switch from lipid to carbohydrate oxidation during insulin stimulation is impaired, which also further contributes to impaired exercise tolerance.Citation192 Obese normoglycemic individuals were shown to have better metabolic flexibility than weight-matched T2D patients.Citation201 However, studies have found that metabolic inflexibility of glucose is largely due to defective glucose transport,Citation201,Citation202 which suggests that skeletal muscle is unable to promote sufficient insulin secretion from type 2 diabetic islet-beta cells via any signalling mechanisms, in which it likely attempts to achieve given its need for increased glucose uptake.Citation3,Citation197 Additionally, given that metabolic inflexibility can be improved by weight loss and exercise, this further supports the notion that impaired glucose uptake largely accounts for this phenomenon, as these activities result in improved insulin sensitivity.Citation203
The phenomenon of metabolic inflexibility has also been observed in individuals with prediabetes, implying that this may play a role in the aetiology/pathogenesis of both prediabetes and T2D.Citation204 Mild muscle atrophy is common in middle-aged T2D patients and becomes more significant during older age and diabetic neuropathy.Citation205,Citation206 Although inflammation and chronic insulin resistance are thought to promote muscle atrophy in T2D patients,Citation206 it is reasonable to assume that the impaired energy utilisation promotes the degradation of intracellular proteins to yield sufficient ATP levels, which would contribute to the observed muscle atrophy. Evidence is also emerging that type 2 diabetic muscle may have detrimental effects on beta-cell function via secretion of myokines into circulation: myokine profiles differ between T2D patients and controls.Citation207 TNFα secretion from T2D subjects was higher than healthy controls,Citation207,Citation208 and one in vitro study has shown that this myokine impairs beta-cell function.Citation208 Decreased numbers of mitochondria have been observed in the muscle cells of T2D patients, which has created the notion that a decreased population of mitochondria induces insulin resistance.Citation200 However, rodent studies have found that an increase in mitochondrial biogenesis occurs concomitantly with the development of muscle insulin resistance, implying that this notion is incorrect.Citation209,Citation210 This increase in mitochondria is postulated to be an early, transient event that is lost as the insulin resistance progresses.Citation211 Furthermore, decreasing the mitochondrial population or disrupting mitochondrial function increases basal and insulin-stimulated glucose uptake into rodent skeletal muscle.Citation200
Decreased insulin secretion is known to result in increased hepatic glucose production, which contributes to hyperglycemia, but type 2 diabetic livers also exhibit hepatic insulin resistance, which further increases glucose production during the fasting and postprandial period, giving the liver a direct role in contributing to the disease phenotype.Citation192,Citation212 Interestingly, insulin-induced suppression of gluconeogenesis and glycogenolysis has also been shown to be impaired in individuals with prediabetes, suggesting that altered liver function is an important component during the initial stages of T2D pathology.Citation191 The diabetic phenotype promotes fat storage in the liver due to elevated fatty acid levels in circulation and ~70% of T2D patients develop non-alcoholic fatty liver disease (NAFLD).Citation213,Citation214 Elevated plasma lipid levels in T2D result in increased delivery of fatty acids to the liver, which is largely caused by reduced suppression of lipolysis in adipose tissue due to decreased insulin action/secretion in diabetes.Citation213 NAFLD is an established risk factor for CVD and so increases the risk of cardiovascular complications in T2D patients.Citation214,Citation215 T2D also increases the risk of NAFLD progressing to non-alcoholic steatohepatitis and subsequent liver cirrhosis.Citation214 The increased fat storage in the liver of T2D patients further promotes hepatic insulin resistance and induces impaired hepatic fatty acid oxidation, which in turn, further increases hepatic fat storage and exacerbation of insulin resistance, which worsens hyperglycemia as a result.Citation213,Citation216
Proinflammatory cytokines, produced by adipose tissue as a consequence of obesity, are known to be cytotoxic to beta-cells, and they likely contribute to T2D pathogenesis and islet dysfunction post-diagnosis.Citation99,Citation217 There is also evidence that these cytokines play a role in the induction of insulin resistance.Citation218 Thus, the adipose tissue also likely contributes to T2D aetiology/pathogenesis, but to what extent remains currently unknown.Citation94 The reduced insulin levels also result in increased lipolysis and decreased fat storage in adipocytes, resulting in hyperlipidaemia and increased deposition of fat into other tissues, such as skeletal muscle and liver.Citation219,Citation220 There is also evidence that altered gut microbiota profiles observed in T2D patients and overweight subjects also promote systemic inflammation and proinflammatory cytokine production, which is thought to encourage the disease pathogenesis and contribute to the disease phenotype post-diagnosis.Citation221 T2D patients exhibit alterations in bone behaviour, which is believed to increase the risk of fracture and promote bone fragility: older adults with T2D have up to an 80% increase in the risk of extremity fracture.Citation222 Decreased levels of the parathyroid hormone have been seen in T2D, indicating decreased bone turnover, and increased levels of sclerostin in patients also indicate inhibited bone formation.Citation222–Citation224 Decreased insulin-like growth factor (IGF)-1 levels in T2D also likely results in decreased bone formation in T2D.Citation222–Citation224
Evidence is now emerging that T2D induces premature ageing of the CNS, as some (but not all) studies have found that patients exhibit impaired cognitive performance and electrophysiological defects in the hippocampus, as well as pathological brain morphological abnormalities – all of which are reminiscent of the changes observed during normal ageing.Citation225 Interestingly, GLP-1R is downregulated in the hypothalamus of T2D patients in comparison to healthy controls, suggesting that the reduced ability of GLP-1 to induce satiety may contribute to the dysfunctional feeding behaviours and metabolic homeostasis observed in these patients.Citation226 Whether or not this downregulation occurs before or after T2D diagnosis is unknown.
Peripheral neuropathy is a common complication of T2D, and an estimated 50% of patients develop diabetic neuropathy after 25 years of being diagnosed.Citation227,Citation228 Neuropathy may result from excessive levels of nutrients in circulation (resulting in complications such as oxidative damage) and impaired cardiovascular function (resulting in complications such as hypoxia), discussed in detail elsewhere.Citation228 Diabetic neuropathy can have a range of consequences depending on the neurons affected: sensory neuropathy results in numbness or pain, motor neuron neuropathy results in impaired muscle movement and autonomic neuropathy results in dysfunctional regulation of involuntary activities, such as internal organ function.Citation229,Citation230
A recent study found that activation of preproglucagon (PPG) neurons in the brainstem of rodents reduced basal hepatic glucose production, enhanced intraperitoneal glucose tolerance, and augmented hepatic insulin sensitivity. This suggests that PPG neuron-mediated circuitry has an important physiological role in promoting glycaemic control and insulin sensitivity and that neuronal activity can promote metabolic homeostasis via extrapancreatic methods.Citation104 Another recent study found that the activation of cholinergic preganglionic neurons in rodents via the melanocortin-4 receptor agonist lorcaserin reduced hepatic glucose production, increased glucose disposal and improved insulin sensitivity.Citation129 Lorcaserin has also been reported to improve glycaemic control and induce weight loss in obese T2D subjects, and this drug was additionally shown to both reduce the risk of prediabetic individuals progressing to T2D and increase the likelihood of these subjects being able to revert to euglycemia.Citation129,Citation231 Given the likely role of these neurons in promoting metabolic homeostasis in humans, it is likely that PPG and cholinergic preganglionic neuronal activity is impaired or absent in T2D, possibly as a result of the disease phenotype, or dysfunction of these neurons could occur at an early stage and play a role in the disease’s aetiology/pathogenesis.
The diabetic phenotype greatly increases the risk of atherosclerotic plaque formation due to dyslipidaemia in patients, which results in increased plasma levels of the small dense atherogenic form of LDL cholesterol, and these molecules can easily penetrate the arterial wall and promote atherosclerosis.Citation232 Hyperglycaemia also promotes atherosclerosis.Citation233 The chronic inflammatory state associated with T2D is also thought to encourage plaque growth and formation.Citation232 The impaired insulin secretion and action induce pathology in the microvasculature, as nitric oxide (NO) production is dependent upon insulin signalling.Citation234 Chronic NO-deficiency in T2D results in a hyper-constricted state of the microvasculature; therefore, the delivery of oxygen and nutrients to tissues is impaired, contributing to diabetic neuropathies and the poor exercise tolerance observed in patients, as well as elevated blood pressure.Citation232,Citation235,Citation236 Diabetic autonomic neuropathy (DAN) also contributes to dysregulated autoregulation of blood flow in the vasculature.Citation237 Also, diabetic patients exhibit capillary basement membrane thickening due to chronic hyperglycemia, further impairing the exchange between the circulation and tissues.Citation232 The excess nutrient levels in the circulation in T2D are also thought to damage cells throughout the cardiovascular system due to excessive ROS production caused by increased metabolism, as a result of increased mitochondrial nutrient supply.Citation232,Citation238 The cardiovascular system in T2D is also in a hypercoagulable state and patients are at increased risk of thrombosis as a result.Citation232 T2D patients are also at increased risk of heart failure due to diabetic cardiomyopathy, where myocardial disease manifests in the absence of any other known CVD, likely due to hyperglycemia, microvascular damage and autonomic neuropathy associated with diabetes.Citation232,Citation239
Diabetic nephropathy (DN) is the most common cause of end-stage renal disease globally, with 10–20% of T2D patients developing this disease.Citation195 Kidney pathology can result due to nerve and blood vessel damage in this organ caused by the diabetic phenotype.Citation195,Citation228,Citation232 The diabetic phenotype also promotes DN by inducing pathology in kidney cells directly involved with glomerular filtration, as, for example, hyperglycemia promotes fibrosis in the kidney via inducing activation of specific intracellular pathways – the mechanisms by which T2D induces DN are reviewed in detail elsewhere.Citation195,Citation240 In addition, urinary tract infections are more common in diabetic patients, which is at least partly thought to be due to increased glucose levels in the urine.Citation241 summarises the pathological effects that T2D has on different organs and systems throughout the body.
Figure 7 A summary of the pathological effects that T2D has on different organs and systems throughout the body, some of which directly contribute to the disease-associated dyslipidaemia/hyperglycaemia and subsequent clinical symptoms. This figure and information in its legend are with data adapted from these studies.Citation94,Citation105,Citation106,Citation111,Citation192–Citation195,Citation206,Citation214,Citation219–Citation222,Citation225,Citation228,Citation232,Citation241,Citation326–Citation328,Citation331
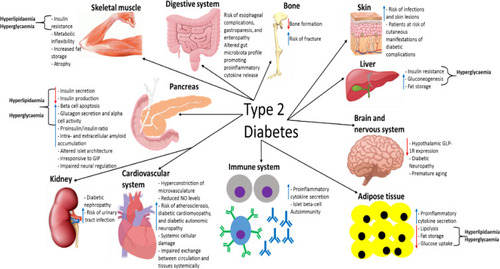
Pharmacological Treatments and Lifestyle Changes for T2D
After the T2D diagnosis, initial treatments usually involve altering diet and lifestyle, as well as increasing physical activity in combination with metformin.Citation3 Metformin decreases hepatic glycogenolysis, decreases peripheral tissue insulin resistance, increases GLP-1 postprandial secretion (which augments insulin secretion), and delays digestion.Citation8 Exercise and weight loss also reduce insulin resistance and reduce the risk of T2D associated post-diagnosis complications.Citation242,Citation243 If this treatment regime is ineffective at promoting normoglycaemia then sulfonylureas and meglitinides are usually the next treatments tried which act as insulin secretagogues.Citation3 Common side effects of insulin secretagogues include weight gain and hypoglycaemia, and these drugs are only usually effective in the short term.Citation8 Insulin therapy, in the form of multiple daily subcutaneous injections, is effective at lowering plasma glucose levels and is usually required in the majority of patients when the aforementioned treatment regimens fail, but there are several side effects, such as weight gain, hypoglycaemia and increased risk of colorectal cancer.Citation3,Citation244 It has been estimated that 50% of patients require insulin therapy within 10 years of T2D diagnosis.Citation245 Recently, rodent and human studies have found that orally administered insulin could be a viable option to treat T2D. This is due to the advent of novel delivery systems that allow oral insulin to resist degradation in the GIT, be transported into circulation, and then act when desired depending on plasma glucose concentration, which is desirable given the complications associated with multiple daily invasive insulin injections.Citation246–Citation249 Modulating GLP-1R activity is the most recent therapy used to treat T2D.Citation3 The first therapy is based on the use of DPP-IV inhibitors to delay GLP-1 breakdown from its active to inactive form.Citation250 DPP-IV inhibitors increase GLP-1 levels by 2–3-fold over 24 hours and have been shown to significantly decrease plasma glucose levels.Citation3 Long-term administration of these drugs will likely have adverse effects as this enzyme is expressed in many tissues and has a variety of functions, but no important side effects have been seen after over 10 years of use.Citation3 The efficacy of GLP-1 analogues is generally greater than DPP-IV inhibitors, due to the supraphysiological concentrations of GLP-1 achieved after administration of the former.Citation251 Hence, the preferred therapy for modulating GLP-1R activity involves the administration of GLP-1 analogues (exenatide and liraglutide, lixisenatide, dulaglutide, albiglutide and semaglutide) to T2D patients, which improve glycaemic control by augmenting insulin secretion and dampening glucagon secretion, as well as delaying gastric emptying.Citation252,Citation253
Exenatide and liraglutide mimic the endogenous GLP-1 activity by binding to GLP-1R on various tissues, but these analogues are resistant to degradation.Citation252 GLP-1 has a half-life of 1–2 minutes, whereas exenatide has a half-life of 3.4–4 hours and liraglutide’s half-life is 11–13 hours.Citation3 Hence, these GLP-1 analogues vastly prolong the GLP-1 response promoting normoglycaemia in T2D patients during fasting and after nutrient ingestion. An advantage of using GLP-1R agonists (and DPP-IV inhibitors) is that they only mediate their insulinotropic effects when glucose levels are elevated, meaning that the risk of hypoglycaemia is minimal, and these therapies have the lowest reported rates of hypoglycaemia for T2D treatments.Citation254 Liraglutide and exenatide (synthetic exendin-4) have 97 and 52%, respectively, homology with GLP-1.Citation255,Citation256 GLP-1R agonists used in T2D treatment are either derivatives of native GLP-1 (liraglutide, albiglutide, semaglutide and dulaglutide), which have been modified to be resistant to DPP-IV inactivation or derivatives of exendin-4 (exenatide, lixisenatide and exenatide-LR).Citation257,Citation258 Exendin-4 was originally isolated from the saliva of the Gila monster lizard and is resistant to the action of DPP-IV.Citation257,Citation259 Key pharmacological and clinical features of clinically available GLP-1R agonists are presented in and compares the amino acid sequence of native GLP-1 and the GLP-1 analogues liraglutide and exenatide.
Table 3 Current Glucagon-Like Peptide 1 Receptor (GLP-1R) Agonists Used in Type 2 Diabetes Therapy
Figure 8 The amino acid sequence of GLP-1, liraglutide and exenatide. Amino acids highlighted green in exenatide indicate altered amino acids from the GLP-1 sequence. The purple amino acids in exenatide indicate amino acids that have been added. The amino acid highlighted red in liraglutide is modified by the addition of palmitic acid enabling binding to albumin which results in an increased half-life of liraglutide in circulation. This figure is with data adapted from Lorenz et al.Citation332
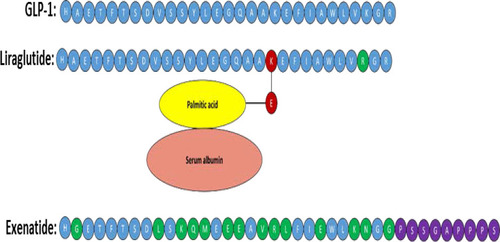
These GLP-1 analogues are effective insulinotropic agents and successfully suppress glucagon secretion in a glucose-dependent manner.Citation260–Citation261 GLP-1 analogues also induce weight loss, which is associated with improved T2D prognosis.Citation252,Citation262,Citation263 GLP-1R activity is thought to mediate weight loss by induction of satiety.Citation3,Citation252 Interestingly, it has been shown that chronic liraglutide treatment in diabetic mice prevents loss of pancreatic beta-cell mass, increases proliferation of the beta-cells and decreases apoptosis after alloxan injection.Citation264 Inhibition of the beta-cell apoptosis in isolated human pancreatic cells was also reported after liraglutide administration, and the beta-cell proliferation rate was increased up to three-fold after incubation for 24 h.Citation265 In streptozocin-induced type 1 diabetic rats and isolated human pancreatic ducts, exendin-4 treatment has also been shown to increase the number of insulin producing cells.Citation266 Side effects of GLP-1 analogue administration are dyspepsia and nausea, but the effects usually subside after continuous administration.Citation3 Additionally, it was implicated that GLP-1 analogue therapy increased the risk of acute pancreatitis, but subsequent studies using larger cohorts did not confirm this finding.Citation8,Citation267 However, there is evidence that GLP-1 analogues increase the risk of medullary thyroid cancer, which is a cause for concern.Citation268
The development of GIP analogues for T2D treatment was initially not a desirable field of research due to GIP action being thought to be lost in T2D and GIPs ability to augment glucagon secretion.Citation3,Citation261 However, unexpectedly, preclinical data from a study in 2013 found that GLP-1/GIP dual agonist treatment induce better glycaemic control and body weight reduction in diet-induced-obesity mice compared to liraglutide only treated controls.Citation269 Co-agonism of GLP-1R and GR was also shown to reduce body weight synergistically in rodent obesity models. These co-agonists also improved glycaemic control in humans.Citation269 Additionally, a recent study found that co-agonism of GLP-1R and GR was reported to ameliorate diet-induced dyslipidemia and atherosclerosis by increasing energy expenditure, reducing liver inflammation and regulating bile homeostasis in rodents.Citation270
A recent study found that exenatide increased adiponectin secretion from adipose tissue in both in vivo and in vitro studies.Citation271 Adiponectin is a peripheral tissue insulin sensitizer, and its levels are known to decrease in obese and T2D patients.Citation272 This study demonstrated that modulating GLP-1R activity has the potential to improve both insulin secretion and action in T2D and that GLP-1 analogues may also be able to induce weight loss by augmenting adiponectin secretion, as well as inducing satiety. GLP-1 analogues are as effective as second-line therapy in improving glycaemic control in patients with T2D.Citation273 Reductions in glycated haemoglobin from baseline with GLP-1 analogues tend to be greater than or comparable to insulin therapy.Citation262,Citation274 GLP-1 analogues are also consistently more effective at inducing weight loss than most oral glucose-lowering drugs and insulin.Citation262,Citation263 GLP-1 analogues were also shown to confer lower hypoglycemia risk versus insulin or sulfonylureas.Citation262 Additionally, GLP-1 analogues also appear to decrease the risk of cardiovascular pathology in patients, according to some studies, and preliminary data suggest they do so to a greater degree than other oral glucose-lowering drugs.Citation262,Citation275–Citation277 However, two GLP-1 analogues tested in two studies demonstrated no advantage in influencing cardiovascular outcome.Citation278,Citation279 Recent systematic reviews and meta-analyses found that GLP-1R agonists reduced major adverse cardiovascular events in T2D patientsCitation280–Citation282 and also had beneficial effects on kidney outcome.Citation283 The reported varying impacts of different GLP-1R agonists on blood glucose levels, weight loss, cardiovascular outcomes and adverse effects should play an integral role when determining which agonist is best to administer to a patient depending on their medical requirements.Citation260
Small Molecule Agonists
Studies have demonstrated that GLP-1R has allosteric agonist binding sites and that these sites are distinct from the orthosteric agonist (GLP-1) binding site.Citation3 The first allosteric agonist identified for GLP-1R was compound 1, which had a low affinity and a low potency for GLP-1R.Citation3 Compound 2 was then produced, which has been shown to be a more potent agonist, and this molecule also increases the affinity of GLP-1R for GLP-1.Citation284 Additionally, compound 2 binding was not inhibited by the exendin (9–39) antagonist (truncated exenatide), demonstrating that this compound binding site on GLP-1R is distinct from the orthosteric GLP-1 binding site.Citation3,Citation284 However, compound 2 does not stimulate insulin secretion to the same extent as GLP-1, liraglutide or exenatide in vivo, and combining liraglutide or exenatide with compound 2 did not improve insulin secretion in mice.Citation285 Compounds A and B have also been shown to demonstrate ago-allosteric properties and induce cyclic adenosine monophosphate (cAMP) signalling, as well as increased insulin secretion in rat islets and animal studies.Citation3,Citation14 One study showed that compound B almost normalised insulin secretion in human islets isolated from a T2D donor.Citation286 Compounds 2 and B bind to a distinct site from the orthosteric binding site, as GLP-1R antagonists exendin (9–39) and JANT4, as well as the V36A mutation in GLP-1R, did not inhibit cAMP production upon compound 2 or B administration.Citation14 Compounds 2 and B induced cAMP production in human GLP-1R expressing cells but caused no intracellular Ca2+ accumulation, ERK phosphorylation or receptor internalisation.Citation14 Prevention of receptor internalisation may allow for continuous downstream signalling of the receptor, which could, in theory, raise insulin levels in T2D patients – this is an area of ongoing research.Citation252 The K334A mutation (which affects the Gαs coupling) of GLP-1R inhibited both GLP-1 and compounds 2 and B induced cAMP production, which demonstrates that GLP-1 and both compounds 2 and B induce similar conformational changes needed for the Gαs activation.Citation14 A recent study showed that compounds 2 and B bind and covalently modify residue 347 in ICL3 of GLP-1R.Citation287 Residues 328, 351 and 335 of GLP-1R were recently demonstrated to be important for compound 2 potency, as mutagenesis of these residues reduced compound 2 activity.Citation288 In this same study, enhanced compound 2 potency was demonstrated by GLP-1R when its residue 332 was mutated to tryptophan, which is in contrast to the effect observed with this mutation on the potency of allosteric antagonists. It was postulated that enhanced compound 2 potency was caused by increased hydrophobic interactions between the receptor and the agonist with this mutation. shows the chemical structures of compounds 2 and B.
Figure 9 The chemical structure of the small molecule allosteric agonists compound 2 and compound B of GLP-1R, which shown schematically with numbered transmembrane domains. This figure is adapted with data from these studies.Citation3,Citation14
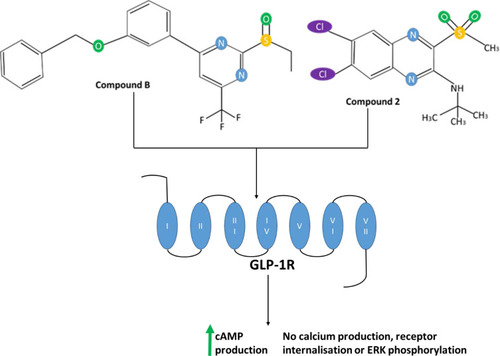
GLP-1R, GIPR and GR Tri-Agonist
In 2015, a triple agonist for GLP-1R, GIPR and GR was developed and tested on diet-induced obese (DIO) mice.Citation289 The tri-agonist lowered body weight in these mice to a greater extent than the GLP-1R/GIPR coagonist after 20 days of treatment. Both the coagonist and tri-agonist did not induce hypoglycaemia and were equally effective at reducing blood glucose levels and improving glucose tolerance, demonstrating that surprisingly chronic GR agonism does not counteract the anti-hyperglycemic effects of GLP-1R and GIPR activity. Interestingly, tri-agonist treated mice had lower plasma insulin levels than the coagonist-treated mice, indicating improved insulin sensitivity. In theory, chronic GR activation should raise plasma glucose levels and insulin secretion would then need to be concomitantly increased to maintain normoglycaemia during tri-agonist treatment, but this was not observed in this study. Given that there was no difference in food intake between wild-type mice treated with the dual-incretin GLP-1/GIP coagonist and those treated with the tri-agonist, the greater difference in weight loss was attributed to significantly enhanced ATP expenditure in tri-agonist-treated DIO mice. The capacity of the tri-agonist to prevent the development of spontaneous diabetes compared with the dual-incretin GLP-1/GIP coagonist was tested in mouse models of T2D. Tri-agonist treatment prevented the excessive weight gain to a greater extent than the coagonist, and this difference was not due to cumulative food intake. Additionally, the tri-agonist protected these mice from fasting hyperglycaemia to a better degree than the coagonist. Interestingly, the tri-agonist also significantly reduced pancreatic alpha-cell infiltration into the core of pancreatic islets, helping to preserve the islet architecture observed in healthy pancreatic islets. The tri-agonist was also demonstrated to delay T2D progression in rodent models of spontaneous diabetes, as glycaemic improvements were maintained in Zucker diabetic fatty rats 3 weeks after treatment cessation, even though they had gained body weight and were comparable in mass to vehicle-treated controls. Given that weight and food intake were not altered in lean mice even after chronic treatment with the tri-agonist, this demonstrates that the effects of the tri-agonist do not manifest in the absence of excessive nutrient storage. Another recent study,Citation290 which investigated the effects of other GLP-1, GIP and GR tri-agonist in rodents, found that this agonist ameliorated diabetic nephropathy-related disorders, as well as reduced body weight, food intake and hyperglycaemia. This suggests that these tri-agonists have the therapeutic potential to alleviate post-diagnosis complications in patients.
Surgical Options for T2D: Bariatric Surgery
Bariatric surgery includes a variety of procedures performed on the GIT in people who are severely obese (BMI ≥40) or in individuals with a BMI of ≥35 that have a condition that can be improved by losing weight such as T2D or high blood pressure.Citation291 Weight loss is achieved by reducing the size of the stomach with a gastric band or by removing a portion of the stomach (sleeve gastrectomy or biliopancreatic diversion with duodenal switch), and resecting and re-routing the small intestine to a small stomach pouch (gastric bypass surgery) is another surgical option to promote weight loss.Citation292 Reduction in the size of the stomach results in patients experiencing satiety with less food and the bypass means that fewer nutrients will be ingested as part of the intestine will no longer be involved in digestion.Citation291,Citation292 This surgery is only used when other options have not been effective for weight reduction, such as diet and lifestyle changes.Citation13,Citation291 The obesity epidemic in recent decades has been parallelled by an increase in bariatric surgery.Citation292 Longitudinal studies have found that this surgery significantly reduces body weight over time, improves glycaemic control and prognosis in T2D patients, and reduces mortality and risk of obesity-related co-morbidities, such as cardiovascular and respiratory pathology.Citation291 Data collected globally revealed that the number of patients undergoing bariatric surgery rose from 40,000 in 1998 to 146,300 in 2003.Citation293
Bariatric surgery is known to be able to induce T2D remission, and one study found that 53% of patients were in complete T2D remission three years post-surgery and another 10% were in partial remission according to the American Diabetes Association criteria, although remission rates vary between the studies, with one study reporting that 34% of patients were in complete remission 23 months post-surgery.Citation294 The consensus in the literature is that complete remission rates decrease with time, as demonstrated in one study where 43% of patients had complete remission after 2 years post-surgery, which dropped to 24% after 5 years.Citation294,Citation295 Recent findings suggest that a persistent impairment in insulin secretion is a crucial factor in failure to sustain T2D remission after bariatric surgery.Citation295–Citation297 The improvements observed in post-surgery were originally postulated to be induced by the subsequent post-surgery weight loss, but it has become apparent that reversal of insulin resistance is induced before any significant weight loss occurs.Citation298 Hence, it is currently unclear how T2D remission is achieved via bariatric surgery, but it is not just due to the weight loss associated with this surgery. In both T2D obese and T2D-free obese patients, bariatric surgery increases postprandial GLP-1 levels (from up to 3-fold pre-surgery and nearly 8-fold vs baseline after gastric bypass).Citation297 Interestingly, gastric bypass and sleeve gastrectomy have been reported to induce similar improvements in glycaemic control, despite inferior GLP-1 levels and BMI decreased in patients that had undergone sleeve gastrectomy.Citation296
A recent study reported that bariatric surgery improves disposition index (DI), a score determined by insulin sensitivity times the amount of insulin secreted in response to blood glucose levels, in diabetes-free subjects such that after 2 years post-surgery, it almost matches general population values.Citation297 However, insulin secretion in response to intravenously administered glucose declines in these individuals, implying that the improved DI is a result of the enhanced post-surgery GLP-1 plasma levels. In the T2D subjects of the same study (91% remission rate at 2 years post-surgery), both disposition indices and insulin secretion in response to intravenously administered glucose improve 2 years post-surgery but are still notably lower than population norms. Fasting plasma glucose levels were reported to revert to normal levels 2 years post-surgery in over 90% of T2D subjects (from 7.30 to 5.24 mmol), and become comparable to non-diabetic subjects (5.00 mmol). Although average peak glucose levels remained similar in the diabetic group 2 years after surgery (from 9.96 to 9.79 mmol/L), peak postprandial glucose levels shifted to an earlier time point and rapidly declined to within the normoglycaemic range, meaning that the time the individual was subjected to hyperglycaemia was greatly reduced and for the majority of 4 hours following nutrient ingestion their plasma glucose levels were in the healthy range. Surprisingly, average peak glucose levels increased in non-diabetic subjects after surgery, but these individuals similarly had peak levels at an earlier time point, with a subsequent more rapid decline in plasma glucose levels. Despite this postprandial hyperglycaemia, the overall reductions in fasting and postprandial glucose exposure following bariatric surgery in nondiabetic individuals are thought to be beneficial due to the reported reduced cardiovascular complications associated with post-surgery groups compared with non-surgical control groups.Citation297 Average peak insulin levels were notably lower in diabetic subjects (771.4 pmol/L) compared to non-diabetic (1221 pmol/L) 2 years post-surgery, although average fasting insulin levels were higher in diabetic subjects (41.5 vs 36.9 pmol/L). In both groups, average peak insulin levels improved 2 years post-surgery: pre-surgery peak insulin levels were 491.9 and 7778.3 pmol/L in the diabetic and non-diabetic groups, respectively. Interestingly, although the average proinsulin: insulin ratio marginally improved 2 years after surgery in diabetic subjects (from 2.2 to 1.8), it remained notably higher than the value observed in the non-diabetic control group (1.1), demonstrating that bariatric surgery does not have much impact on defective insulin processing by islet beta-cells. Interestingly, average peak postprandial glucagon levels increased in both diabetic (from 102.2 to 112.3ng/L) and non-diabetic (from 99.2 to 109.9 ng/L) groups after surgery, implying a beneficial role of this hormone in promoting post-surgical weight loss, and the opposite was the case for fasting glucagon levels.
Plasma GLP-1 and GIP responses, as well as insulin levels, were augmented in a pattern that mirrored the accelerated postprandial glucose appearance and disappearance following surgery in both diabetic and nondiabetic groups. Despite the notable benefits and reported metabolic improvements, this surgery is expensive and not always successful in inducing weight loss. Further, there are numerous post-surgery complications, such as vomiting and dumping syndrome, iron and B12 deficiency, and secondary hyperparathyroidism.Citation13 The majority of patients develop vomiting and dumping syndrome post-surgery but this usually subsides within a year due to patients’ GIT and feeding behaviours adjusting, and medication is available to treat this syndrome if necessary.Citation13 After one year post-surgery, ~30% of patients need B12 supplementation to maintain normal plasma levels of B12.Citation299 The prevalence of B12 deficiency increases in the post-surgery population yearly, it has even been reported to be as high as 70% in one study in the long term.Citation300 B12 supplementation usually treats this post-surgery complication, but a minority of patients require monthly intramuscular B12 injections.Citation13 Iron deficiency is usually only seen in menstruating women after one year of post-surgery, and this complication can be overcome by intravenous iron infusion several times a year.Citation13,Citation301 During weight loss after surgery, many patients develop gallstones, and patients that develop gallstone-related symptoms undergo surgery to remove the gall bladder.Citation13 Secondary hyperparathyroidism is characterised by elevated levels of parathyroid hormone caused by excessive activity and hyperplasia of the parathyroid glands.Citation302 This hormone has catabolic effects on bone tissue and increases vitamin D synthesis, which raises the plasma calcium levels.Citation303 Bariatric surgery patients often develop secondary hyperparathyroidism due to malabsorption of calcium from the GIT as a result of the surgery.Citation13 Vitamin D and calcium supplementation are effective treatments for this post-surgical complication.Citation304 It has been estimated that ~29% of patients that undergo bariatric surgery develop secondary hyperparathyroidism.Citation13 Thus, it is desirable for a treatment to be developed to mimic the effects of bariatric surgery but without the aforementioned post-surgery complications.
Future Perspectives
Studies have found evidence that GLP-1 exerts multiple beneficial extrapancreatic actions on both tissues positive and negative for GLP-1R expression,Citation252 and clinical trials have found that some GLP-1 mimetic-based therapies alleviate cardiovascular pathology associated with T2D.Citation305 This implies that the GLP-1R agonists may be able to alleviate the systemic pathology that is associated with T2D,Citation306–Citation308 and not just be able to augment insulin secretion from islet beta-cells of T2D patients.
In addition to augmenting insulin secretion from islet beta-cells, both in vivo and in vitro studies have produced evidence that GLP-1 action improves islet beta-cell survival and proliferation,Citation252,Citation259 which is also a relevant area of research as it is well established that T2D pathology is associated with an increased rate of islet beta-cell apoptosis.Citation309 Given the reduced population of these cells in patients,Citation310 enhanced proliferation would, in theory, be desirable to increase plasma insulin levels in patients to promote normoglycaemia and normolipidaemia. Therefore, comparing how GLP-1R orthosteric and allosteric agonists influence islet beta-cell survival is a desirable future area of research. Despite controversial evidence that islet alpha-cells express GLP-1R, there is substantial evidence to suggest that GLP-1 action suppresses glucagon secretion, which is also important for alleviating T2D pathology, as T2D is now considered to be a bi-hormonal disorder.Citation142 Therefore, determining how GLP-1R orthosteric and allosteric agonists modulate islet alpha-cell activity is another area of future research. Finally, to obtain further insight into how orthosteric and allosteric GLP-1R agonists can modulate islet beta-cells, it will be necessary to determine how these agonists influence other cells in the pancreatic islets, such as alpha-cells and determine how these cells, in turn, may subsequently modulate beta-cell activity via paracrine signalling.
GLP-1R orthosteric agonism has been shown to be an effective treatment for T2D, and the therapeutic potential of GLP-1R allosteric agonism to provide more efficacious treatment is an ongoing area of research. Although not discussed here, modulating the activity of other cell receptors via allosteric agonism could also yield more efficacious therapeutic options for T2D. Modulating the activity of multiple receptors (including GLP-1R) simultaneously for hormones involved in metabolic homeostasis using a unimolecular agonist appears to be a promising new therapeutic option for future T2D treatment, and is an area of ongoing research.Citation289,Citation290 Extrapancreatic pathology associated with T2D is discussed in this review to highlight that this disease has systemic consequences on the body, and ideally, treatments should aim to alleviate this systemic pathology via direct mechanisms as well as by improving insulin secretion/action; given that GLP-1 targets multiple organs, GLP-1-based therapies could achieve this.Citation252 A recent longitudinal study demonstrated the potential of GLP-1R agonism to prevent T2D manifestation in obese individuals and individuals with a BMI of >27 that had hypertension or dyslipidaemia: after 160 weeks, the liraglutide treated group (n=1472) had a smaller percentage of the population diagnosed with T2D (2% vs 6%) than the placebo-treated control group (n=738).Citation311 Additionally, time for T2D onset during the 160-week study was found to be 2.7x longer in the liraglutide treated group than in the placebo control group. The results of this study have important future implications, as GLP-1R agonism may not only be an effective treatment for T2D but also prevent or delay the disease manifestation in at-risk individuals. A recent study also found that a new GLP-1R agonist, peptide 8, had a greater insulinotropic effect (at 30nM) than GLP-1 on rat islets, and this agonist was also shown to have effects similar to exendin-4 in mice during an OGTT.Citation312 However, peptide 8 had a significant glucose-lowering effect even when it was administered 30 min before the OGTT, in contrast to exendin-4, due to its extended duration of action. The clinical potential of peptide 8 is another area of future research, given its enhanced insulinotropic activity over GLP-1 and other GLP-1R orthosteric agonists tested in this study. Given the different structures of GLP-1R agonists, it is plausible that how they bind to GLP-1R differs, which subsequently produces differential activation of the receptor, explaining the observations from studies.Citation288,Citation312 Further understanding mechanistically how GLP-1R can be activated differently by different orthosteric and allosteric agonists may have clinical relevance in the future. Given the conflicting findings observed between human and animal studies investigating the activity of hormones involved in regulating metabolic homeostasis (especially GLP-1), it is important to note that observations from animal studies may have limited clinical implications for developing new T2D therapies for humans.Citation257,Citation305
Interestingly, over recent years, it has become increasingly recognised that the aetiology of T2D encompasses autoimmune activity, similar to T1D.Citation313 Strikingly, studies have demonstrated that islet-reactive T-cells can be found in phenotypic human T2D patients, and many studies have found that a subset of T2D patients test positive for islet beta-cell autoantibodies - these T-cells and antibodies are usually associated with T1D pathogenesis.Citation313 One study found that 12% of T2D patients screened for islet autoantibodies were positive for at least one islet autoantibody.Citation314 Staggeringly, islet reactive T-cells were found in 72% of phenotypic T2D patients that participated in one study, which was an unexpected finding, especially as great care was taken to select patients that had minimal chance of being misdiagnosed with T2D and have T1D - all participants were obese or had an increased waist-to-hip ratio, had no history of ketoacidosis, and had not received insulin treatment.Citation315 Convincing evidence of autoimmunity being involved in T2D pathogenesis was also provided by a study that induced B lymphocyte deficiency in New Zealand obese mice (an obese polygenic mouse model of obesity and T2D), as B lymphocyte deficient mice were found to not develop T2D - this implies that B cells may be vital for T2D pathogenesis.Citation316 Further evidence of B lymphocyte involvement in T2D pathogenesis has been provided by the observation that the B cells of T2D patients fail to secrete IL-10 (an important negative immunoregulatory molecule) after stimulation.Citation317 The notion that T1D and T2D may not have such distinct aetiologies as previously thought is certainly an area for future research and is of great interest. If it becomes elucidated that T2D does have an autoimmune aetiology then it can be classified as a less severe variant of T1D, as T2D patients do not exhibit the absolute insulin deficiency seen in T1D. Additionally, if both T1D and T2D have similar autoimmune aetiologies, understanding why T2D patients do not have the T1D clinical phenotype is an important area of future research.
A recent study found by using live-cell imaging that the ability of insulin vesicles to successfully dock onto the plasma membrane correlates with insulin secretion from human islet beta-cells and this was shown to be reduced in T2D.Citation318 This study found that defective insulin secretion in T2D islet beta-cells was caused by a drastically decreased rate of successful docking events (docking events were found to be 5-fold more frequent in non-diabetic beta-cells compared to diabetic beta-cells), rather than reduced delivery of insulin vesicles to the plasma membrane, which was found to be similar between non-diabetic and diabetic beta-cells. Expression analysis found that the expression of proteins involved in granule docking was downregulated in T2D. Hence, this suggests in T2D that defective molecular attachment of insulin vesicles to the release site results in inadequate plasma insulin levels to promote normoglycaemia, and that the diabetic phenotype is not as a result of intracellular insulin content and/or the number of insulin vesicles being insufficient in diabetic islet beta-cells. Improving islet beta-cell granule docking, therefore, represents an attractive and new therapeutic target for the development of antidiabetic therapies.
A recent systematic review and meta-analysis reported that using hematopoietic stem cells and mesenchymal stem cells derived from bone marrow, placenta or umbilical cord tissue to treat T2D is generally safe. The stem cell therapy also improves one or more of the following for T2D patients: levels of C-peptide, HbA1c, quality of life score, and insulin requirements.Citation319–Citation321 However, there have been inconsistencies with these findings generated in other studies and one study reported severe infections in T1D patients after the stem cell therapy.Citation319,Citation322–Citation324 Additionally, the mechanism by which these stem cells exert any positive effects is debated. Also, the studies that were used to generate these findings had several design flaws with regard to deducing accurate conclusions and the total sample size was not large enough.Citation319 The encouraging preliminary results from these studies warrant validation in larger, randomized, double-blind studies, as well as longer follow-up periods to establish the possibility of stem cell-based therapies for future T2D treatment.
Conclusion
A better understanding of metabolic homeostasis in healthy individuals and the altered metabolic phenotype in T2D will likely lead to the development of better treatments for T2D. The role of the nervous system, genetics, hormones involved in metabolic homeostasis (such as insulin, glucagon, GLP-1 and GIP), glucolipotoxicity diets and feeding behaviours, sedentary lifestyles, altered islet architecture, the immune system, altered islet-cell behaviour, UCP2, altered extrapancreatic behaviour and risk factors (such as psychological stress) have in T2D aetiology and pathogenesis remains to be mechanistically understood. Given that T2D is a multifactorial disease involving an array of hormones, their receptors and subsequent intracellular activity, future therapeutic research needs to take into account how the action of all of these hormones interact synergistically in T2D to produce the altered metabolic phenotype, and also how treatments such as GLP-1R activation-based therapies can influence this hormonal synergism to produce a metabolic phenotype more similar to that of a healthy individual. GLP-1R agonists are an attractive target to generate more effective therapies for T2D given that they have been reported to have beneficial effects on multiple organs in the body, which are involved in disease pathology.
Abbreviations
ADP/ATP, adenosine diphosphate/triphosphate; BMI, Body mass index; cAMP, cyclic adenosine monophosphate; CVD, Cardiovascular disease; CNS, Central nervous system; DN, Diabetic nephropathy; DIO, Diet-induced obese; DPP, Dipeptidyl peptidase; GIP, Gastric inhibitory peptide; GIPR, Gastric inhibitory peptide receptor; GIT, Gastrointestinal tract; GLP-1, Glucagon-like peptide-1 GLP-1, glucagon-like peptide-1; GLP-1R, GLP-1 receptor; GSIS, glucose-stimulated insulin secretion; GR, Glucagon receptor; HDL, High-density lipoprotein; IAPP, Islet amyloid polypeptide; IFG, Impaired fasting glucose; IGF, Insulin-like growth factor; IR, Insulin receptor; LDL, Low-density lipoprotein; NAFLD, Non-alcoholic fatty liver disease; NO, Nitric Oxide; PPG, Preproglucagon; PMF, Proton motive force; ROS, Reactive oxygen species; T1D/T2D, type 1/2 diabetes; UCP1/2, Uncoupler protein 1/2; WHO, World Health Organisation.
Acknowledgments
The work in VK’s laboratory was supported by grants from Biotechnology and Biological Sciences Research Council (BBSRC) UK (BB/F017596/1, BB/C515455/2 and BB/S019588/1) and Medical Research Council (MRC) UK (G0401232). JR was a recipient of the Knowledge Economy Skills Scholarship (KESS) II UK PhD studentship. We thank the members of the VK laboratory.
Disclosure
The authors report no conflicts of interest in this work.
References
- Guariguata L , Whiting DR , Hambleton I , Beagley J , Linnenkamp U , Shaw JE . Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract . 2014;103(2):137–149. doi:10.1016/j.diabres.2013.11.002 24630390
- Ozougwu JC , Obimba KC , Belonwu CD , Unakalamba CB . The pathogenesis and pathophysiology of type 1 and type 2 diabetes mellitus. J Physiol Pathophysiol . 2013;4:46–57. doi:10.5897/JPAP2013.0001
- Thompson A , Kanamarlapudi V . Type 2 Diabetes mellitus and glucagon like peptide-1 receptor signalling. Clin Exp Pharmacol . 2013;1:3.
- Maahs DM , West NA , Lawrence JM , Mayer-Davis EJ . Chapter 1: epidemiology of Type 1 Diabetes. Endocrinol Metab Clin North Am . 2010;39(3):481–497. doi:10.1016/j.ecl.2010.05.011 20723815
- Kahn SE , Cooper ME , Del Prato S . Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet . 2014;383(9922):1068–1083. doi:10.1016/S0140-6736(13)62154-6 24315620
- Leahy JL . Pathogenesis of Type 2 Diabetes Mellitus. Arch Med Res . 2005;36(3):197–209. doi:10.1016/j.arcmed.2005.01.003 15925010
- Chen L , Magliano DJ , Zimmet PZ . The worldwide epidemiology of type 2 diabetes mellitus[mdash]present and future perspectives. Nat Rev Endocrinol . 2012;8(4):228–236. doi:10.1038/nrendo.2011.183
- Raz I . Guideline approach to therapy in patients with newly diagnosed type 2 diabetes. Diabetes Care . 2013;36(Suppl 2):S139–S144. doi:10.2337/dcS13-2035 23882038
- Golay A , Ybarra J . Link between obesity and type 2 diabetes. Best Pract Res Clin Endocrinol Metab . 2005;19(4):649–663. doi:10.1016/j.beem.2005.07.010 16311223
- Haslam D . Obesity and diabetes: the links and common approaches. Prim Care Diabetes . 2010;4(2):105–112. doi:10.1016/j.pcd.2010.04.002 20447884
- Horton ES , Silberman C , Davis KL , Berria R . Weight Loss, Glycemic Control, and Changes in Cardiovascular Biomarkers in Patients With Type 2 Diabetes Receiving Incretin Therapies or Insulin in a Large Cohort Database. Diabetes Care . 2010;33(8):1759–1765. doi:10.2337/dc09-2062 20460445
- Wilding JPH . The importance of weight management in type 2 diabetes mellitus. Int J Clin Pract . 2014;68(6):682–691. doi:10.1111/ijcp.12384 24548654
- Fujioka K . Follow-up of nutritional and metabolic problems after bariatric surgery. Diabetes Care . 2005;28(2):481–484. doi:10.2337/diacare.28.2.481 15677821
- Thompson A , Stephens JW , Bain SC , Kanamarlapudi V . Molecular characterisation of small molecule agonists effect on the human glucagon like peptide-1 receptor internalisation. PLoS One . 2016;11(4):e0154229–e0154229. doi:10.1371/journal.pone.0154229 27100083
- Kyrou IRH , Weickert MO . Clinical Problems Caused by Obesity . South Dartmouth: Endotext; 2010.
- Nguyen DM , El-Serag HB . The Epidemiology of Obesity. Gastroenterol Clin North Am . 2010;39(1):1–7. doi:10.1016/j.gtc.2009.12.014 20202574
- Paz-Filho G , Mastronardi C , Delibasi T , Wong M-L , Licinio J . Congenital leptin deficiency: diagnosis and effects of leptin replacement therapy. Arq Bras Endocrinol Metabol . 2010;54(8):690–697. doi:10.1590/S0004-27302010000800005 21340154
- WHO. Obesity and overweight; 2014. Available from: http://www.who.int/mediacentre/factsheets/fs311/en/. Accessed 7 12, 2021.
- Malnick SDH , Knobler H . The medical complications of obesity. QJM . 2006;99(9):565–579. doi:10.1093/qjmed/hcl085 16916862
- Must A . The Disease Burden Associated with Overweight and Obesity . South Dartmouth: Endotext; 2012.
- Bagdade JD , Bierman EL , Porte D . The significance of basal insulin levels in the evaluation of the insulin response to glucose in diabetic and nondiabetic subjects. J Clin Invest . 1967;46(10):1549–1557. doi:10.1172/JCI105646 6061732
- Ferrannini E , Natali A , Bell P , Cavallo-Perin P , Lalic N , Mingrone G . Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). J Clin Invest . 1997;100(5):1166–1173. doi:10.1172/JCI119628 9303923
- Gerald R . All obese individuals are not created equal: insulin resistance is the major determinant of cardiovascular disease in overweight/obese individuals. Diabetes Vascular Dis Res . 2005;2(3):105–112. doi:10.3132/dvdr.2005.017
- Templeman NM , Skovsø S , Page MM , Lim GE , Johnson JD . A causal role for hyperinsulinemia in obesity. J Endocrinol . 2017;2017:58.
- Weng J , Ji L , Jia W , et al. Standards of care for type 2 diabetes in China. Diabetes Metab Res Rev . 2016;32(5):442–458. doi:10.1002/dmrr.2827 27464265
- Hegazi R , El-Gamal M , Abdel-Hady N , Hamdy O . Epidemiology of and risk factors for type 2 diabetes in egypt. Ann Global Health . 2015;81(6):814–820. doi:10.1016/j.aogh.2015.12.011
- Wild SH , Byrne CD . Commentary: sub-types of diabetes—what’s new and what’s not. Int J Epidemiol . 2013;42(6):1600–1602. doi:10.1093/ije/dyt201 24415600
- Hu FB . Globalization of Diabetes. Role Diet Lifestyle Genes . 2011;34(6):1249–1257.
- D’Alessio D . The role of dysregulated glucagon secretion in type 2 diabetes. Diabetes Obes Metab . 2011;13:126–132. doi:10.1111/j.1463-1326.2011.01449.x 21824266
- Lin Y , Sun Z . Current views on type 2 diabetes. J Endocrinol . 2010;204(1):1. doi:10.1677/JOE-09-0260 19770178
- Okon EB , Chung AWY , Zhang H , Laher I , van Breemen C . Hyperglycemia and hyperlipidemia are associated with endothelial dysfunction during the development of type 2 diabetes. Can J Physiol Pharmacol . 2007;85(5):562–567. doi:10.1139/Y07-026 17632592
- Voet D , Voet J . Biochemistry . 4th ed. New York: Wiley & Sons; 2011.
- Whitmore C . Type 2 diabetes and obesity in adults. Br J Nursing . 2010;19(14):880–886. doi:10.12968/bjon.2010.19.14.49041
- Zhou X , Ji L , Ran X , et al. Prevalence of Obesity and Its Influence on Achievement of Cardiometabolic Therapeutic Goals in Chinese Type 2 Diabetes Patients: an Analysis of the Nationwide, Cross-Sectional 3B Study. PLoS One . 2016;11(1):e0144179. doi:10.1371/journal.pone.0144179 26726883
- Mathews AEW , Mathews CE . Inherited β-Cell Dysfunction in Lean Individuals With Type 2 Diabetes. Diabetes . 2012;61(7):1659–1660. doi:10.2337/db12-0373 22723272
- Ali O . Genetics of type 2 diabetes. World J Diabetes . 2013;4(4):114–123. doi:10.4239/wjd.v4.i4.114 23961321
- Prasad RB , Groop L . Genetics of Type 2 Diabetes—Pitfalls and Possibilities. Genes . 2015;6(1):87–123. doi:10.3390/genes6010087 25774817
- Faerch KHA , Solomon TP . Heterogeneity of Pre-diabetes and Type 2 Diabetes: implications for Prediction, Prevention and Treatment Responsiveness. Curr Diabetes Rev . 2016;12:30–41. doi:10.2174/1573399811666150416122903 25877695
- Karalliedde J , Gnudi L . Diabetes mellitus, a complex and heterogeneous disease, and the role of insulin resistance as a determinant of diabetic kidney disease. Nephrol Dialysis Transplant . 2016;31(2):206–213.
- Brownlee M . Biochemistry and molecular cell biology of diabetic complications. Nature . 2001;414(6865):813–820. doi:10.1038/414813a 11742414
- Holman N , Young RJ , Jeffcoate WJ . Variation in the recorded incidence of amputation of the lower limb in England. Diabetologia . 2012;55(7):1919–1925. doi:10.1007/s00125-012-2468-6 22398645
- Akter K , Lanza EA , Martin SA , Myronyuk N , Rua M , Raffa RB . Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment? Br J Clin Pharmacol . 2011;71(3):365–376. doi:10.1111/j.1365-2125.2010.03830.x 21284695
- Hu G , Jousilahti P , Bidel S , Antikainen R , Tuomilehto J . Type 2 Diabetes and the Risk of Parkinson’s Disease. Diabetes Care . 2007;30(4):842–847. doi:10.2337/dc06-2011 17251276
- Ali MK , Narayan V , Tandon N . Diabetes & coronary heart disease: current perspectives. Indian J Med Res . 2010;132(5):584–597.21150011
- Leal J , Gray AM , Clarke PM . Development of life-expectancy tables for people with type 2 diabetes. Eur Heart J . 2009;30(7):834–839. doi:10.1093/eurheartj/ehn567 19109355
- Mazzola N . Review of current and emerging therapies in type 2 diabetes mellitus. Am J Manag Care . 2012;18:17–26.22435745
- Olokoba AB , Obateru OA , Olokoba LB . Type 2 Diabetes Mellitus: a Review of Current Trends. Oman Med J . 2012;27(4):269–273. doi:10.5001/omj.2012.68 23071876
- Amiel SA , Dixon T , Mann R , Jameson K . Hypoglycaemia in Type 2 diabetes. Diabetic Med . 2008;25(3):245–254. doi:10.1111/j.1464-5491.2007.02341.x 18215172
- Fonseca VA . Defining and Characterizing the Progression of Type 2 Diabetes. Diabetes Care . 2009;32(Suppl 2):S151–S156. doi:10.2337/dc09-S301 19875543
- Leibowitz G , Kaiser N , Cerasi E . β‐Cell failure in type 2 diabetes. J Diabetes Investig . 2011;2(2):82–91. doi:10.1111/j.2040-1124.2010.00094.x
- Forbes JM , Cooper ME . Mechanisms of Diabetic Complications. Physiol Rev . 2013;93(1):137–188. doi:10.1152/physrev.00045.2011 23303908
- Bhurosy T , Jeewon R . Overweight and Obesity Epidemic in Developing Countries: a Problem with Diet, Physical Activity, or Socioeconomic Status? Scientific World J . 2014;2014:7. doi:10.1155/2014/964236
- Shaw JE , Sicree RA , Zimmet PZ . Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract . 2010;87(1):4–14. doi:10.1016/j.diabres.2009.10.007 19896746
- Onge ES , Miller SA , Motycka C , DeBerry A . A review of the treatment of type 2 diabetes in children. J Pediatric Pharmacol Therapeutics . 2015;20(1):4–16. doi:10.5863/1551-6776-20.1.4
- Reinehr T . Type 2 diabetes mellitus in children and adolescents. World J Diabetes . 2013;4(6):270–281. doi:10.4239/wjd.v4.i6.270 24379917
- Atkinson MA . The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med . 2012;2(11):a007641. doi:10.1101/cshperspect.a007641 23125199
- Turner R , Stratton I , Horton V , et al. UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes. Lancet . 1997;350(9087):1288–1293. doi:10.1016/S0140-6736(97)03062-6 9357409
- Cizza G , Brown RJ , Rothe KI . Rising incidence and challenges of childhood diabetes. A mini review. J Endocrinol Invest . 2012;35(5):541–546. doi:10.3275/8411 22572768
- Kaufman FR . Type 2 Diabetes in Children and Young Adults: a “New Epidemic”. Clin Diabetes . 2002;20(4):217–218. doi:10.2337/diaclin.20.4.217
- D’Adamo E , Caprio S . Type 2 diabetes in youth: epidemiology and pathophysiology. Diabetes Care . 2011;34(Suppl 2):S161–S165. doi:10.2337/dc11-s212 21525449
- Liao C , Zhang D , Mungo C , Tompkins DA , Zeidan AM . Is diabetes mellitus associated with increased incidence and disease-specific mortality in endometrial cancer? A systematic review and meta-analysis of cohort studies. Gynecol Oncol . 2014;135(1):163–171. doi:10.1016/j.ygyno.2014.07.095 25072931
- Saeedi P , Petersohn I , Salpea P , et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract . 2019;2:157.
- Ginter E , Type VS . 2 diabetes mellitus, pandemic in 21st century. Adv Exp Med Biol . 2012;771:42–50.23393670
- Mi H . Diabetes in America . New York: U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES* Public Health Service* National Institutes of Health; 1985.
- Kaveeshwar SA , Cornwall J . The current state of diabetes mellitus in India. Australas Med J . 2014;7(1):45–48. doi:10.4066/AMJ.2014.1979 24567766
- Sosale A , Prasanna Kumar KM , Sadikot SM , et al. Chronic complications in newly diagnosed patients with Type 2 diabetes mellitus in India. Indian J Endocrinol Metab . 2014;18(3):355–360. doi:10.4103/2230-8210.131184 24944931
- Burke JP , Williams K , Gaskill SP , Hazuda HP , Haffner SM , Stern MP . Rapid rise in the incidence of type 2 diabetes from 1987 to 1996: results from the san antonio heart study. Arch Intern Med . 1999;159(13):1450–1456. doi:10.1001/archinte.159.13.1450 10399896
- Bos M , Agyemang C . Prevalence and complications of diabetes mellitus in Northern Africa, a systematic review. BMC Public Health . 2013;13:387. doi:10.1186/1471-2458-13-387 23617762
- Ardisson Korat AV , Willett WC , Hu FB . Diet, lifestyle, and genetic risk factors for type 2 diabetes: a review from the Nurses’ Health Study, Nurses’ Health Study 2, and Health Professionals’ Follow-up Study. Curr Nutr Rep . 2014;3(4):345–354. doi:10.1007/s13668-014-0103-5 25599007
- Nguyen NT , Nguyen X-MT , Lane J , Wang P . Relationship Between Obesity and Diabetes in a US Adult Population: findings from the National Health and Nutrition Examination Survey, 1999–2006. Obes Surg . 2011;21(3):351–355. doi:10.1007/s11695-010-0335-4 21128002
- George AM , Jacob AG , Fogelfeld L . Lean diabetes mellitus: an emerging entity in the era of obesity. World J Diabetes . 2015;6(4):613–620. doi:10.4239/wjd.v6.i4.613 25987958
- Ma RCW , Chan JCN . Type 2 diabetes in East Asians: similarities and differences with populations in Europe and the United States. Ann N Y Acad Sci . 2013;1281(1):64–91. doi:10.1111/nyas.12098 23551121
- Gastaldelli A . Abdominal fat: does it predict the development of type 2 diabetes? Am J Clin Nutr . 2008;87(5):1118–1119. doi:10.1093/ajcn/87.5.1118 18469227
- Noble JA , Erlich HA . Genetics of Type 1 Diabetes. Cold Spring Harb Perspect Med . 2012;2(1):a007732. doi:10.1101/cshperspect.a007732 22315720
- Ahlqvist E , Ahluwalia TS , Groop L . Genetics of Type 2 Diabetes. Clin Chem . 2011;57(2):241–254. doi:10.1373/clinchem.2010.157016 21119033
- Hyttinen V , Kaprio J , Kinnunen L , Koskenvuo M , Tuomilehto J . Genetic Liability of Type 1 Diabetes and the Onset Age Among 22,650 Young Finnish Twin Pairs. Nationwide Follow Up Study . 2003;52(4):1052–1055.
- Poulsen P , Grunnet LG , Pilgaard K , et al. Increased Risk of Type 2 Diabetes in Elderly Twins. Diabetes . 2009;58(6):1350–1355. doi:10.2337/db08-1714 19336677
- Mozaffarian D , Kamineni A , Carnethon M , Djoussé L , Mukamal KJ , Siscovick D . Lifestyle risk factors and new-onset diabetes mellitus in older adults: the cardiovascular health study. Arch Intern Med . 2009;169(8):798–807. doi:10.1001/archinternmed.2009.21 19398692
- Kelly SJIM . Stress and type 2 diabetes: a review of how stress contributes to the development of type 2 diabetes. Annu Rev Public Health . 2015;18:441–462. doi:10.1146/annurev-publhealth-031914-122921
- Kautzky-Willer A , Harreiter J , Pacini G . Sex and Gender Differences in Risk, Pathophysiology and Complications of Type 2 Diabetes Mellitus. Endocr Rev . 2016;37(3):278–316.27159875
- Sattar N . Gender aspects in type 2 diabetes mellitus and cardiometabolic risk. Best Pract Res Clin Endocrinol Metab . 2013;27(4):501–507. doi:10.1016/j.beem.2013.05.006 24054927
- Doró P , Benkő R , Matuz M , Soós G . Seasonality in the Incidence of Type 2 Diabetes. Diabetes Care . 2006;29(1):173. doi:10.2337/diacare.29.01.06.dc05-1839 16373925
- Si J , Yu C , Guo Y , et al. Season of birth and the risk of type 2 diabetes in adulthood: a prospective cohort study of 0.5 million Chinese adults. Diabetologia . 2017;60(5):836–842. doi:10.1007/s00125-016-4200-4 28064359
- Corkey BE . Diabetes: have We Got It All Wrong?: insulin hypersecretion and food additives: cause of obesity and diabetes? Diabetes Care . 2012;35(12):2432–2437. doi:10.2337/dc12-0825 23173132
- Alonso-Magdalena P , Quesada INA . Endocrine disruptors in the etiology of type 2 diabetes mellitus. Nat Rev Endocrinol . 2011;7:346–353. doi:10.1038/nrendo.2011.56 21467970
- Sun Y , Pei W , Wu Y , Yang Y . An Association of Herpes Simplex Virus Type 1 Infection With Type 2 Diabetes. Diabetes Care . 2005;28(2):435. doi:10.2337/diacare.28.2.435 15677810
- Wang C-S , Wang S-T , Yao W-J , Chang -T-T , Chou P . Hepatitis C Virus Infection and the Development of Type 2 Diabetes in a Community-based Longitudinal Study. Am J Epidemiol . 2007;166(2):196–203. doi:10.1093/aje/kwm061 17496314
- Bose SK , Ray R . Hepatitis C virus infection and insulin resistance. World J Diabetes . 2014;5(1):52–58. doi:10.4239/wjd.v5.i1.52 24567801
- Torres DM , Harrison SA . Hepatitis C virus and insulin resistance/diabetes mellitus. Gastroenterol Hepatol (N Y) . 2008;4(8):568–570.21960938
- Bansal N . Prediabetes diagnosis and treatment: a review. World J Diabetes . 2015;6(2):296–303. doi:10.4239/wjd.v6.i2.296 25789110
- Tabák AG , Herder C , Rathmann W , Brunner EJ , Kivimäki M . Prediabetes: a high-risk state for developing diabetes. Lancet . 2012;379(9833):2279–2290. doi:10.1016/S0140-6736(12)60283-9 22683128
- Tuso P . Prediabetes and lifestyle modification: time to prevent a preventable disease. Permanente J Summer . 2014;18(3):88–93. doi:10.7812/TPP/14-002
- Davidson MB , Schriger DL , Peters AL , Lorber B . Revisiting the oral glucose tolerance test criterion for the diagnosis of diabetes. J Gen Intern Med . 2000;15(8):551–555. doi:10.1046/j.1525-1497.2000.08024.x 10940146
- Affourtit C . Mitochondrial involvement in skeletal muscle insulin resistance: a case of imbalanced bioenergetics. Biochimica Et Biophysica Acta . 2016;1857(10):1678–1693.27473535
- Muoio DM , Newgard CB . Molecular and metabolic mechanisms of insulin resistance and [beta]-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol . 2008;9(3):193–205. doi:10.1038/nrm2327 18200017
- Linnemann AK , Baan M , Davis DB . Pancreatic β-Cell Proliferation in Obesity. Adv Nutrition . 2014;5(3):278–288. doi:10.3945/an.113.005488
- Mehran Arya E , Templeman Nicole M , Brigidi GS , et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab . 2012;16(6):723–737. doi:10.1016/j.cmet.2012.10.019 23217255
- Boland BB , Rhodes CJ , Grimsby JS . The dynamic plasticity of insulin production in β-cells. Mol Metab . 2017;6(9):958–973.28951821
- Cerf ME . Beta Cell Dysfunction and Insulin Resistance. Front Endocrinol (Lausanne) . 2013;4:37. doi:10.3389/fendo.2013.00037 23542897
- Achenbach P , Hummel M , Thümer L , Boerschmann H , Höfelmann D , Ziegler AG . Characteristics of rapid vs slow progression to type 1 diabetes in multiple islet autoantibody-positive children. Diabetologia . 2013;56(7):1615–1622. doi:10.1007/s00125-013-2896-y 23539116
- Breuer TGK , Menge BA , Banasch M , et al. Proinsulin levels in patients with pancreatic diabetes are associated with functional changes in insulin secretion rather than pancreatic β-cell area. Eur J Endocrinol . 2010;163(4):551–558. doi:10.1530/EJE-10-0330 20679359
- Pfützner A , Hermanns I , Ramljak S , et al. Elevated Intact proinsulin levels during an oral glucose challenge indicate progressive cell dysfunction and may be predictive for development of type 2 diabetes. J Diabetes Sci Technol . 2015;9(6):1307–1312. doi:10.1177/1932296815607862 26420624
- Vangipurapu J , Stančáková A , Kuulasmaa T , Kuusisto J , Laakso M . Both Fasting and Glucose-Stimulated Proinsulin Levels Predict Hyperglycemia and Incident Type 2 Diabetes: a Population-Based Study of 9,396 Finnish Men. PLoS One . 2015;10(4):e0124028. doi:10.1371/journal.pone.0124028 25853252
- Cernea S , Dobreanu M . Diabetes and beta cell function: from mechanisms to evaluation and clinical implications. Biochem Med (Zagreb) . 2013;23(3):266–280. doi:10.11613/BM.2013.033 24266296
- Wilcox G . Insulin and Insulin Resistance. Clin Biochemist Rev . 2005;26(2):19–39.
- Perilli G , Saraceni C , Daniels MN , Ahmad A . Diabetic ketoacidosis: a review and update. Curr Emerg Hosp Med Rep . 2013;1(1):10–17. doi:10.1007/s40138-012-0001-3
- Woerle HJ , Carneiro L , Derani A , Göke B , Schirra J . The role of endogenous incretin secretion as amplifier of glucose-stimulated insulin secretion in healthy subjects and patients with type 2 diabetes. Diabetes . 2012;61(9):2349–2358. doi:10.2337/db11-1701 22721966
- Atkinson MA , Eisenbarth GS , Michels AW . Type 1 diabetes. Lancet . 2014;383(9911):69–82. doi:10.1016/S0140-6736(13)60591-7 23890997
- White H , Venkatesh B . Clinical review: ketones and brain injury. Critical Care . 2011;15(2):219. doi:10.1186/cc10020 21489321
- Gosmanov AR , Gosmanova EO , Dillard-Cannon E . Management of adult diabetic ketoacidosis. Diabetes Metab Syndrome Obesity . 2014;7:255–264. doi:10.2147/DMSO.S50516
- Olson AL . Regulation of GLUT4 and Insulin-Dependent Glucose Flux. ISRN Molecular Biol . 2012;2012:12. doi:10.5402/2012/856987
- Hardy OT , Czech MP , Corvera S . What causes the insulin resistance underlying obesity? Curr Opin Endocrinol Diabetes Obes . 2012;19(2):81–87. doi:10.1097/MED.0b013e3283514e13 22327367
- Goodpaster BH , He J , Watkins S , Kelley DE . Skeletal Muscle Lipid Content and Insulin Resistance: evidence for a Paradox in Endurance-Trained Athletes. J Clin Endocrinol Metab . 2001;86(12):5755–5761. doi:10.1210/jcem.86.12.8075 11739435
- van Loon LJC , Koopman R , Manders R , van der Weegen W , van Kranenburg GP , Keizer HA . Intramyocellular lipid content in type 2 diabetes patients compared with overweight sedentary men and highly trained endurance athletes. Am J Physiol . 2004;287(3):E558–E565. doi:10.1152/ajpendo.00464.2003
- Vaag A , Lund SS . Non-obese patients with type 2 diabetes and prediabetic subjects: distinct phenotypes requiring special diabetes treatment and (or) prevention? Applied Physiol Nutrition Metab . 2007;32(5):912–920. doi:10.1139/H07-100 18059616
- Chang SA , Kim HS , Yoon KH , et al. Body mass index is the most important determining factor for the degree of insulin resistance in non-obese type 2 diabetic patients in Korea. Metab Clin Exp . 2004;53(2):142–146. doi:10.1016/S0026-0495(03)00314-7 14767863
- Khaodhiar L , Cummings S , Apovian CM . Treating diabetes and prediabetes by focusing on obesity management. Curr Diab Rep . 2009;9(5):348–354. doi:10.1007/s11892-009-0055-0 19793504
- Madeira FB , Silva AA , Veloso HF , et al. Normal Weight Obesity Is Associated with Metabolic Syndrome and Insulin Resistance in Young Adults from a Middle-Income Country. PLoS One . 2013;8(3):e60673. doi:10.1371/journal.pone.0060673 23556000
- Lim EL , Hollingsworth KG , Aribisala BS , Chen MJ , Mathers JC , Taylor R . Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia . 2011;54(10):2506–2514. doi:10.1007/s00125-011-2204-7 21656330
- Taylor R . Pathogenesis of type 2 diabetes: tracing the reverse route from cure to cause. Diabetologia . 2008;51(10):1781–1789. doi:10.1007/s00125-008-1116-7 18726585
- DeLany JP , Dubé JJ , Standley RA , et al. Racial Differences In Peripheral Insulin Sensitivity and Mitochondrial Capacity in the Absence of Obesity. J Clin Endocrinol Metab . 2014;99(11):4307–4314. doi:10.1210/jc.2014-2512 25105736
- Khoo CM , Leow MK-S , Sadananthan SA , et al. Body Fat Partitioning Does Not Explain the Interethnic Variation in Insulin Sensitivity Among Asian Ethnicity: the Singapore Adults Metabolism Study. Diabetes . 2014;63(3):1093–1102. doi:10.2337/db13-1483 24353181
- Yoshino J , Almeda-Valdes P , Patterson BW , et al. Diurnal Variation in Insulin Sensitivity of Glucose Metabolism Is Associated With Diurnal Variations in Whole-Body and Cellular Fatty Acid Metabolism in Metabolically Normal Women. J Clin Endocrinol Metab . 2014;99(9):E1666–E1670. doi:10.1210/jc.2014-1579 24878055
- Butler AE , Janson J , Bonner-Weir S , Ritzel R , Rizza RA , Butler PC . β-Cell Deficit and Increased β-Cell Apoptosis in Humans With Type 2 Diabetes. Diabetes . 2003;52(1):102–110. doi:10.2337/diabetes.52.1.102 12502499
- Biarnés M , Montolio M , Nacher V , Raurell M , Soler J , Montanya E . β-Cell Death and Mass in Syngeneically Transplanted Islets Exposed to Short- and Long-Term Hyperglycemia. Diabetes . 2002;51(1):66–72. doi:10.2337/diabetes.51.1.66 11756324
- Bretherton-Watt D , Ghatei MA , Bloom SR , et al. Altered islet amyloid polypeptide (amylin) gene expression in rat models of diabetes. Diabetologia . 1989;32(12):881–883. doi:10.1007/BF00297454 2612759
- Mulder H , Ahrén B , Sundler F . Islet amyloid polypeptide (amylin) and insulin are differentially expressed in chronic diabetes induced by streptozotocin in rats. Diabetologia . 1996;39(6):649–657. doi:10.1007/BF00418536 8781760
- Gupta D , Leahy JL . Islet amyloid and type 2 diabetes: overproduction or inadequate clearance and detoxification? J Clin Invest . 2014;124(8):3292–3294. doi:10.1172/JCI77506 25036704
- Alrefai HA , Latif KA , Hieronymus LB , Weakley CR , Moss RJ . Pramlintide: clinical Strategies for Success. Diabetes Spectrum . 2010;23(2):124–130. doi:10.2337/diaspect.23.2.124
- Howard CJ . Diabetes mellitus: relationships of nonhuman primates and other animal models to human forms of diabetes. Adv Vet Sci Comp Med . 1984;28:115–149.6395671
- Matveyenko AV , Butler PC . Islet Amyloid Polypeptide (IAPP) Transgenic Rodents as Models for Type 2 Diabetes. ILAR J . 2006;47(3):225–233. doi:10.1093/ilar.47.3.225 16804197
- Shigihara N , Fukunaka A , Hara A , et al. Human IAPP–induced pancreatic β cell toxicity and its regulation by autophagy. J Clin Invest . 2014;124(8):3634–3644. doi:10.1172/JCI69866 25036706
- Cnop M , Welsh N , Jonas J-C , Jörns A , Lenzen S , Eizirik DL . Mechanisms of Pancreatic β-Cell Death in Type 1 and Type 2 Diabetes. Many Differences Few Similarities . 2005;54(suppl2):S97–S107.
- Costanzo P , Cleland JF , Pellicori P , et al. The obesity paradox in type 2 diabetes mellitus: relationship of body mass index to prognosis: a cohort study. Ann Intern Med . 2015;162(9):610–618. doi:10.7326/M14-1551 25938991
- Hainer V , Aldhoon-Hainerová I . Obesity Paradox Does Exist. Diabetes Care . 2013;36(Supplement 2):S276–S281. doi:10.2337/dcS13-2023 23882059
- Doehner W , Erdmann E , Cairns R , et al. Inverse relation of body weight and weight change with mortality and morbidity in patients with type 2 diabetes and cardiovascular co-morbidity: an analysis of the PROactive study population. Int J Cardiol . 2012;162(1):20–26. doi:10.1016/j.ijcard.2011.09.039 22037349
- Sohn M-W , Budiman-Mak E , Oh EH , et al. Obesity Paradox in Amputation Risk Among Nonelderly Diabetic Men. Obesity . 2012;20(2):460–462. doi:10.1038/oby.2011.301 21996669
- Coleman NJ , Miernik J , Philipson L , Fogelfeld L . Lean versus obese diabetes mellitus patients in the United States minority population. J Diabetes Complications . 2013;28(4):500–505. doi:10.1016/j.jdiacomp.2013.11.010 24581791
- Kim JY , Song EH , Lee HJ , et al. Chronic Ethanol Consumption-induced Pancreatic β-Cell Dysfunction and Apoptosis through Glucokinase Nitration and Its Down-regulation. J Biol Chem . 2010;285(48):37251–37262. doi:10.1074/jbc.M110.142315 20855893
- Gerich JE . Insulin Resistance Is Not Necessarily an Essential Component of Type 2 Diabetes1. J Clin Endocrinol Metab . 2000;85(6):2113–2115.10852436
- Brereton MF , Vergari E , Zhang Q , Clark A . Alpha-, Delta- and PP-cells: are They the Architectural Cornerstones of Islet Structure and Co-ordination? J Histochem Cytochemistry . 2015;63(8):575–591. doi:10.1369/0022155415583535
- Moon JS , Won KC . Pancreatic α-Cell Dysfunction in Type 2 Diabetes: old Kids on the Block. Diabetes Metab J . 2015;39(1):1–9. doi:10.4093/dmj.2015.39.1.1 25729706
- Sherwin RS , Fisher M , Hendler R , Felig P . Hyperglucagonemia and Blood Glucose Regulation in Normal, Obese and Diabetic Subjects. N Eng J Med . 1976;294(9):455–461. doi:10.1056/NEJM197602262940901
- Ivan Quesada ET , Ripoll C , Nadal Á . Physiology of the pancreatic α-cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol . 2008;2:68–96.
- Dinneen S , Alzaid A , Turk D , Rizza R . Failure of glucagon suppression contributes to postprandial hyperglycaemia in IDDM. Diabetologia . 1995;38(3):337–343. doi:10.1007/BF00400639 7758881
- Reaven GM , Chen YDI , Golay A , Swislocki ALM , Jaspan JB . Documentation of Hyperglucagonemia Throughout the Day in Nonobese and Obese Patients with Noninsulin-Dependent Diabetes Mellitus*. J Clin Endocrinol Metab . 1987;64(1):106–110. doi:10.1210/jcem-64-1-106 3536980
- Shah P , Vella A , Basu A , Basu R , Schwenk WF , Rizza RA . Lack of Suppression of Glucagon Contributes to Postprandial Hyperglycemia in Subjects with Type 2 Diabetes Mellitus1. J Clin Endocrinol Metab . 2000;85(11):4053–4059.11095432
- Baron AD , Schaeffer L , Shragg P , Kolterman OG . Role of Hyperglucagonemia in Maintenance of Increased Rates of Hepatic Glucose Output in Type II Diabetics. Diabetes . 1987;36(3):274–283. doi:10.2337/diab.36.3.274 2879757
- Kawamori D , Kurpad AJ , Hu J , et al. Insulin Signaling in α-cells Modulates Glucagon Secretion in vivo. Cell Metab . 2009;9(4):350–361. doi:10.1016/j.cmet.2009.02.007 19356716
- Elder DA , Prigeon RL , Wadwa RP , Dolan LM , D’Alessio DA . β-Cell Function, Insulin Sensitivity, and Glucose Tolerance in Obese Diabetic and Nondiabetic Adolescents and Young Adults. J Clin Endocrinol Metab . 2006;91(1):185–191. doi:10.1210/jc.2005-0853 16263830
- Kilimnik G , Zhao B , Jo J , et al. Altered Islet Composition and Disproportionate Loss of Large Islets in Patients with Type 2 Diabetes. PLoS One . 2011;6(11):e27445. doi:10.1371/journal.pone.0027445 22102895
- Brereton MF , Iberl M , Shimomura K , et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat Commun . 2014;5:4639. doi:10.1038/ncomms5639 25145789
- Clark A , Wells CA , Buley ID , et al. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res . 1988;9:151–159.3073901
- Rahier J , Goebbels RM , Henquin JC . Cellular composition of the human diabetic pancreas. Diabetologia . 1983;24(5):366–371. doi:10.1007/BF00251826 6347784
- Guardado Mendoza R , Perego C , Finzi G , et al. Delta cell death in the islet of Langerhans and the progression from normal glucose tolerance to type 2 diabetes in non-human primates (baboon, Papio hamadryas). Diabetologia . 2015;58(8):1814–1826. doi:10.1007/s00125-015-3625-5 26049399
- Talchai C , Xuan S , Lin HV , Sussel L , Accili D . Pancreatic β-Cell Dedifferentiation As Mechanism Of Diabetic β-Cell Failure. Cell . 2012;150(6):1223–1234. doi:10.1016/j.cell.2012.07.029 22980982
- Ahrén B . Incretin dysfunction in type 2 diabetes: clinical impact and future perspectives. Diabetes Metab . 2013;39:195–201. doi:10.1016/j.diabet.2013.03.001 23643349
- Meier JJ , Nauck MA . Is the Diminished Incretin Effect in Type 2 Diabetes Just an Epi-Phenomenon of Impaired β-Cell Function? Diabetes . 2010;59(5):1117–1125. doi:10.2337/db09-1899 20427697
- Pratley RE . GIP: an Inconsequential Incretin or Not? Diabetes Care . 2010;33(7):1691–1692. doi:10.2337/dc10-0704 20587731
- Nauck MA , Heimesaat MM , Orskov C , Holst JJ , Ebert R , Creutzfeldt W . Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest . 1993;91(1):301–307. doi:10.1172/JCI116186 8423228
- Højberg PV , Vilsbøll T , Rabøl R , et al. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia . 2009;52(2):199. doi:10.1007/s00125-008-1195-5 19037628
- Højberg PV , Vilsbøll T , Zander M , et al. Four weeks of near-normalization of blood glucose has no effect on postprandial GLP-1 and GIP secretion, but augments pancreatic B-cell responsiveness to a meal in patients with Type 2 diabetes. Diabetic Med . 2008;25(11):1268–1275. doi:10.1111/j.1464-5491.2008.02579.x 19046215
- Thorens B . Neural regulation of pancreatic islet cell mass and function. Diabetes Obes Metab . 2014;16(S1):87–95. doi:10.1111/dom.12346 25200301
- Thorens B . Brain glucose sensing and neural regulation of insulin and glucagon secretion. Diabetes Obes Metab . 2011;13:82–88. doi:10.1111/j.1463-1326.2011.01453.x 21824260
- Kiba T . Relationships between the autonomic nervous system and the pancreas including regulation of regeneration and apoptosis: recent developments. Pancreas . 2004;29:51–58. doi:10.1097/00006676-200408000-00019
- Tarussio D , Metref S , Seyer P , et al. Nervous glucose sensing regulates postnatal β cell proliferation and glucose homeostasis. J Clin Invest . 2014;124(1):413–424. doi:10.1172/JCI69154 24334455
- Poitout V , Amyot J , Semache M , Zarrouki B , Hagman D , Fontés G . Glucolipotoxicity of the Pancreatic Beta Cell. Biochim Biophys Acta . 2010;1801(3):289–298. doi:10.1016/j.bbalip.2009.08.006 19715772
- Kim J-W , Yoon K-H . Glucolipotoxicity in Pancreatic β-Cells. Diabetes Metab J . 2011;35(5):444–450. doi:10.4093/dmj.2011.35.5.444 22111034
- Prentki M , Nolan CJ . Islet β cell failure in type 2 diabetes. J Clin Invest . 2006;116(7):1802–1812. doi:10.1172/JCI29103 16823478
- Mason TM , Goh T , Tchipashvili V , et al. Prolonged elevation of plasma free fatty acids desensitizes the insulin secretory response to glucose in vivo in rats. Diabetes . 1999;48(3):524–530. doi:10.2337/diabetes.48.3.524 10078552
- Kashyap S , Belfort R , Gastaldelli A , et al. A Sustained Increase in Plasma Free Fatty Acids Impairs Insulin Secretion in Nondiabetic Subjects Genetically Predisposed to Develop Type 2 Diabetes. Diabetes . 2003;52(10):2461–2474. doi:10.2337/diabetes.52.10.2461 14514628
- Barlow J , Affourtit C . Novel insights into pancreatic β-cell glucolipotoxicity from real-time functional analysis of mitochondrial energy metabolism in INS-1E insulinoma cells. Biochemical J . 2013;456(3):417–426. doi:10.1042/BJ20131002
- Barlow J , Jensen Verena H , Jastroch M , Affourtit C . Palmitate-induced impairment of glucose-stimulated insulin secretion precedes mitochondrial dysfunction in mouse pancreatic islets. Biochemical J . 2016;473(4):487–496. doi:10.1042/BJ20151080
- Hogan AE , Tobin AM , Ahern T , et al. Glucagon-like peptide-1 (GLP-1) and the regulation of human invariant natural killer T cells: lessons from obesity, diabetes and psoriasis. Diabetologia . 2011;54(11):2745–2754. doi:10.1007/s00125-011-2232-3 21744074
- Wang Z , Thurmond DC . Mechanisms of biphasic insulin-granule exocytosis – roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci . 2009;122(7):893. doi:10.1242/jcs.034355 19295123
- Athauda D , Maclagan K , Skene SS , et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet . 2017;390(10103):1664–1675. doi:10.1016/S0140-6736(17)31585-4 28781108
- Lynn FC , Pamir N , Ng EHC , McIntosh CHS , Kieffer TJ , Pederson RA . Defective Glucose-Dependent Insulinotropic Polypeptide Receptor Expression in Diabetic Fatty Zucker Rats. Diabetes . 2001;50(5):1004–1011. doi:10.2337/diabetes.50.5.1004 11334402
- Affourtit C , Brand martin D . Uncoupling protein-2 contributes significantly to high mitochondrial proton leak in INS-1E insulinoma cells and attenuates glucose-stimulated insulin secretion. Biochemical J . 2008;409(1):199–204. doi:10.1042/BJ20070954
- Affourtit C , Brand MD . On the role of uncoupling protein-2 in pancreatic beta cells. Biochimica et Biophysica Acta . 2008;1777(7–8):973–979. doi:10.1016/j.bbabio.2008.03.022 18433713
- Affourtit C , Jastroch M , Brand MD . Uncoupling protein-2 attenuates glucose-stimulated insulin secretion in INS-1E insulinoma cells by lowering mitochondrial reactive oxygen species. Free Radic Biol Med . 2011;50(5):609–616. doi:10.1016/j.freeradbiomed.2010.12.020 21172424
- Chan CB , De Leo D , Joseph JW , et al. Increased uncoupling protein-2 levels in β-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes . 2001;50(6):1302–1310. doi:10.2337/diabetes.50.6.1302 11375330
- Chan CB , MacDonald PE , Saleh MC , Johns DC , Marbàn E , Wheeler MB . Overexpression of uncoupling protein 2 inhibits glucose-stimulated insulin secretion from rat islets. Diabetes . 1999;48(7):1482–1486. doi:10.2337/diabetes.48.7.1482 10389858
- Pi J , Collins S . Reactive oxygen species and uncoupling protein 2 in pancreatic β‐cell function. Diabetes Obes Metab . 2010;12(s2):141–148. doi:10.1111/j.1463-1326.2010.01269.x 21029311
- Robson-Doucette CA , Sultan S , Allister EM , et al. β-Cell Uncoupling Protein 2 Regulates Reactive Oxygen Species Production, Which Influences Both Insulin and Glucagon Secretion. Diabetes . 2011;60(11):2710–2719. doi:10.2337/db11-0132 21984579
- Schrauwen P , Hesselink M . UCP2 and UCP3 in muscle controlling body metabolism. J Exp Biol . 2002;205(15):2275. doi:10.1242/jeb.205.15.2275 12110661
- Bensellam M , Laybutt DR , Jonas J-C . The molecular mechanisms of pancreatic β-cell glucotoxicity: recent findings and future research directions. Mol Cell Endocrinol . 2012;364(1–2):1–27.22885162
- Hirschberg Jensen V , Affourtit C . Mitochondrial uncoupling protein-2 is not involved in palmitate-induced impairment of glucose-stimulated insulin secretion in INS-1E insulinoma cells and is not needed for the amplification of insulin release. Biochemistry Biophysics Rep . 2015;1:8–15. doi:10.1016/j.bbrep.2015.03.008
- Vozza A , Parisi G , De Leonardis F , et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci U S A . 2014;111(3):960–965. doi:10.1073/pnas.1317400111 24395786
- Mailloux RJ , Fu A , Robson-Doucette C , et al. Glutathionylation State of Uncoupling Protein-2 and the Control of Glucose-stimulated Insulin Secretion. J Biol Chem . 2012;287(47):39673–39685. doi:10.1074/jbc.M112.393538 23035124
- Mailloux RJ , Seifert EL , Bouillaud F , Aguer C , Collins S , Harper M-E . Glutathionylation Acts as a Control Switch for Uncoupling Proteins UCP2 and UCP3. J Biol Chem . 2011;286(24):21865–21875. doi:10.1074/jbc.M111.240242 21515686
- Basu R , Barosa C , Jones J , et al. Pathogenesis of Prediabetes: role of the Liver in Isolated Fasting Hyperglycemia and Combined Fasting and Postprandial Hyperglycemia. J Clin Endocrinol Metab . 2013;98(3):E409–E417. doi:10.1210/jc.2012-3056 23345093
- Phielix E , Mensink M . Type 2 Diabetes Mellitus and Skeletal Muscle Metabolic Function. Physiol Behav . 2008;94(2):252–258. doi:10.1016/j.physbeh.2008.01.020 18342897
- De Macedo G , Maria C , Nunes S , Barreto T . Skin disorders in diabetes mellitus: an epidemiology and physiopathology review. Diabetol Metab Syndr . 2016;8(1):63. doi:10.1186/s13098-016-0176-y 27583022
- Krishnan B , Babu S , Walker J , Walker AB , Pappachan JM . Gastrointestinal complications of diabetes mellitus. World J Diabetes . 2013;4(3):51–63. doi:10.4239/wjd.v4.i3.51 23772273
- Sharaf El Din A , Abdulazim DO . Diabetic nephropathy: time to withhold development and progression - A review. J Adv Res . 2017;8(4):363–373. doi:10.1016/j.jare.2017.04.004 28540086
- Sanad E , ElFangary M , Sorour N , ElNemisy N . Skin manifestations in Egyptian diabetic patients: a case series study. Egypt J Dermatol Venerology . 2013;33(2):56–62. doi:10.4103/1110-6530.123941
- DeFronzo RA , Tripathy D . Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care . 2009;32(Suppl 2):S157–S163. doi:10.2337/dc09-S302 19875544
- Blaak EE , van Aggel-leijssen DP , Wagenmakers AJ , Saris WH , van Baak MA . Impaired oxidation of plasma-derived fatty acids in type 2 diabetic subjects during moderate-intensity exercise. Diabetes . 2000;49(12):2102. doi:10.2337/diabetes.49.12.2102 11118013
- Mensink M , Blaak EE , van Baak MA , Wagenmakers AJM , Saris WHM . Plasma Free Fatty Acid Uptake and Oxidation Are Already Diminished in Subjects at High Risk for Developing Type 2 Diabetes. Diabetes . 2001;50(11):2548. doi:10.2337/diabetes.50.11.2548 11679433
- Holloszy JO . “Deficiency” of Mitochondria in Muscle Does Not Cause Insulin Resistance. Diabetes . 2013;62(4):1036. doi:10.2337/db12-1107 23520283
- van de Weijer T , Sparks LM , Phielix E , et al. Relationships between Mitochondrial Function and Metabolic Flexibility in Type 2 Diabetes Mellitus. PLoS One . 2013;8(2):e51648. doi:10.1371/journal.pone.0051648 23418416
- Galgani JE , Heilbronn LK , Azuma K , et al. Metabolic Flexibility in Response to Glucose Is Not Impaired in People With Type 2 Diabetes After Controlling for Glucose Disposal Rate. Diabetes . 2008;57(4):841–845. doi:10.2337/db08-0043 18285553
- Goodpaster BH , Katsiaras A , Kelley DE . Enhanced Fat Oxidation Through Physical Activity Is Associated With Improvements in Insulin Sensitivity in Obesity. Diabetes . 2003;52(9):2191. doi:10.2337/diabetes.52.9.2191 12941756
- Færch K , Vaag A . Metabolic inflexibility is a common feature of impaired fasting glycaemia and impaired glucose tolerance. Acta Diabetol . 2011;48(4):349–353. doi:10.1007/s00592-010-0245-x 21207234
- Park SW , Goodpaster BH , Lee JS , et al. Excessive Loss of Skeletal Muscle Mass in Older Adults With Type 2 Diabetes. Diabetes Care . 2009;32(11):1993–1997. doi:10.2337/dc09-0264 19549734
- Perry BD , Caldow MK , Brennan-Speranza TC , et al. Muscle atrophy in patients with Type 2 Diabetes Mellitus: roles of inflammatory pathways, physical activity and exercise. Exerc Immunol Rev . 2016;22:94–109.26859514
- Ciaraldi TP , Ryan AJ , Mudaliar SR , Henry RR . Altered Myokine Secretion Is an Intrinsic Property of Skeletal Muscle in Type 2 Diabetes. PLoS One . 2016;11(7):e0158209. doi:10.1371/journal.pone.0158209 27453994
- Bouzakri K , Plomgaard P , Berney T , Donath MY , Pedersen BK , Halban PA . Bimodal Effect on Pancreatic β-Cells of Secretory Products From Normal or Insulin-Resistant Human Skeletal Muscle. Diabetes . 2011;60(4):1111–1121. doi:10.2337/db10-1178 21378173
- Hancock CR , Han D-H , Chen M , et al. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc National Acad Sci . 2008;105(22):7815–7820. doi:10.1073/pnas.0802057105
- Turner N , Bruce CR , Beale SM , et al. Excess Lipid Availability Increases Mitochondrial Fatty Acid Oxidative Capacity in Muscle. Diabetes . 2007;56(8):2085. doi:10.2337/db07-0093 17519422
- Sreekumar R , Unnikrishnan J , Fu A , et al. Impact of high-fat diet and antioxidant supplement on mitochondrial functions and gene transcripts in rat muscle. Am J Physiol Endocrinol Metabo . 2002;282(5):E1055–E1061. doi:10.1152/ajpendo.00554.2001
- Beck-Nielsen H , Hother-Nielsen O , Staehr P . Is hepatic glucose production increased in type 2 diabetes mellitus? Curr Diab Rep . 2002;2(3):231–236. doi:10.1007/s11892-002-0088-0 12643178
- Bhatt HB , Smith RJ . Fatty liver disease in diabetes mellitus. Hepatobiliary Surg Nutr . 2015;4(2):101–108. doi:10.3978/j.issn.2304-3881.2015.01.03 26005676
- Mazzotti A , Caletti MT , Marchignoli F , Forlani G , Marchesini G . Which treatment for type 2 diabetes associated with non-alcoholic fatty liver disease? Digestive Liver Dis . 2017;49(3):235–240. doi:10.1016/j.dld.2016.12.028
- Targher G , Day CP , Bonora E . Risk of Cardiovascular Disease in Patients with Nonalcoholic Fatty Liver Disease. N Eng J Med . 2010;363(14):1341–1350. doi:10.1056/NEJMra0912063
- Rijkelijkhuizen JM , Doesburg T , Girman CJ , et al. Hepatic fat is not associated with beta-cell function or postprandial free fatty acid response. Metab Clin Exp . 2009;58(2):196–203. doi:10.1016/j.metabol.2008.09.013 19154952
- Gurgul-Convey E , Mehmeti I , Lortz S , Lenzen S . Cytokine toxicity in insulin-producing cells is mediated by nitro-oxidative stress-induced hydroxyl radical formation in mitochondria. J Mol Med . 2011;89(8):785–798. doi:10.1007/s00109-011-0747-1 21487676
- Makki K , Froguel P , Wolowczuk I . Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: cells, Cytokines, and Chemokines. ISRN Inflammation . 2013;2013:139239. doi:10.1155/2013/139239 24455420
- Frayn KN . Adipose tissue and the insulin resistance syndrome. Proc Nutrition Soc . 2007;60(3):375–380. doi:10.1079/PNS200195
- Blaak EE . Fatty acid metabolism in obesity and type 2 diabetes mellitus. Proc Nutrition Soc . 2007;62(3):753–760. doi:10.1079/PNS2003290
- Moreno-Indias I , Cardona F , Tinahones FJ , Queipo-Ortuño MI . Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front Microbiol . 2014;5:190. doi:10.3389/fmicb.2014.00190 24808896
- Rubin MR , Patsch JM . Assessment of bone turnover and bone quality in type 2 diabetic bone disease: current concepts and future directions. Bone Res . 2016;4:16001. doi:10.1038/boneres.2016.1 27019762
- Dobnig H , Piswanger-Sölkner JC , Roth M , et al. Type 2 Diabetes Mellitus in Nursing Home Patients: effects on Bone Turnover, Bone Mass, and Fracture Risk. J Clin Endocrinol Metab . 2006;91(9):3355–3363. doi:10.1210/jc.2006-0460 16735485
- Yamamoto M , Yamaguchi T , Nawata K , Yamauchi M , Sugimoto T . Decreased PTH Levels Accompanied by Low Bone Formation Are Associated with Vertebral Fractures in Postmenopausal Women with Type 2 Diabetes. J Clin Endocrinol Metab . 2012;97(4):1277–1284. doi:10.1210/jc.2011-2537 22337915
- Wrighten SA , Piroli GG , Grillo CA , Reagan LP . A look inside the diabetic brain: contributors to diabetes-induced brain aging. Biochim Biophys Acta . 2009;1792(5):444–453. doi:10.1016/j.bbadis.2008.10.013 19022375
- Ten Kulve JS , van Bloemendaal L , Balesar R , et al. Decreased Hypothalamic Glucagon-Like Peptide-1 Receptor Expression in Type 2 Diabetes Patients. J Clin Endocrinol Metab . 2016;101(5):2122–2129. doi:10.1210/jc.2015-3291 26672638
- Pirart J . Diabetes mellitus and its degenerative complications: a prospective study of 4400 patients observed. Diabetes Care . 1978;1:168–188. doi:10.2337/diacare.1.3.168
- Yagihashi S , Mizukami H , Sugimoto K . Mechanism of diabetic neuropathy: where are we now and where to go? J Diabetes Investig . 2011;2(1):18–32. doi:10.1111/j.2040-1124.2010.00070.x
- Duby JJ , Campbell RK , Setter SM . Diabetic neuropathy: an intensive review. Am J Health Sys Pharmacy . 2004;61(2):160. doi:10.1093/ajhp/61.2.160
- Tracy JA , Dyck PJB . The Spectrum of Diabetic Neuropathies. Phys Med Rehabil Clin N Am . 2008;19(1):1–26. doi:10.1016/j.pmr.2007.10.010 18194747
- Nesto R , Fain R , Li Y , Shanahan W . Evaluation of lorcaserin on progression of prediabetes to type 2 diabetes and reversion to euglycemia. Postgrad Med . 2016;128(4):364–370. doi:10.1080/00325481.2016.1178590 27116910
- Dokken BB . The Pathophysiology of Cardiovascular Disease and Diabetes: beyond Blood Pressure and Lipids. Diabetes Spectrum . 2008;21(3):160. doi:10.2337/diaspect.21.3.160
- Funk SD , Ory AW . Hyperglycemia and Endothelial Dysfunction in Atherosclerosis: lessons from Type 1 Diabetes. Int J Vasc Med . 2012;2012. doi:10.1155/2012/569654
- Wu G , Meininger CJ . Nitric oxide and vascular insulin resistance. BioFactors . 2009;35(1):21–27. doi:10.1002/biof.3 19319842
- Lastra G , Syed S , Kurukulasuriya LR , Manrique C , Sowers JR . Type 2 diabetes mellitus and hypertension: an update. Endocrinol Metab Clin North Am . 2014;43(1):103–122. doi:10.1016/j.ecl.2013.09.005 24582094
- Tate M , Chong A , Robinson E , Green BD , Grieve DJ . Selective targeting of glucagon-like peptide-1 signalling as a novel therapeutic approach for cardiovascular disease in diabetes. Br J Pharmacol . 2015;172(3):721–736. doi:10.1111/bph.12943 25231355
- Verrotti A , Prezioso G , Scattoni R , Chiarelli F . Autonomic Neuropathy in Diabetes Mellitus. Front Endocrinol (Lausanne) . 2014;5:205. doi:10.3389/fendo.2014.00205 25520703
- Nourooz-Zadeh J , Rahimi A , Tajaddini-Sarmadi J , et al. Relationships between plasma measures of oxidative stress and metabolic control in NIDDM. Diabetologia . 1997;40(6):647–653. doi:10.1007/s001250050729 9222643
- Kawaguchi M , Techigawara M , Ishihata T , et al. A comparison of ultrastructural changes on endomyocardial biopsy specimens obtained from patients with diabetes mellitus with and without hypertension. Heart Vessels . 1997;12(6):267–274. doi:10.1007/BF02766802 9860193
- Cao Z , Cooper ME . Pathogenesis of diabetic nephropathy. J Diabetes Investig . 2011;2(4):243–247. doi:10.1111/j.2040-1124.2011.00131.x
- Nitzan O , Elias M , Chazan B , Saliba W . Urinary tract infections in patients with type 2 diabetes mellitus: review of prevalence, diagnosis, and management. Diabetes Metab Syndrome Obesity . 2015;8:129–136. doi:10.2147/DMSO.S51792
- Colberg SR , Sigal RJ , Fernhall B , et al. Exercise and Type 2 Diabetes: the American College of Sports Medicine and the American Diabetes Association: joint position statement. Diabetes Care . 2010;33(12):e147–e167. doi:10.2337/dc10-9990 21115758
- Keshel TE , Coker RH . Exercise Training and Insulin Resistance: a Current Review. J Obes Weight Loss Ther . 2015;5(5):S5–003.26523243
- Chiasson J-L . Early Insulin Use in Type 2 Diabetes: what are the cons? Diabetes Care . 2009;32(Suppl 2):S270–S274. doi:10.2337/dc09-S321 19875563
- Taylor R . Type 2 Diabetes. Diabetes Care . 2013;36(4):1047. doi:10.2337/dc12-1805 23520370
- Zhou X , Wu H , Long R , et al. Oral delivery of insulin with intelligent glucose-responsive switch for blood glucose regulation. J Nanobiotechnology . 2020;18(1):96. doi:10.1186/s12951-020-00652-z 32664978
- Xiao Y , Tang Z , Wang J , et al. Oral Insulin Delivery Platforms: strategies To Address the Biological Barriers. Angewandte Chemie Int Edition . 2020;59(45):19787–19795. doi:10.1002/anie.202008879
- Eldor ROY , Fleming GA , Neutel J , Homer KE , Kidron M , Rosenstock J . 105-LB: evening Oral Insulin (ORMD-0801) Glycemic Effects in Uncontrolled T2DM Patients. Diabetes . 2020;69(Supplement 1):105–LB. doi:10.2337/db20-105-LB
- Bahman F , Taurin S , Altayeb D , Taha S , Bakhiet M , Greish K . Oral Insulin Delivery Using Poly (Styrene Co-Maleic Acid) Micelles in a Diabetic Mouse Model. Pharmaceutics . 2020;12(11):1026. doi:10.3390/pharmaceutics12111026
- Gallwitz B . The evolving place of incretin-based therapies in type 2 diabetes. Pediatric Nephrol . 2010;25(7):1207–1217. doi:10.1007/s00467-009-1435-z
- Brunton S . GLP-1 receptor agonists vs. DPP-4 inhibitors for type 2 diabetes: is one approach more successful or preferable than the other? Int J Clin Pract . 2014;68(5):557–567. doi:10.1111/ijcp.12361 24499291
- De Graaf C , Donnelly D , Wootten D , et al. Glucagon-Like Peptide-1 and Its Class B G Protein–Coupled Receptors: a Long March to Therapeutic Successes. Pharmacol Rev . 2016;68(4):954–1013. doi:10.1124/pr.115.011395 27630114
- Holst JJ . The Physiology of Glucagon-like Peptide 1. Physiol Rev . 2007;87(4):1409–1439. doi:10.1152/physrev.00034.2006 17928588
- Prasad-Reddy L , Isaacs D . A clinical review of GLP-1 receptor agonists: efficacy and safety in diabetes and beyond. Drugs Context . 2015;4:212283. doi:10.7573/dic.212283 26213556
- Edavalath M , Stephens JW . Liraglutide in the treatment of type 2 diabetes mellitus: clinical utility and patient perspectives. Patient Prefer Adherence . 2010;4:61–68. doi:10.2147/ppa.s6358 20361006
- Eng J , Kleinman W , Singh L , Singh G , Raufman J . Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem . 1992;267:7402–7405. doi:10.1016/S0021-9258(18)42531-8 1313797
- Reed J , Bain S , Kanamarlapudi V . Recent advances in understanding the role of glucagon-like peptide 1 [version 1; peer review: 2 approved]. F1000Research . 2020;9:239. doi:10.12688/f1000research.20602.1
- Wang M , Yao P , Gao M , Jin J , Yu Y . Novel fatty chain-modified GLP-1R G-protein biased agonist exerts prolonged anti-diabetic effects through targeting receptor binding sites. RSC Adv . 2020;10(14):8044–8053. doi:10.1039/C9RA10593J
- Reed J , Kanamarlapudi V . GLP-1. In: Choi S , editor. Encyclopedia of Signaling Molecules . Cham: Springer International Publishing; 2018:2098–2106.
- Trujillo JM , Nuffer W , Smith BA . GLP-1 receptor agonists: an updated review of head-to-head clinical studies. Ther Adv Endocrinol Metab . 2021;12:2042018821997320. doi:10.1177/2042018821997320 33767808
- Seino Y , Fukushima M , Yabe D . GIP and GLP‐1, the two incretin hormones: similarities and differences. J Diabetes Investig . 2010;1(1–2):8–23. doi:10.1111/j.2040-1124.2010.00022.x
- Levin PA , Nguyen H , Wittbrodt ET , Kim SC . Glucagon-like peptide-1 receptor agonists: a systematic review of comparative effectiveness research. Diabetes Metab Syndrome Obesity . 2017;10:123–139. doi:10.2147/DMSO.S130834
- Potts JE , Gray LJ , Brady EM , Khunti K , Davies MJ , Bodicoat DH . The Effect of Glucagon-Like Peptide 1 Receptor Agonists on Weight Loss in Type 2 Diabetes: a Systematic Review and Mixed Treatment Comparison Meta-Analysis. PLoS One . 2015;10(6):e0126769. doi:10.1371/journal.pone.0126769 26121478
- Tamura K , Minami K , Kudo M , Iemoto K , Takahashi H , Seino S . Liraglutide improves pancreatic Beta cell mass and function in alloxan-induced diabetic mice. PLoS One . 2015;10(5):e0126003–e0126003. doi:10.1371/journal.pone.0126003 25938469
- Garber AJ . Incretin effects on β-cell function, replication, and mass: the human perspective. Diabetes Care . 2011;34 Suppl 2(Suppl2):S258–S263. doi:10.2337/dc11-s230 21525465
- Xu G , Kaneto H , Lopez-Avalos MD , Weir GC , Bonner-Weir S . GLP-1/exendin-4 facilitates β-cell neogenesis in rat and human pancreatic ducts. Diabetes Res Clin Pract . 2006;73(1):107–110. doi:10.1016/j.diabres.2005.11.007 16406191
- Drucker DJ , Sherman SI , Gorelick FS , Bergenstal RM , Sherwin RS , Buse JB . Incretin-Based Therapies for the Treatment of Type 2 Diabetes: evaluation of the Risks and Benefits. Diabetes Care . 2010;33(2):428–433. doi:10.2337/dc09-1499 20103558
- Nauck MA , Friedrich N . Do GLP-1–Based Therapies Increase Cancer Risk? Diabetes Care . 2013;36(Supplement 2):S245–S252. doi:10.2337/dcS13-2004 23882053
- Finan B , Ma T , Ottaway N , et al. Unimolecular Dual Incretins Maximize Metabolic Benefits in Rodents, Monkeys, and Humans. Sci Transl Med . 2013;5(209):209ra151–209ra151. doi:10.1126/scitranslmed.3007218
- Patel V , Joharapurkar A , Kshirsagar S , et al. Coagonist of GLP-1 and glucagon decreases liver inflammation and atherosclerosis in dyslipidemic condition. Chem Biol Interact . 2018;282:13–21. doi:10.1016/j.cbi.2018.01.004 29325849
- Wang A , Li T , An P , et al. Exendin-4 Upregulates Adiponectin Level in Adipocytes via Sirt1/Foxo-1 Signaling Pathway. PLoS One . 2017;12(1):e0169469. doi:10.1371/journal.pone.0169469 28122026
- Lihn AS , Pedersen SB , Richelsen B . Adiponectin: action, regulation and association to insulin sensitivity. Obesity Rev . 2005;6(1):13–21. doi:10.1111/j.1467-789X.2005.00159.x
- Tomkin GH . Treatment of type 2 diabetes, lifestyle, GLP1 agonists and DPP4 inhibitors. World J Diabetes . 2014;5(5):636–650. doi:10.4239/wjd.v5.i5.636 25317241
- Aroda VR . A review of GLP-1 receptor agonists: evolution and advancement, through the lens of randomised controlled trials. Diabetes Obes Metab . 2018;20(Suppl 1):22–33. doi:10.1111/dom.13162 29364586
- Marso SP , Bain SC , Consoli A , et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Eng J Med . 2016;375(19):1834–1844. doi:10.1056/NEJMoa1607141
- Marso SP , Daniels GH , Brown-Frandsen K , et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Eng J Med . 2016;375(4):311–322. doi:10.1056/NEJMoa1603827
- Forst T , Matthias MW , Andreas P . Cardiovascular Benefits of GLP-1-Based Therapies in Patients with Diabetes Mellitus Type 2: effects on Endothelial and Vascular Dysfunction beyond Glycemic Control. Exp Diabetes Res . 2012;2012. doi:10.1155/2012/635472
- Holman RR , Bethel MA , Mentz RJ , et al. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N Eng J Med . 2017;377(13):1228–1239. doi:10.1056/NEJMoa1612917
- Pfeffer MA , Claggett B , Diaz R , et al. Lixisenatide in Patients with Type 2 Diabetes and Acute Coronary Syndrome. N Eng J Med . 2015;373(23):2247–2257. doi:10.1056/NEJMoa1509225
- Strain W , Griffiths J . A systematic review and meta-analysis of the impact of GLP-1 receptor agonists and SGLT-2 inhibitors on cardiovascular outcomes in biologically healthy older adults. Br J Diabetes . 2021;21:30–35. doi:10.15277/bjd.2021.292
- Karagiannis T , Tsapas A , Athanasiadou E , et al. GLP-1 receptor agonists and SGLT2 inhibitors for older people with type 2 diabetes: a systematic review and meta-analysis. Diabetes Res Clin Pract . 2021;174:108737. doi:10.1016/j.diabres.2021.108737 33705820
- Marsico F , Paolillo S , Gargiulo P , et al. Effects of glucagon-like peptide-1 receptor agonists on major cardiovascular events in patients with Type 2 diabetes mellitus with or without established cardiovascular disease: a meta-analysis of randomized controlled trials. Eur Heart J . 2020;41(35):3346–3358. doi:10.1093/eurheartj/ehaa082 32077924
- Kristensen SL , Rørth R , Jhund PS , et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol . 2019;7(10):776–785. doi:10.1016/S2213-8587(19)30249-9 31422062
- Knudsen LB , Kiel D , Teng M , et al. Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc Natl Acad Sci U S A . 2007;104(3):937–942. doi:10.1073/pnas.0605701104 17213325
- Irwin N , Flatt PR , Patterson S , Green BD . Insulin-releasing and metabolic effects of small molecule GLP-1 receptor agonist 6,7-dichloro-2-methylsulfonyl-3-N-tert-butylaminoquinoxaline. Eur J Pharmacol . 2010;628(1–3):268–273. doi:10.1016/j.ejphar.2009.11.022 19917278
- Sloop KW , Willard FS , Brenner MB , et al. Novel Small Molecule Glucagon-Like Peptide-1 Receptor Agonist Stimulates Insulin Secretion in Rodents and From Human Islets. Diabetes . 2010;59(12):3099. doi:10.2337/db10-0689 20823098
- Nolte WM , Fortin J-P , Stevens BD , et al. A potentiator of orthosteric ligand activity at GLP-1R acts via covalent modification. Nat Chem Biol . 2014;10(8):629–631. doi:10.1038/nchembio.1581 24997604
- Song G , Yang D , Wang Y , et al. Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators. Nature . 2017;546:312. doi:10.1038/nature22378 28514449
- Finan B , Yang B , Ottaway N , et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med . 2015;21(1):27–36. doi:10.1038/nm.3761 25485909
- Cui J , Shang A , Wang W , Chen W . Rational design of a GLP-1/GIP/Gcg receptor triagonist to correct hyperglycemia, obesity and diabetic nephropathy in rodent animals. Life Sci . 2020;260:118339. doi:10.1016/j.lfs.2020.118339 32841660
- Elder KA , Wolfe BM . Bariatric Surgery: a Review of Procedures and Outcomes. Gastroenterology . 2007;132(6):2253–2271. doi:10.1053/j.gastro.2007.03.057 17498516
- Eldar S , Heneghan HM , Brethauer SA , Schauer PR . Bariatric surgery for treatment of obesity. Int J Obes . 2011;35(S3):S16–S21. doi:10.1038/ijo.2011.142
- Buchwald H , Williams SE . Bariatric Surgery Worldwide 2003. Obes Surg . 2004;14(9):1157–1164. doi:10.1381/0960892042387057 15527627
- Singh AK , Singh R , Kota SK . Bariatric surgery and diabetes remission: who would have thought it? Indian J Endocrinol Metab . 2015;19(5):563–576. doi:10.4103/2230-8210.163113 26425464
- Brethauer SA , Aminian A , Romero-Talamás H , et al. Can Diabetes Be Surgically Cured?: long-Term Metabolic Effects of Bariatric Surgery in Obese Patients with Type 2 Diabetes Mellitus. Ann Surg . 2013;258(4):628–637. doi:10.1097/SLA.0b013e3182a5034b 24018646
- Koliaki C , Liatis S , le Roux CW , Kokkinos A . The role of bariatric surgery to treat diabetes: current challenges and perspectives. BMC Endocr Disord . 2017;17(1):50. doi:10.1186/s12902-017-0202-6 28797248
- Shah A , Laferrère B . Diabetes after Bariatric Surgery. Can J Diabetes . 2017;41(4):401–406. doi:10.1016/j.jcjd.2016.12.009 28457649
- Khalaf KI , Taegtmeyer H . Clues from Bariatric Surgery: reversing Insulin Resistance to Heal the Heart. Curr Diab Rep . 2013;13(2):245–251. doi:10.1007/s11892-013-0364-1 23354680
- Provenzale D , Reinhold RB , Golner B , et al. Evidence for diminished B12 absorption after gastric bypass: oral supplementation does not prevent low plasma B12 levels in bypass patients. J Am Coll Nutr . 1992;11:29–35. doi:10.1080/07315724.1992.10718193 1541791
- Halverson J . Micronutrient deficiencies after gastric bypass for morbid obesity. Am Surg . 1986;52:594–598.3777703
- Avinoah EOA , Charuzi I . Nutritional status seven years after Roux-en-Y gastric bypass surgery. Surgery . 1992;111:137–142.1736382
- Saliba W , El-Haddad B . Secondary Hyperparathyroidism: pathophysiology and Treatment. J Am Board Family Med . 2009;22(5):574–581. doi:10.3122/jabfm.2009.05.090026
- Potts JT . Parathyroid hormone: past and present. J Endocrinol . 2005;187(3):311–325. doi:10.1677/joe.1.06057 16423810
- Toh SY , Zarshenas N , Jorgensen J . Prevalence of nutrient deficiencies in bariatric patients. Nutrition . 2009;25(11):1150–1156. doi:10.1016/j.nut.2009.03.012 19487104
- Reed J , Kanamarlapudi V , Bain S . Mechanism of cardiovascular disease benefit of glucagon-like peptide 1 agonists. Cardiovascular Endocrinol Metab . 2018;7(1):18–23.
- Alicic RZ , Rooney MT , Tuttle KR . Diabetic Kidney Disease. Clin J Am Soc Nephrol . 2017;12(12):2032. doi:10.2215/CJN.11491116 28522654
- Duh EJ , Sun JK , Stitt AW . Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight . 2017;2(14):e93751. doi:10.1172/jci.insight.93751
- Leon BM , Maddox TM . Diabetes and cardiovascular disease: epidemiology, biological mechanisms, treatment recommendations and future research. World J Diabetes . 2015;6(13):1246–1258. doi:10.4239/wjd.v6.i13.1246 26468341
- Tomita T . Apoptosis in pancreatic β-islet cells in Type 2 diabetes. Bosn J Basic Med Sci . 2016;16(3):162–179. doi:10.17305/bjbms.2016.919 27209071
- Cho J-H , Kim J-W , Shin J-A , Shin J , Yoon K-H . β-cell mass in people with type 2 diabetes. J Diabetes Investig . 2011;2(1):6–17. doi:10.1111/j.2040-1124.2010.00072.x
- le Roux CW , Astrup A , Fujioka K , et al. 3 years of liraglutide versus placebo for type 2 diabetes risk reduction and weight management in individuals with prediabetes: a randomised, double-blind trial. Lancet . 2017;389(10077):1399–1409. doi:10.1016/S0140-6736(17)30069-7 28237263
- Jazayeri A , Rappas M , Brown AJH , et al. Corrigendum: crystal structure of the GLP-1 receptor bound to a peptide agonist. Nature . 2017;548:122. doi:10.1038/nature23311
- Itariu BK , Stulnig TM . Autoimmune Aspects of Type 2 Diabetes Mellitus - A Mini-Review. Gerontology . 2014;60(3):189–196. doi:10.1159/000356747 24457898
- Pietropaolo M , Barinas-Mitchell E , Pietropaolo SL , Kuller LH , Trucco M . Evidence of islet cell autoimmunity in elderly patients with type 2 diabetes. Diabetes . 2000;49(1):32–38. doi:10.2337/diabetes.49.1.32 10615947
- Brooks-Worrell BM , Reichow JL , Goel A , Ismail H , Palmer JP . Identification of Autoantibody-Negative Autoimmune Type 2 Diabetic Patients. Diabetes Care . 2011;34(1):168–173. doi:10.2337/dc10-0579 20855551
- Haskell BDFK , Duffy TM , Sargent EE , Leiter EH . The diabetes-prone NZO/HlLt strain. I. Immunophenotypic comparison to the related NZB/BlNJ and NZW/LacJ strains. Lab Invest . 2002;82:833–842. doi:10.1097/01.LAB.0000018915.53257.00 12118085
- Jagannathan M , McDonnell M , Liang Y , et al. Toll-like receptors regulate B cell cytokine production in patients with diabetes. Diabetologia . 2010;53(7):1461–1471. doi:10.1007/s00125-010-1730-z 20383694
- Gandasi NR , Yin P , Omar-Hmeadi M , Ottosson laakso E , Vikman P , Glucose-Dependent Granule BS . Docking Limits Insulin Secretion and Is Decreased in Human Type 2 Diabetes. Cell Metab . 2018;27(2):470–478.e474. doi:10.1016/j.cmet.2017.12.017 29414688
- Zhang Y , Chen W , Feng B , Cao H . The Clinical Efficacy and Safety of Stem Cell Therapy for Diabetes Mellitus: a Systematic Review and Meta-Analysis. Aging Dis . 2020;11(1):141–153. doi:10.14336/AD.2019.0421 32010488
- El-Badawy A , El-Badri N . Clinical Efficacy of Stem Cell Therapy for Diabetes Mellitus: a Meta-Analysis. PLoS One . 2016;11(4):e0151938. doi:10.1371/journal.pone.0151938 27073927
- Bhansali A , Upreti V , Khandelwal N , et al. Efficacy of autologous bone marrow-derived stem cell transplantation in patients with type 2 diabetes mellitus. Stem Cells Dev . 2009;18(10):1407–1416. doi:10.1089/scd.2009.0164 19686048
- D’Addio F , Valderrama Vasquez A , Ben Nasr M , et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in new-onset type 1 diabetes: a multicenter analysis. Diabetes . 2014;63(9):3041–3046. doi:10.2337/db14-0295 24947362
- Rahim F , Arjmand B , Shirbandi K , Payab M , Larijani B . Stem cell therapy for patients with diabetes: a systematic review and meta-analysis of metabolomics-based risks and benefits. Stem Cell Investig . 2018;5:40. doi:10.21037/sci.2018.11.01
- Bhansali S , Dutta P , Kumar V , et al. Efficacy of Autologous Bone Marrow-Derived Mesenchymal Stem Cell and Mononuclear Cell Transplantation in Type 2 Diabetes Mellitus: a Randomized, Placebo-Controlled Comparative Study. Stem Cells Dev . 2017;26(7):471–481. doi:10.1089/scd.2016.0275 28006991
- World Health Organization. Body Mass Index (BMI). Available from: https://www.who.int/data/gho/data/themes/theme-details/GHO/body-mass-index-(bmi)?introPage=intro_3.html. Accessed August 03, 2021.
- Dimitriadis G , Mitrou P , Lambadiari V , Maratou E , Raptis SA . Insulin effects in muscle and adipose tissue. Diabetes Res Clin Pract . 2011;93:S52–S59. doi:10.1016/S0168-8227(11)70014-6 21864752
- Huang S , Czech MP . The GLUT4 Glucose Transporter. Cell Metab . 2007;5(4):237–252. doi:10.1016/j.cmet.2007.03.006 17403369
- Steiner DJ , Kim A , Miller K , Hara M . Pancreatic islet plasticity: interspecies comparison of islet architecture and composition. Islets . 2010;2(3):135–145. doi:10.4161/isl.2.3.11815 20657742
- Henquin J-C . The dual control of insulin secretion by glucose involves triggering and amplifying pathways in beta-cells. Diabetes Res Clin Pract . 2011;93:S27–S31. doi:10.1016/S0168-8227(11)70010-9 21864748
- Mailloux RJ , Harper M-E . Mitochondrial proticity and ROS signaling: lessons from the uncoupling proteins. Trends Endocrinol Metab . 2012;23(9):451–458. doi:10.1016/j.tem.2012.04.004 22591987
- Reed J , Kanamarlapudi V . GLP-1R. In: Choi S , editor. Encyclopedia of Signaling Molecules . New York: Springer New York; 2017:2106–2117.
- Lorenz M , Evers A , Wagner M . Recent progress and future options in the development of GLP-1 receptor agonists for the treatment of diabesity. Bioorg Med Chem Lett . 2013;23(14):4011–4018. doi:10.1016/j.bmcl.2013.05.022 23743288