
Abstract
Type 2 diabetes (T2D) is a complex metabolic disorder characterized by hyperglycemia in the context of insulin resistance, which precedes insulin deficiency as a result of β-cell failure. Accumulating evidence indicates that β-cell loss in T2D results as a response to the combination of oxidative stress and endoplasmic reticulum (ER) stress. Failure of the ER’s adaptive capacity and further activation of the unfolded protein response may trigger macroautophagy (hereafter referred as autophagy) as a process of self-protection and inflammation. Many studies have shown that inflammation plays a very important role in the pathogenesis of T2D. Inflammatory mechanisms and cytokine production activated by stress via the inflammasome may further alter the normal structure of β-cells by inducing pancreatic islet cell apoptosis. Thus, the combination of oxidative and ER stress, together with autophagy insufficiency and inflammation, may contribute to β-cell death or dysfunction in T2D. Therapeutic approaches aimed at ameliorating stress and inflammation may therefore prove to be promising targets for the development of new diabetes treatment methods. Here, we discuss different mechanisms involved in stress and inflammation, and the role of antioxidants, endogenous and chemical chaperones, and autophagic pathways, which may shift the tendency from ER stress and apoptosis toward cell survival. Strategies targeting cell survival can be essential for relieving ER stress and reestablishing homeostasis, which may diminish inflammation and prevent pancreatic β-cell death associated with T2D.
Introduction
Type 2 diabetes (T2D) is characterized by hyperglycemia in the context of insulin resistance and β-cell dysfunction.Citation1 Over time, islet β-cell function compensates for the insulin resistance existing in peripheral tissues, resulting in defects in insulin secretion that impair the regulation of blood glucose levels.Citation1–Citation3 Moreover, postmortem studies on β-cell loss in T2D have concluded that there is a marked reduction in β-cell mass,Citation4–Citation6 which is probably due to an increase in apoptosis rather than a decrease in β-cell replication. In addition to the increased β-cell workload in response to the abnormally high demand induced by insulin resistance, several factors likely play a role in this process. For example, high levels of glucose and saturated fatty acids in the blood, increased expression of islet amyloid polypeptide (IAPP), which is mainly responsible for amyloid deposits in the pancreas,Citation7,Citation8 as well as inflammatory cytokines released from visceral adipose tissue,Citation9 may be involved as inductors of oxidative stress and endoplasmic reticulum (ER) stress. These factors, together with the activation of the local inflammatory response signal the pathways leading to β-cell exhaustion and death.
A growing number of studies implicate ER stress in the loss and death of β-cells during the evolution of T2D.Citation10,Citation11 The ER is considered a vital organelle for protein synthesis and maturation, quality control, and secretion;Citation12,Citation13 however, these processes require a stable environment for balancing ER protein load and ER folding capacity. A variety of factors can disturb the proper functioning of the ER, leading to ER stress and inflammation as well as the induced synthesis of proinflammatory cytokines, including tumor necrosis factor-α and interleukin (IL)-6, via inflammasome activation.Citation11 In addition, the unfolded protein response (UPR) activates other pathways, such as oxidative stress and autophagyCitation10,Citation14,Citation15 which eventually lead to cell death or cell survival, depending on the balance of such factors in the cellular milieu.
In this review, we address the central mechanisms underlying ER stress, oxidative stress, autophagy, and inflammation, as well as the pathways that contribute to pancreatic β-cell death in the framework of T2D.
The link between stress and inflammation in pancreatic β-cells
ER stress and the UPR response
Providing a high-fidelity quality control system, the ER has developed an elaborate adaptive response known as the UPR, in which there is a perfect recognition of misfolded proteins and an efficient removal of these proteins from the ER lumen in order to protect and alleviate cells from ER stress. The UPR attempts to reestablish homeostasis and restore ER function by diminishing protein translation and activating a series of mechanisms that increase the biosynthetic capacity of the secretory pathway, such as ER chaperones. For this, a complex signaling network is initiated by three ER transmembrane kinases: protein kinase R-like endoplasmic reticulum kinase (PERK); inositol-requiring enzyme 1 (IRE1); and activating transcription factor (ATF)6 (). Chaperone 78 kDa glucose-regulated protein (GRP78), also referred to as BiP (immunoglobulin heavy chain binding protein), is a central regulator of ER stress due to its controlling of the activation of transmembrane ER stress sensors (PERK, IRE1, and ATF6) through a binding-release mechanism.Citation11
Figure 1 The link between β-cell stress and inflammation in type 2 diabetes.
Abbreviations: hIAPP, human islet amyloid polypeptide; ER, endoplasmic reticulum; ATF, activating transcription factor; PERK, protein kinase R-like endoplasmic reticulum kinase; IRE1, inositol-requiring enzyme 1; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; JNK, c-Jun N-terminal kinases; ROS, reactive oxygen species; IL, interleukin; MCP, monocyte chemotactic protein; TNF, tumor necrosis factor.
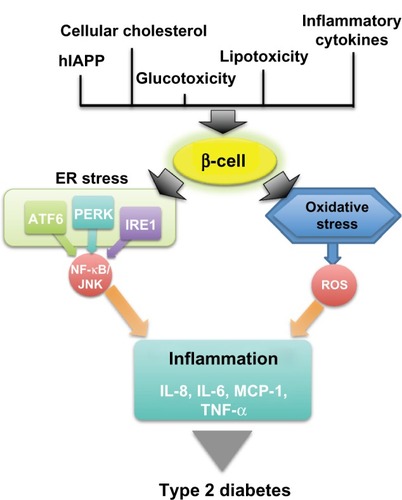
A reduction in protein translation and in ER workload are the first responses to counteract ER stress. This is mediated by PERK, which phosphorylates the α subunit of eukaryotic translation initiation factor 2, reducing global protein synthesis and inducing the translation of ATF4 messenger (m)RNA. This transcription factor activates the translation of ATF3 and CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP). The loss of PERK expression in humans and mice has been associated with a failure to properly regulate the UPR response, underlying dysfunction in the ER stress and UPR pathways, which can result in increased cell death and diabetes.Citation16 Although PERK expression in adult β-cells does not appear to be required for maintaining β-cell function, mutations in PERK result in the elevation of ER stress markers, leading to a form of permanent neonatal diabetes in humans,Citation17–Citation19 suggesting that PERK may play an important role in controlling ER stress during fetal development.
IRE1, the second pathway of the UPR, is highly expressed in the pancreas and is considered a central regulator of ER stress signaling, playing a crucial function in the regulation of protein biosynthesis.Citation20 A previous study has shown that IRE1 signaling knockdown in vitro decreases insulin biosynthesis at the translation and protein-folding level.Citation21 Once activated, IRE1 cleaves the mRNA encoding X-box binding protein 1 (XBP1), leading to an activated version of the transcription factor spliced XBP1. Once translocated to the nucleus, the spliced XBP1 protein initiates several transcriptional programs that upregulate ER expansion and biogenesis, increase protein entry into the ER for maturation, and degrade misfolded proteins.Citation22 Lee et al demonstrated that β-cell-specific XBP1-deficient mice elicited an impairment in β-cell proliferation, proinsulin processing, and insulin secretion, along with a hyperactivation of IRE1,Citation23 suggesting that XBP1 is critical in achieving optimal insulin secretion and glucose control and thus may be considered a key regulator of the UPR.
The third pathway of the UPR initiates with the activation of the basic leucine zipper domain protein ATF6. ATF6 activation stimulates its own translocation to the Golgi, where site-1 protease and site-2 proteases are cleaved and conducted to the nucleus to target transcription chaperones, elements of the ER-associated degradation pathway, and the upregulation of XBP1. Under chronic ER stress, ATF6 attempts to suppress the apoptotic UPR signaling cascade by upregulation of the PERK and IRE1 pathways. Recent reports have tried to elucidate the role of ATF6 in β-cell function. No association has been found between ATF6 polymorphisms in the general population and cohorts of type 2 diabetic patients;Citation24 however, ATF6 knockdown in insulinoma cells showed a decrease in ER chaperones and induced cell apoptosis without any changes in the PERK and IRE1 pathways.Citation25 Similarly, ER stress-induced activation of ATF6 has been shown to suppress insulin gene expression,Citation26 suggesting that ATF6 plays an important role in β-cell dysfunction.
ER stress and the inflammatory signal
The three branches of UPR response can trigger inflammatory signals through different branches that converge in signaling pathways involving c-Jun N-terminal kinases (JNKs) and the nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) (). The activation of these kinases highlights the overlap of metabolic and immune pathways, since these are the same kinases that are activated by innate immune responses.Citation27,Citation28 JNKs are considered to play an important role in ER stress in mouse models of diabetes. For instance, an increase in JNK activity promotes insulin resistance in peripheral tissues and in pancreatic β-cells without affecting cell viability.Citation29,Citation30 The importance of the JNK pathway in stress has also been observed in knockout mice, in which suppression of the JNK pathway protects β-cells against oxidative stress induction.Citation31
The pathway of the UPR involving IRE1 can, by different mechanisms, trigger an inflammatory signaling pathway through the activation of JNKs.Citation32 In addition, through multiple mechanisms, both the IRE1 and PERK pathways can also lead to the activation of the NF-κB pathway,Citation11 which also plays a critical role in the induction of multiple inflammatory mediators and has been implicated in insulin resistance.Citation11,Citation33 ATF6 has also been linked to inflammatory signaling. Genetic and pharmacological inhibition of ATF6 significantly suppresses NF-κB activation, which can transcriptionally regulate many other inflammatory genes.Citation34 Activation of either JNKs or NF-κB pathways in pancreatic β-cells has been reported to cause increased expression of proinflammatory molecules, such as IL-8, IL-6, monocyte chemotactic protein-1, and tumor necrosis factor-α,Citation35 that have a detrimental effect on cell survival and function.Citation36–Citation38 Local chemokine and cytokine release can also contribute to the inflammatory milieu, attracting host macrophages to the pancreatic β-cells, which further propagate local inflammation.Citation39,Citation40 In addition, the NF-κB pathway has been shown to activate the NLRP3 inflammasome, a multi-protein, cytosolic molecular platform that controls the activation of caspase 1, and the secretion of proinflammatory cytokines interleukin IL-1β and IL-18 in metabolic stress.Citation41,Citation42 Inflammation induced by inflammasome-dependent proinflammatory cytokines may produce insulin resistance or cause the death of pancreatic β-cells, leading to development of diabetes ().Citation42
Oxidative stress and the inflammatory signal
In T2D, when insulin demand is constantly elevated, reactive oxygen species (ROS) generation is increased by mitochondrial respiration, which saturates the neutralizing capacity of antioxidants, resulting in oxidative stress. β-cells are more susceptible to oxidative stress when compared to other cell types, probably due to their low antioxidant capacity. β-cells have low levels of antioxidant enzyme expressions, such as catalase and glutathione peroxidase, making β-cells more vulnerable to free radical damage when exposed to oxidative stress.Citation43 In addition, β-cells display highly efficient glucose uptake when exposed to high glucose concentrations, due to the expression of glucose transporter 2. Thus, it is likely that oxidative stress plays a major role in β-cell dysfunction in T2D. Exposure of rat islets to high concentrations of glucose resulted in an increased production of intracellular ROS.Citation44 Furthermore, when human islets were incubated with high glucose concentrations, the levels of intracellular peroxide were increased within the islets.Citation45 Similarly, elevated markers of oxidative stress have been found in the plasma and urine of type 2 diabetic patients, as well as decreased levels of antioxidant molecule glutathione in their blood cells.Citation46 In β-cells, elevated ROS levels lead to impaired insulin secretion and contribute to insulin resistance in T2D.Citation47,Citation48 Under normal conditions, ROS are finely regulated to avoid oxidative damage to cellular processes.Citation49 Prolonged ER stress can also accumulate ROS through a PERK-mediated pathway, promoting a state of oxidative stress.Citation50 To protect itself from the highly toxic radicals, the β-cell must metabolize ROS by using cellular antioxidants, including glutathione peroxidase, catalase, thioredoxin, and superoxide dismutase, among others.Citation51 In addition, glutathione, the primary intracellular antioxidant, has been reported to be low in diabetic patients.Citation52,Citation53 After treatment with sulfonylurea, a reduction in oxidative stress was observed, with patients showing a decrease in lipid peroxidation and an increase in glutathione circulating levels, with levels almost reaching those found in a nondiabetic control group.Citation53
Inflammation resulting from oxidative stress is a key component for many human diseases, such as metabolic syndrome or T2D.Citation27 ROS production is capable of acting as a signaling molecule, but also inflicts oxidative damage by oxidizing fatty acids, DNA, RNA, amino acids, and cofactors.Citation54 ROS have been shown to play an important role in various cellular processes, including differentiation, autophagy, metabolic adaptation, and inflammation.Citation55 High ROS levels activate several inflammatory signaling cascades, leading to the transcription of the NF-κB pathway, monocyte chemotactic protein-1, cellular adhesion molecules, nitric oxide, transforming growth factor-β, connective tissue growth factor, and ILs.Citation27,Citation56,Citation57 Consequently, expression of proinflammatory molecules might attract inflammatory cells such as macrophages to the site and further exacerbate the local inflammation. ROS production in adipocytes can also lead to an increased production of proinflammatory cytokines that can affect β-cells in a paracrine manner.Citation58
Stress, inflammation, and the activation of apoptotic signaling
Linkers of stress and apoptosis
Metabolically stressed human β-cells display markers of ER stress and activation of inflammation and apoptosis pathways.Citation10,Citation11,Citation59–Citation62 ER stress and oxidative stress are intricately related and represent possible mediators that link toxic stimuli with target molecules in the apoptotic cascade. As far as we know, the apoptotic pathways that may be activated by ER stress are also activated by oxidative stress and inflammatory signals (). At least three parallel pathways are involved in the stress-mediated apoptosis: activation of CHOP; activation of the IRE1–JNK pathway; and activation of caspase 12 ().Citation63,Citation64
CHOP signaling is activated in β-cells under conditions of metabolic stress, and the deletion of CHOP has been demonstrated to enhance β-cell function and mass in several mouse models of diabetes.Citation65 In this regard, islets from CHOP knockout mice have fewer apoptotic cells and show an increased expression of UPR genes. Furthermore, CHOP deletion delays the onset and severity of the diabetic phenotype.Citation65,Citation66 Additional mechanisms of apoptotic induction have been associated with particular branches of the ER stress pathway. ATF4 can promote apoptosis by suppression of B-cell lymphoma 2.Citation67 IRE1 promotes JNK signaling through a mitogen-activated protein kinase 1 pathway.Citation67,Citation68 IRE1 also promotes the activation of caspase-12. Procaspase-12 is localized to ER membranes and undergoes cleavage during ER stress in murine cells, promoting the downstream cleaving of caspase-3, the last effector caspase of the apoptotic cascade.Citation68,Citation69 Pharmacologically induced ER stress has given additional insight into mechanisms by which ER stress may promote apoptosis.Citation69 Such agents cause mitochondrial cytochrome-c release and loss of mitochondrial transmembrane potential, causing ER stress-induced apoptosis. Moreover, perturbed ER Ca2+ homeostasis may contribute to apoptosis following induction of ER stress,Citation69 since knockout mice lacking the ER Ca2+ channel Wsf1 are particularly susceptible to ER stress-induced apoptosis.Citation70 Overall, when ER stress-induced apoptosis causes the loss of a large number of β-cells, insulin secretory capacity is impaired, resulting in T2D ().
Triggers of stress, inflammation, and apoptosis
Multiple physiological and pathological conditions, including the accumulation of misfolded proteins, such as insulin or human IAPP (hIAPP), are responsible for the loss of ER homeostasis in β-cells ().Citation11,Citation71 In some cases, protein overexpression in cells of transgenic mice can trigger ER stress and apoptosis due to a high biosynthetic misfolded load.Citation60,Citation72,Citation73 For example, studies in the Akita mouse have shown that ER stress, secondary to the misfolding of mutated insulin, leads to β-cell death and glucose intolerance.Citation73 The loss of β-cell mass in diabetes is exacerbated by islet amyloid deposits that correlate with the severity of the disease in humans. β-cell apoptosis is also observed in human pancreatic sections and postmortem islet grafts in correlation with amyloid deposition levels.Citation71,Citation74,Citation75 Oligomers of human IAPP have been shown to increase inflammation in β-cells via the inflammasome.Citation76 Comparably, hIAPP can form proinflammatory oligomers and fibrils that contribute to islet inflammation by recruiting and activating macrophages in vivo.Citation39,Citation76 Nevertheless, the role of hIAPP and ER stress still needs to be elucidated. Some reports show that ER stress-mediated apoptosis is exacerbated in rodent cells expressing amyloidogenic isoforms of hIAPP in β-cells, leading to a reduction of β-cell mass in hIAPP transgenic mice and rats.Citation61,Citation71 In addition, Casas et al demonstrated that extracellular hIAPP aggregation is associated with ER stress responses in mouse β-cells, by an intracellular signaling that involves downstream inhibition of the ubiquitin–proteasome pathway, contributing to β-cell apoptosis.Citation77,Citation78 Nevertheless, in a rat pancreatic β-cell line overexpressing hIAPP, the detection of toxic intracellular oligomers, which lead to defective insulin and IAPP secretion levels in response to glucose, did not change the expression of genes involved in ER stress.Citation79 These results agree with other findings with hIAPP transgenic mice, in which the authors demonstrated that amyloid formation was not associated with significant increases in the expression of ER stress markers.Citation80 The discrepancy in these results may be explained by differences in the ratio of IAPP and insulin produced by the different models used, ranging from low to significantly high levels of IAPP.
The synergistic toxic effect of hyperglycemia and hyperlipidemia is now well recognized as a contributor toward β-cell death.Citation81 Exposure of islets to high glucose concentrations induces a significant increase in apoptosis.Citation81 Similarly, high glucose concentrations increased IL-1β, followed by NF-κB activation from nondiabetic islet donors.Citation82 Increased concentrations of saturated fatty acids are also toxic to islets. Saturated fatty acids impair insulin gene expression and glucose-induced insulin secretion.Citation83–Citation85 Palmitate induces ER stress via NF-κB activation, contributing to islet inflammation.Citation86 In addition, fatty acids can have a toxic effect on β-cells, since treatment of human or mouse islets with palmitate induces apoptosis.Citation87 Importantly, it has been demonstrated that lipotoxicity only occurs in the presence of concomitantly elevated glucose levels.Citation84 Other mediators include islet cholesterol accumulation as an important cause of lipotoxic stress in β-cells. Cellular cholesterol homeostasis is important for normal β-cell function. Thus, disruption of cholesterol transport by decreased function of the Adenosine triphosphate (ATP)-binding cassette (ABC) transporter Adenosine triphosphate-binding cassette transporter 1 (ABCA1), a cholesterol efflux regulatory protein responsible for cholesterol transport in β-cells, results in impaired insulin secretion.Citation88–Citation90 In addition, the combined deficiency of ABCA1 and ABCG1 also results in significant islet inflammation, as indicated by the increased expression of IL-1β and macrophage infiltration.Citation91
The role of stress and autophagy in apoptosis induction
The ER stress pathway is directly involved in the induction of autophagy. Autophagy is a degradation pathway responsible for the large turnover of intracellular proteins and organelles via lysosomal degradation.Citation92 Autophagy is a highly regulated process that can either be involved in the turnover of long-lived proteins and whole organelles in a generalized fashion, or specifically target distinct organelles.Citation92 Thus, autophagy, together with apoptosis, is a process through which damaged or aged cells or organelles can be eliminated.Citation92 Autophagy may be either a mechanism to avoid apoptosis (by eliminating old or damaged organelles) or a mechanism to induce apoptosis by autophagic-induced cell death.Citation93 Autophagy and apoptosis may be triggered by common upstream signals, resulting in combined autophagy and apoptosis. For example, autophagy can destroy large proportions of the cytosol or organelles, causing irreversible cellular collapse.Citation93 Autophagy can also be a response to stress stimuli by triggering apoptosis or necrotic cell death. Likewise, several signal transduction pathways related to cellular stress (such as oxidative or ER stress pathways) can elicit both autophagy and apoptosis.Citation93 Autophagy can also be induced by proteasome inhibition under conditions of ER stress, demonstrating that autophagy and apoptosis share many common inducers.Citation93
Several reports have studied the role of autophagy in pancreatic β-cells. Diabetic db/db or nondiabetic C57Bl/6 mice fed with a high-fat diet have shown upregulated autophagosome formation in β-cells.Citation94,Citation95 Furthermore, genetic ablation of Autophagy-related protein 7 (ATG7) (an essential gene for autophagosome formation) in β-cells was shown to have resulted in the degeneration of islets and impaired glucose tolerance with reduced insulin secretion.Citation94,Citation96 These findings were associated with an increased level of apoptosis and a decreased proliferation, which contributed to the loss of β-cell mass. It has been described that increased expression of hIAPP in transgenic mice and rats leads to an impaired autophagy pathway, due to the disruption of lysosome-dependent degradation.Citation97 In addition, inhibition of lysosomal degradation increases the vulnerability of β-cells to hIAPP-induced toxicity and, conversely, stimulation of autophagy protects β-cells from hIAPP-induced apoptosis.Citation97 In humans, the autophagy pathway also declines with age and is impaired in β-cells in type 2 diabetic patients.Citation98,Citation99 These studies suggest that autophagy is necessary to maintain the structure, mass, and function of pancreatic β-cells, and its impairment may participate in the mechanisms that cause β-cell failure and T2D.
Moving the balance toward homeostasis and promoting β-cell survival
Pancreatic β-cells need to increase protein synthesis during acute or chronic stimulation. This causes a burden on the ER that may activate autophagy and the UPR response, which may lead to pancreatic cell death. Together, cell survival and cell death factors represent key opposing forces underlying stress response (). As previously seen, many factors can mediate the respective outcome of this antagonistic process. If a prolonged imbalance persists, the response system initiates proapoptotic mechanisms that eventually will lead to pancreatic cell death and dysfunction associated with T2D (). Thus, therapeutic interventions that target molecules of the UPR component or reduce ER stress, such as increasing cellular antioxidants, chaperone capacity, or autophagy levels, may bring the balance toward homeostasis and provide promising strategies for treating ER stress-related human diseases such as T2D ().
Figure 2 Moving the balance toward prosurvival strategies.
Abbreviation: C/EBP, CCAAT/enhancer-binding protein.
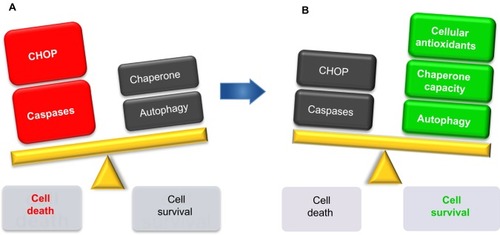
The inhibition of intracellular free radical formation may represent one therapeutic strategy for preventing oxidative stress. Antioxidants act at different levels, inhibiting the formation of ROS, scavenging free radicals or increasing their own defense enzyme capabilities. Therapeutic measures designed to increase intrinsic antioxidant activity within the islet may also protect it against the oxidative stress associated with glucose toxicity (). The transduction of islets with adenovirus encoding for the antioxidant enzyme, glutathione peroxidase, has been shown to protect islets against the intra-islet peroxide levels produced by high glucose concentrations.Citation100 Furthermore, exogenous antioxidants can compensate for the lower plasma antioxidant levels often observed in T2D. Antioxidants such as N-acetylcysteine, vitamin C, and α-lipoic acid are effective in reducing diabetic complications,Citation101,Citation102 indicating that antioxidants may prove an essential tool in the investigation of oxidative stress-related diabetic pathologies.
An increasing body of evidence suggests that chaperones exert important protective effects in the decrease of ER stress, protein aggregation, and the pathophysiology of amyloid deposition. Overexpression of certain particular ER chaperones in cell systems can protect cells against cell death caused by disturbances of ER homeostasis.Citation103–Citation106 Of interest, transgenic mice overexpressing the molecular chaperone GRP78/BiP specifically in β-cells are protected against the injury of obesity-induced T2D, maintaining β-cell function and improving glucose homeostasis.Citation104 Overexpression of BiP attenuates fatty acid-induced ER stress and apoptosis in hepatocytes.Citation106 Furthermore, BiP is one of the chaperones responsible for trafficking hIAPP through the ER and Golgi in human β-cells.Citation107 Efforts to understand the impact of chaperones may provide insights into the formation of misfolded hIAPP, which, consequently, might be a speculative approach for preventing amyloid formation, which may lead to inflammation and β-cell apoptosis in T2D (). Few investigations have been performed on inhibiting the aggregation of IAPP. The small interfering RNA-mediated suppression of human amyloid polypeptide expression inhibits islet amyloid formation and enhances the survival of human islets in culture.Citation108 Similarly, peptide-based amyloid inhibitors have been seen to enhance the survival of cultured human islets.Citation109 Thus, inhibitors of IAPP synthesis or aggregation may have therapeutic value in diminishing amyloid formation in T2D.
Recent reports suggest that pharmacological agents can directly activate or deactivate UPR components and can potentially be useful in treating T2D. A promising approach is the use of pharmacological agents, such as orally active chemical chaperones, which can stabilize protein conformation, improve ER folding capacity, and facilitate the trafficking of mutant proteins.Citation110–Citation113 Ozcan et al have shown that chemical chaperones, such as 4-phenyl butyric acid and taurine-conjugated ursodeoxycholic acid, reduce ER stress and restore glucose homeostasis in a mouse model of T2D.Citation114 In this model, the oral chemical chaperone treatment of obese diabetic mice resulted in the normalization of hyperglycemia and restoration of peripheral insulin sensitivity, thus acting as a potential antidiabetic agent.Citation114 Chemical chaperones have also been tested in obese human subjects;Citation115,Citation116 4-phenyl butyric acid, for instance, may provide health benefits by ameliorating insulin resistance and pancreatic β-cell dysfunction in obese subjects.Citation115 The ability of endogenous and chemical chaperones to alleviate ER stress in transgenic and obese mice models strongly supports the ER stress-based mechanistic model of T2D and demonstrates the feasibility of targeting ER function for therapeutic goals.
Several studies have focused on the prosurvival role of autophagy. As previously discussed, autophagy is activated in response to ER stress and helps cellular adaptation to stress, via clearance of misfolded proteins.Citation117 Thus, stimulation of autophagy may improve ER stress in diabetes. Treatment with rapamycin, an autophagy inducer, used in diabetic Akita mice, improved diabetes, increased pancreatic insulin content, and prevented β-cell apoptosis.Citation118 In contrast, the same study showed that inhibition of autophagy exacerbated stress and abolished the anti-ER stress effects of rapamycin.Citation118 In a similar manner, increasing autophagy by overexpression of scaffold protein p62, which delivers polyubiquitinated proteins to autophagy, confers a protective role against hIAPP-induced apoptosis by sequestrating protein targets for degradation.Citation97 Such evidence highlights an important role for autophagy in protection against toxic oligomer-induced apoptosis in β-cells. Thus, strategies that target ER stress in β-cells will promote β-cell survival and function in T2D.
Summary
A great variety of stimuli, such as IAPP, cytokines, cellular cholesterol, or high glucose and lipids levels in the blood, can disturb ER homeostasis, leading to oxidative and ER stress, inflammation, and pancreatic β-cell death. Elucidating the cellular mechanisms of stress, inflammation, and cell death has contributed toward our understanding of these processes, which may lead to new therapeutic agents for treating T2D. Strategies targeting the balance toward prosurvival have proven to be essential for the shift from stress to homeostasis, which may prevent pancreatic β-cell death associated with T2D.
Acknowledgments
JM is a recipient of an IDIBAPS Postdoctoral Fellowship-BIOTRACK, supported by the European Community’s Seventh Framework Programme (ECFP7/2007–2013) under grant agreement number 229673. LC was a recipient of Fundação da Ciência e Tecnologia (FCT-PhD) fellowship SFRH/BD/65645/2009 financed by POPH-QREN. This work was supported by grants from FIS (PI08/0088 and PI1100679) and Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM). The authors wish to acknowledge Kimberly Katte for technical manuscript correction.
Disclosure
The authors report no conflicts of interest in this work.
References
- Unger RH Lipid overload and overflow: metabolic trauma and the metabolic syndrome Trends Endocrinol Metab 2003 14 9 398 403 14580758
- Kasuga M Insulin resistance and pancreatic beta cell failure J Clin Invest 2006 116 7 1756 1760 16823472
- Prentki M Nolan CJ Islet beta cell failure in type 2 diabetes J Clin Invest 2006 116 7 1802 1812 16823478
- Jin W Patti ME Genetic determinants and molecular pathways in the pathogenesis of type 2 diabetes Clin Sci (Lond) 2009 116 2 99 111 19076063
- Butler AE Janson J Bonner-Weir S Ritzel R Rizza RA Butler PC Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes Diabetes 2003 52 1 102 110 12502499
- Donath MY Halban PA Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications Diabetologia 2004 47 3 581 589 14767595
- Novials A Sarri Y Casamitjana R Rivera F Gomis R Regulation of islet amyloid polypeptide in human pancreatic islets Diabetes 1993 42 10 1514 1519 8375592
- Gasa R Gomis R Casamitjana R Novials A High glucose concentration favors the selective secretion of islet amyloid polypeptide through a constitutive secretory pathway in human pancreatic islets Pancreas 2001 22 3 307 310 11291934
- Hotamisligil GS Inflammation and metabolic disorders Nature 2006 444 7121 860 867 17167474
- Oslowski CM Hara T O’Sullivan-Murphy B Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome Cell Metab 2012 16 2 265 273 22883234
- Hotamisligil GS Endoplasmic reticulum stress and the inflammatory basis of metabolic disease Cell 2010 140 6 900 917 20303879
- Contreras JL Smyth CA Bilbao G Coupling endoplasmic reticulum stress to cell death program in isolated human pancreatic islets: effects of gene transfer of Bcl-2 Transpl Int 2003 16 7 537 542 12819863
- Harding HP Ron D Endoplasmic reticulum stress and the development of diabetes: a review Diabetes 2002 51 Suppl 3 S455 S461 12475790
- Gotoh T Endo M Oike Y Endoplasmic reticulum stress-related inflammation and cardiovascular diseases Int J Inflam 2011 2011 259462 21755026
- Zhang K Kaufman RJ From endoplasmic-reticulum stress to the inflammatory response Nature 2008 454 7203 455 462 18650916
- Harding HP Zeng H Zhang Y Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival Mol Cell 2001 7 6 1153 1163 11430819
- Zhang W Feng D Li Y Iida K McGrath B Cavener DR PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis Cell Metab 2006 4 6 491 497 17141632
- Boyce M Bryant KF Jousse C A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress Science 2005 307 5711 935 939 15705855
- Wang R McGrath BC Kopp RF Insulin secretion and Ca2+ dynamics in beta-cells are regulated by PERK (EIF2AK3) in concert with calcineurin J Biol Chem 2013 288 47 33824 33836 24114838
- Cardozo AK Ortis F Storling J Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells Diabetes 2005 54 2 452 461 15677503
- Haze K Okada T Yoshida H Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response Biochem J 2001 355 Pt 1 19 28 11256944
- Lee AH Chu GC Iwakoshi NN Glimcher LH XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands EMBO J 2005 24 24 4368 4380 16362047
- Lee AH Heidtman K Hotamisligil GS Glimcher LH Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion Proc Natl Acad Sci U S A 2011 108 21 8885 8890 21555585
- Chu WS Das SK Wang H Activating transcription factor 6 (ATF6) sequence polymorphisms in type 2 diabetes and pre-diabetic traits Diabetes 2007 56 3 856 862 17327457
- Teodoro T Odisho T Sidorova E Volchuk A Pancreatic β-cells depend on basal expression of active ATF6α-p50 for cell survival even under nonstress conditions Am J Physiol Cell Physiol 2012 302 7 C992 C1003 22189555
- Seo HY Kim YD Lee KM Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases insulin gene expression via up-regulation of orphan nuclear receptor small heterodimer partner Endocrinology 2008 149 8 3832 3841 18450959
- Wellen KE Hotamisligil GS Inflammation, stress, and diabetes J Clin Invest 2005 115 5 1111 1119 15864338
- Medzhitov R Toll-like receptors and innate immunity Nat Rev Immunol 2001 1 2 135 145 11905821
- Lanuza-Masdeu J Arévalo MI Vila C Barberà A Gomis R Caelles C In vivo JNK activation in pancreatic β-cells leads to glucose intolerance caused by insulin resistance in pancreas Diabetes 2013 62 7 2308 2317 23349497
- Hirosumi J Tuncman G Chang L A central role for JNK in obesity and insulin resistance Nature 2002 420 6913 333 336 12447443
- Kaneto H Matsuoka TA Nakatani Y Kawamori D Matsuhisa M Yamasaki Y Oxidative stress and the JNK pathway in diabetes Curr Diabetes Rev 2005 1 1 65 72 18220583
- Urano F Wang X Bertolotti A Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1 Science 2000 287 5453 664 666 10650002
- Deng J Lu PD Zhang Y Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2 Mol Cell Biol 2004 24 23 10161 10168 15542827
- Yamazaki H Hiramatsu N Hayakawa K Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response J Immunol 2009 183 2 1480 1487 19561103
- Li Y Schwabe RF DeVries-Seimon T Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis J Biol Chem 2005 280 23 21763 21772 15826936
- Marselli L Dotta F Piro S Th2 cytokines have a partial, direct protective effect on the function and survival of isolated human islets exposed to combined proinflammatory and Th1 cytokines J Clin Endocrinol Metab 2001 86 10 4974 4978 11600573
- Wu JJ Chen X Cao XC Baker MS Kaufman DB Cytokine-induced metabolic dysfunction of MIN6 beta cells is nitric oxide independent J Surg Res 2001 101 2 190 195 11735275
- Zaitseva II Hultcrantz M Sharoyko V Flodstrom-Tullberg M Zaitsev SV Berggren PO Suppressor of cytokine signaling-1 inhibits caspase activation and protects from cytokine-induced beta cell death Cell Mol Life Sci 2009 66 23 3787 3795 19763396
- Westwell-Roper C Dai DL Soukhatcheva G IL-1 blockade attenuates islet amyloid polypeptide-induced proinflammatory cytokine release and pancreatic islet graft dysfunction J Immunol 2011 187 5 2755 2765 21813778
- Ehses JA Perren A Eppler E Increased number of islet-associated macrophages in type 2 diabetes Diabetes 2007 56 9 2356 2370 17579207
- Zhou R Yazdi AS Menu P Tschopp J A role for mitochondria in NLRP3 inflammasome activation Nature 2011 469 7329 221 225 21124315
- Dixit VD Nlrp3 inflammasome activation in type 2 diabetes: is it clinically relevant? Diabetes 2013 62 1 22 24 23258906
- Tiedge M Lortz S Drinkgern J Lenzen S Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells Diabetes 1997 46 11 1733 1742 9356019
- Ihara Y Toyokuni S Uchida K Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes Diabetes 1999 48 4 927 932 10102716
- Tanaka Y Tran PO Harmon J Robertson RP A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity Proc Natl Acad Sci U S A 2002 99 19 12363 12368 12218186
- Robertson RP Harmon JS Pancreatic islet beta-cell and oxidative stress: the importance of glutathione peroxidase FEBS Lett 2007 581 19 3743 3748 17433304
- Sakai K Matsumoto K Nishikawa T Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells Biochem Biophys Res Commun 2003 300 1 216 222 12480546
- Houstis N Rosen ED Lander ES Reactive oxygen species have a causal role in multiple forms of insulin resistance Nature 2006 440 7086 944 948 16612386
- Evans JL Goldfine ID Maddux BA Grodsky GM Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes Endocr Rev 2002 23 5 599 622 12372842
- Cullinan SB Diehl JA Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway Int J Biochem Cell Biol 2006 38 3 317 332 16290097
- Robertson RP Harmon J Tran PO Tanaka Y Takahashi H Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection Diabetes 2003 52 3 581 587 12606496
- Murakami K Kondo T Ohtsuka Y Fujiwara Y Shimada M Kawakami Y Impairment of glutathione metabolism in erythrocytes from patients with diabetes mellitus Metabolism 1989 38 8 753 758 2569661
- Sharma A Kharb S Chugh SN Kakkar R Singh GP Evaluation of oxidative stress before and after control of glycemia and after vitamin E supplementation in diabetic patients Metabolism 2000 49 2 160 162 10690938
- Freeman BA Crapo JD Biology of disease: free radicals and tissue injury Lab Invest 1982 47 5 412 426 6290784
- Sena LA Chandel NS Physiological roles of mitochondrial reactive oxygen species Mol Cell 2012 48 2 158 167 23102266
- Hung JH Su IJ Lei HY Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase J Biol Chem 2004 279 45 46384 46392 15319438
- Chambers KT Unverferth JA Weber SM Wek RC Urano F Corbett JA The role of nitric oxide and the unfolded protein response in cytokine-induced beta-cell death Diabetes 2008 57 1 124 132 17928398
- Lin Y Berg AH Iyengar P The hyperglycemia-induced inflammatory response in adipocytes: the role of reactive oxygen species J Biol Chem 2005 280 6 4617 4626 15536073
- Eizirik DL Cardozo AK Cnop M The role for endoplasmic reticulum stress in diabetes mellitus Endocr Rev 2008 29 1 42 61 18048764
- Huang CJ Lin CY Haataja L High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes Diabetes 2007 56 8 2016 2027 17475933
- Huang CJ Haataja L Gurlo T Induction of endoplasmic reticulum stress-induced beta-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide Am J Physiol Endocrinol Metab 2007 293 6 E1656 E1662 17911343
- Marchetti P Bugliani M Lupi R The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients Diabetologia 2007 50 12 2486 2494 17906960
- Oyadomari S Mori M Roles of CHOP/GADD153 in endoplasmic reticulum stress Cell Death Differ 2004 11 4 381 389 14685163
- Morishima N Nakanishi K Takenouchi H Shibata T Yasuhiko Y An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12 J Biol Chem 2002 277 37 34287 34294 12097332
- Song B Scheuner D Ron D Pennathur S Kaufman RJ Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes J Clin Invest 2008 118 10 3378 3389 18776938
- Oyadomari S Koizumi A Takeda K Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes J Clin Invest 2002 109 4 525 532 11854325
- Herbert TP PERK in the life and death of the pancreatic beta-cell Biochem Soc Trans 2007 35 Pt 5 1205 1207 17956313
- Araki E Oyadomari S Mori M Endoplasmic reticulum stress and diabetes mellitus Intern Med 2003 42 1 7 14 12583611
- Breckenridge DG Germain M Mathai JP Nguyen M Shore GC Regulation of apoptosis by endoplasmic reticulum pathways Oncogene 2003 22 53 8608 8618 14634622
- Ueda K Kawano J Takeda K Endoplasmic reticulum stress induces Wfs1 gene expression in pancreatic beta-cells via transcriptional activation Eur J Endocrinol 2005 153 1 167 176 15994758
- Montane J Klimek-Abercrombie A Potter KJ Westwell-Roper C Bruce Verchere C Metabolic stress, IAPP and islet amyloid Diabetes Obes Metab 2012 14 Suppl 3 68 77 22928566
- Shameli A Yamanouchi J Thiessen S Santamaria P Endoplasmic reticulum stress caused by overexpression of islet-specific glucose-6-phosphatase catalytic subunit-related protein in pancreatic beta-cells Rev Diabet Stud 2007 4 1 25 32 17565413
- Pearson T Shultz LD Lief J A new immunodeficient hyperglycaemic mouse model based on the Ins2Akita mutation for analyses of human islet and beta stem and progenitor cell function Diabetologia 2008 51 8 1449 1456 18563383
- Potter KJ Abedini A Marek P Islet amyloid deposition limits the viability of human islet grafts but not porcine islet grafts Proc Natl Acad Sci U S A 2010 107 9 4305 4310 20160085
- Andersson A Bohman S Borg LA Amyloid deposition in transplanted human pancreatic islets: a conceivable cause of their long-term failure Exp Diabetes Res 2008 2008 562985 19277203
- Masters SL Dunne A Subramanian SL Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes Nat Immunol 2010 11 10 897 904 20835230
- Casas S Gomis R Gribble FM Altirriba J Knuutila S Novials A Impairment of the ubiquitin-proteasome pathway is a downstream endoplasmic reticulum stress response induced by extracellular human islet amyloid polypeptide and contributes to pancreatic beta-cell apoptosis Diabetes 2007 56 9 2284 2294 17563070
- Casas S Novials A Reimann F Gomis R Gribble FM Calcium elevation in mouse pancreatic beta cells evoked by extracellular human islet amyloid polypeptide involves activation of the mechanosensitive ion channel TRPV4 Diabetologia 2008 51 12 2252 2262 18751967
- Soty M Visa M Soriano S Carmona Mdel C Nadal Á Novials A Involvement of ATP-sensitive potassium (K(ATP)) channels in the loss of beta-cell function induced by human islet amyloid polypeptide J Biol Chem 2011 286 47 40857 40866 21984830
- Hull RL Zraika S Udayasankar J Aston-Mourney K Subramanian SL Kahn SE Amyloid formation in human IAPP transgenic mouse islets and pancreas, and human pancreas, is not associated with endoplasmic reticulum stress Diabetologia 2009 52 6 1102 1111 19352619
- Poitout V Robertson RP Minireview: secondary beta-cell failure in type 2 diabetes – a convergence of glucotoxicity and lipotoxicity Endocrinology 2002 143 2 339 342 11796484
- Maedler K Sergeev P Ris F Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets J Clin Invest 2002 110 6 851 860 12235117
- McGarry JD Dobbins RL Fatty acids, lipotoxicity and insulin secretion Diabetologia 1999 42 2 128 138 10064091
- Jacqueminet S Briaud I Rouault C Reach G Poitout V Inhibition of insulin gene expression by long-term exposure of pancreatic beta cells to palmitate is dependent on the presence of a stimulatory glucose concentration Metabolism 2000 49 4 532 536 10778881
- Milburn JLJr Hirose H Lee YH Pancreatic beta-cells in obesity. Evidence for induction of functional, morphologic, and metabolic abnormalities by increased long chain fatty acids J Biol Chem 1995 270 3 1295 1299 7836394
- Igoillo-Esteve M Marselli L Cunha DA Palmitate induces a proinflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes Diabetologia 2010 53 7 1395 1405 20369226
- Wali JA Masters SL Thomas HE Linking metabolic abnormalities to apoptotic pathways in beta cells in type 2 diabetes Cells 2013 2 266 283
- Brunham LR Kruit JK Pape TD Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment Nat Med 2007 13 3 340 347 17322896
- Brunham LR Kruit JK Hayden MR Verchere CB Cholesterol in beta-cell dysfunction: the emerging connection between HDL cholesterol and type 2 diabetes Curr Diab Rep 2010 10 1 55 60 20425068
- Kruit JK Brunham LR Verchere CB Hayden MR HDL and LDL cholesterol significantly influence beta-cell function in type 2 diabetes mellitus Curr Opin Lipidol 2010 21 3 178 185 20463468
- Kruit JK Wijesekara N Westwell-Roper C Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired beta-cell function Diabetes 2012 61 3 659 664 22315310
- Maiuri MC Zalckvar E Kimchi A Kroemer G Self-eating and self-killing: crosstalk between autophagy and apoptosis Nat Rev Mol Cell Biol 2007 8 9 741 752 17717517
- Kroemer G Marino G Levine B Autophagy and the integrated stress response Mol Cell 2010 40 2 280 293 20965422
- Ebato C Uchida T Arakawa M Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet Cell Metab 2008 8 4 325 332 18840363
- Bartolome A Guillen C Benito M Autophagy plays a protective role in endoplasmic reticulum stress-mediated pancreatic beta cell death Autophagy 2012 8 12 1757 1768 22951927
- Quan W Hur KY Lim Y Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice Diabetologia 2012 55 2 392 403 22075916
- Rivera JF Gurlo T Daval M Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic beta-cells: protective role of p62-positive cytoplasmic inclusions Cell Death Differ 2011 18 3 415 426 20814419
- Masini M Bugliani M Lupi R Autophagy in human type 2 diabetes pancreatic beta cells Diabetologia 2009 52 6 1083 1086 19367387
- Costes S Langen R Gurlo T Matveyenko AV Butler PC β-cell failure in type 2 diabetes: a case of asking too much of too few? Diabetes 2013 62 2 327 335 23349537
- Robertson RP Tanaka Y Takahashi H Tran PO Harmon JS Prevention of oxidative stress by adenoviral overexpression of glutathione-related enzymes in pancreatic islets Ann N Y Acad Sci 2005 1043 513 520 16037273
- Feskens EJ Virtanen SM Räsänen L Dietary factors determining diabetes and impaired glucose tolerance. A 20-year follow-up of the Finnish and Dutch cohorts of the Seven Countries Study Diabetes Care 1995 18 8 1104 1112 7587845
- Karunakaran U Park KG A systematic review of oxidative stress and safety of antioxidants in diabetes: focus on islets and their defense Diabetes Metab J 2013 37 2 106 112 23641350
- Sun FC Wei S Li CW Chang YS Chao CC Lai YK Localization of GRP78 to mitochondria under the unfolded protein response Biochem J 2006 396 1 31 39 16433633
- Teodoro-Morrison T Schuiki I Zhang L Belsham DD Volchuk A GRP78 overproduction in pancreatic beta cells protects against high-fat-diet-induced diabetes in mice Diabetologia 2013 56 5 1057 1067 23475366
- Chien V Aitken JF Zhang S The chaperone proteins HSP70, HSP40/DnaJ and GRP78/BiP suppress misfolding and formation of β-sheet-containing aggregates by human amylin: a potential role for defective chaperone biology in type 2 diabetes Biochem J 2010 432 1 113 121 20735358
- Gu X Li K Laybutt DR Bip overexpression, but not CHOP inhibition, attenuates fatty-acid-induced endoplasmic reticulum stress and apoptosis in HepG2 liver cells Life Sci 2010 87 23–26 724 732 20970436
- Wu X Huang A Haataja L Butler PC Chaperone proteins Hsp70 and BiP fold and traffic islet amyloid polypeptide in humans, candidates for increased β-cell apoptosis in type 2 diabetes Poster presented at: 71st Scientific Sessions of American Diabetes Association June 24, 2011 San Diego, CA
- Marzban L Tomas A Becker TC Small interfering RNA-mediated suppression of proislet amyloid polypeptide expression inhibits islet amyloid formation and enhances survival of human islets in culture Diabetes 2008 57 11 3045 3055 18694973
- Potter KJ Scrocchi LA Warnock GL Amyloid inhibitors enhance survival of cultured human islets Biochim Biophys Acta 2009 1790 6 566 574 19264107
- Welch WJ Brown CR Influence of molecular and chemical chaperones on protein folding Cell Stress Chaperones 1996 1 2 109 115 9222596
- Ozcan L Tabas I Role of endoplasmic reticulum stress in metabolic disease and other disorders Annu Rev Med 2012 63 317 328 22248326
- Ozcan U Cao Q Yilmaz E Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes Science 2004 306 5695 457 461 15486293
- Schneeberger M Dietrich MO Sebastián D Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance Cell 2013 155 1 172 187 24074867
- Ozcan U Yilmaz E Ozcan L Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes Science 2006 313 5790 1137 1140 16931765
- Xiao C Giacca A Lewis GF Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans Diabetes 2011 60 3 918 924 21270237
- Kars M Yang L Gregor MF Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women Diabetes 2010 59 8 1899 1905 20522594
- Ogata M Hino S Saito A Autophagy is activated for cell survival after endoplasmic reticulum stress Mol Cell Biol 2006 26 24 9220 9231 17030611
- Bachar-Wikstrom E Wikstrom JD Ariav Y Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes Diabetes 2013 62 4 1227 1237 23274896