
Abstract
Introduction
Diabetes mellitus (DM) is characterized by chronic hyperglycemia and diabetic complications. Exacerbated cortical neuronal degeneration was observed in Alzheimer’s disease (AD) patients with DM. In fact, DM is now considered a risk factor of AD, as DM-induced activation of stress responses in the central nervous system (CNS) such as oxidative stress and neuroinflammation may lead to various neurodegenerative disorders. Methylglyoxal (MG) is one of the most reactive advanced glycation end-product (AGE) precursors. Abnormal accumulation of MG is observed in the serum of diabetic patients. As MG is reported to promote brain cells impairment in the CNS, and it is found that AGEs are abnormally increased in the brains of AD patients. Therefore, the effect of MG causing subsequent symptoms of AD was investigated.
Methods
5-week-old C57BL/6 mice were intraperitoneally injected with MG solution for 11 weeks. The Morris water maze (MWM) was used to examine the spatial learning ability and cognition of mice. After MG treatment, MTT assay, real-time PCR analyses, and Western blot were performed to assess the harvested astrocytes and hippocampi.
Results
Significantly longer escape latency and reduced percentage time spent in the target quadrant were observed in the 9-week-MG-treated mice. We have found in both in vitro and in vivo models that MG induced astrogliosis, pro-inflammatory cytokines, AD-related markers, and ERK activation. Further, trend of normalization of the tested markers mRNA expressions were observed after ERK inhibition.
Conclusion
Our in vivo results suggested that MG could induce AD symptoms and in vitro results implied that ERK may regulate the promotion of inflammation and Aβ formation in MG-induced reactive astrocytes. Taken together, MG may participate in the dysfunction of brain cells resulting in possible diabetes-related neurodegeneration by promoting astrogliosis, Aβ production, and neuroinflammation through the ERK pathway. Our findings provide insight of targeting ERK as a therapeutic application for diabetes-induced AD.
Introduction
Diabetes mellitus (DM) is a chronic disease marked by metabolic disorders that result in chronic hyperglycemia. Diabetic complications due to constantly high blood glucose levels may cause serious enduring dysfunction, failure, and destruction in numerous organs and systems including the brain.Citation1 Cognitive dysfunction is becoming more widely recognized as a significant comorbidity of diabetes. DM patients have a markedly higher chance of developing the neurodegenerative disorder, mainly Alzheimer’s disease (AD), marked by amyloid plaques and neurofibrillary tangles (NFTs),Citation2 with gradual cognitive decline and memory impairment caused by neuroinflammation and progressive neuronal loss in the brain.Citation3 The heavy burden of diabetes-associated cognitive dysfunction keeps raising. Yet, there is limited knowledge of the underlying pathogenic mechanisms of the progression of AD from DM. The involvement of DM in neurodegeneration and their relationship has not been fully understood. Comparing with neurons, there are much fewer studies targeting astrocytes as a significant candidate participating in the pathological progression of AD. Yet, increasing evidence shows the important role of astrocytes in the central nervous system (CNS) and the development of neuronal diseases.Citation4 This gives us a reasonable foundation in investigating the participation of astrocytes in diabetes-related neurodegeneration.
Astrocytes are the most numerous specialized glial type cells in the CNS, and that their number is five times more than that of the neuron. They have multiple essential complicated functions that maintain the normal function of the brain. Apart from secreting nutrients and maintaining a microenvironment to support and protect neurons, healthy astrocytes regulate extracellular ion concentrations, monitor synaptic remodeling, maintain protective barriers, including glia limitans, glial scars, and blood-brain barrier.Citation5 They also play a major role in controlling the inflammatory response in the brain.Citation6 Astrocytes, however, become activated by various pathological conditions in the CNS. In the development of neurodegenerative disorders, reactive astrocytes, known as astrogliosis, lose their supportive function and gain toxic function.Citation7,Citation8 Reactive astrocytes lead to neuroinflammation and neuronal dysfunction by becoming neurotoxic due to their production of reactive oxygen species (ROS) and pro-inflammatory cytokine.Citation9 Increase of reactive astrocytes is often correlated with cognitive decline.Citation10 The study confirmed that astrocytes also contribute to the formation of AD’s hallmarks.Citation11 Moreover, astrogliosis appears in multiple areas in the brain with DM and AD.Citation12,Citation13 Apart from hyperglycemia, there is increasing evidence showing methylglyoxal contributes to astrogliosis and neurodegeneration leading to cognitive dysfunction.
Methylglyoxal (MG) is a highly reactive metabolite. It is a precursor to advanced glycation end products (AGEs), as it causes non-enzymatic glycation of proteins resulting in irreversible AGEs. MG is produced from the anaerobic glycolysis from glyceraldehyde-3-phosphate.Citation14 It is maintained at a low level in healthy individuals. Increased formation and accumulation of MG often occur under metabolic disorders, including DM which is associated with hyperglycemia, also with aging.Citation15 In the plasma of diabetic individuals, there is a significantly elevated MG level.Citation16 MG induces cross-linking of protein, cellular damage and plays an important role in the pathogenesis of many neurodegenerative diseases through inducing oxidative stress and ROS production.Citation17,Citation18 Elevated MG level in AD patients was also observed.Citation19 Recently, only a few research have examined the involvement of MG in the modulation of animal spatial learning, memory, and cognition.Citation20–24 Animal studies and solid evidence of the role of MG in the progression of DM-induced AD are limited which warrants further investigation.
Our previous studies have demonstrated that MG induces neuroinflammatory responses and JNK activation in astrocytes.Citation25 However, the JNK pathway was found to only contribute to part of the development of neuroinflammation, via TNF-α, among the tested inflammatory markers implying that other mechanisms are also involved. Growing evidence show that ERK pathway is also a critical regulator of pro-inflammatory activation.Citation26 More importantly, it was suggested that ERK1/2 was the only MAPK pathway signaling protein that correlated strongly with neuropathology.Citation27,Citation28 Therefore, in this study, consistent with the hypothesis of hyperglycemia-induced MG would damage astrocytes causing neuroinflammation thus leading to neurodegenerative disorder, we further investigated the involvement of the ERK pathway. Also, a more clinically comparable in vivo model will be presented showing the effect of MG on spatial learning ability and cognition. In this report, we demonstrated that MG induces the behavioural pattern of AD.Citation29 In line with our previous study, our results further suggested that the ERK pathway plays an important role in diabetes-related neurodegeneration. Essential AD-related markers were found altered in astrocytes after being challenged by MG. Moreover, ERK inhibition was observed to have a normalization effect on the tested markers. These findings implicate the close linkage between DM and AD through the MG-activated ERK pathway. Further investigation of the underlying mechanism in detail could benefit the development of therapeutic strategies regarding diabetes-induced AD.
Materials and Methodology
Animals
Five-week-old male C57BL/6 mice were obtained from the Chinese University of Hong Kong. All techniques and animal handling followed the National Institutes of Health protocol for the care and use of laboratory animals, as well as the Animals (Control of Experiments) Ordinance, Hong Kong, China. The Hong Kong Department of Health and the Hong Kong Baptist University Committee on the Use of Human and Animal Subjects in Teaching and Research both, COA. No. (21–36) in DH/HT&A/8/2/6 Pt.3, approved the use of animals, and every effort was taken to reduce the number of animals used and their suffering.
Chemicals
Methylglyoxal, 3-[4, 5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT), PD 98059 were acquired from Sigma (St. Louis, MO, USA). All primary antibodies were acquired from Cell Signaling Technology (Beverly, MA, USA): β-actin antibody (Cat. #4967), glial fibrillary acidic protein (GFAP) antibody (Cat. #80788). Unless otherwise noted, all of the chemicals were acquired from Sigma and were of reagent-grade quality (St. Louis, MO, USA).
DITNC1 Cell Cultures and Treatments
Rat astrocytic cell line DITNC1 (ATCC, cat. number CCL-2005) were cultured in Dulbecco’s Modified Eagle Medium, supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic at 37°C humidified atmosphere with 5% carbon dioxide (CO2). Cells were seeded on 96 and 6 well plates for 24 hrs. For 96 well plates, different concentrations of MG were added to the cells for 24 hrs to conduct an MTT assay. For 6 wells plates, different concentrations of MG were added to the cells, and cells were collected for 24 hrs. To evaluate ERK inhibition, cells were incubated with the ERK upstream kinase MEK inhibitor PD98059 (20μM, Sigma) for 2hrs before MG treatment. The proteins and mRNA were isolated to be studied further.
Intraperitoneal Injection of Methylglyoxal
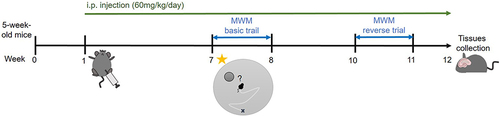
Mice were kept in an animal house with 12 hours of darkness and 12 hours of light. The mice were housed for seven days after they arrived. MG solution (60 mg/kg) or corresponding volume of sham control were administrated into the mice through intraperitoneal (ip) injection 6 days/week for 11 weeks. Hippocampi were dissected out and homogenized using various reagents for further analysis ().
Figure 1 Schematic diagram of treatment and behavioral experiments. MG solution (60 mg/kg/day) or corresponding volume of sham control were administrated into the mice daily through ip injection. After 6 weeks treatment, the MWM basic trial was conducted. And after 9 weeks treatment, the reverse trial was conducted. Hippocampi were dissected out and homogenized using various reagents for further analysis at week 12.
Morris Water Maze Test
After 6- and 9-week MG treatment, the MWM basic trial and reverse trial were conducted respectively ().Citation30 After the 4 training trials with a visible platform on day 0, mice were placed in the pool, filled with opaque painted water, with a hidden platform for platform searching on days 1–5 (4 trials/day, 3 different starting points). Probe trials were performed on day 6. Mice were placed in the pool without a platform for 1 minute. The percentage of time spent in the target quadrant was recorded. A probe trial with a visible platform was also performed to ensure mice in both groups have normal vision. The footprint and escape latency of each mouse was recorded by the tracking device. Data were collected for analysis.
Immunohistochemistry (IHC) and Immunofluorescence (IF) Staining
The half brains of the mice were fixed in formaldehyde for 24 hrs then stored in ethanol. The treated tissues were further dehydrated and embedded in paraffin. 4µm thick coronal brain sections were obtained. Deparaffinization, rehydration, and antigen retrieval were then performed. After blocking, sections were incubated with different primary antibodies overnight at 4°C and then with conjugated secondary antibodies for 1 hr at room temperature. For IHC, hematoxylin staining was performed to visualize the nucleus. For IF, 4-, 6-diamidino-2-phenylindole (DAPI) was used as a nuclear counterstain before microscopic analysis.
MTT Assay
To evaluate the cellular survival of DITNC1 cells following exposure to different concentrations of methylglyoxal treatment, 0.5mg/mL of MTT solution was added to the culture medium and incubated at 37°C for 4 hrs. Formazan salts in cells were dissolved by DMSO and absorbance was measured at 570 nm by a microplate reader (Bio-Rad, Hercules, CA, USA).
Western Blot Analysis
The protein of cells and hippocampus were extracted with RIPA buffer (Cell Signaling, Beverly, MA, USA), containing protease inhibitors 2mM PMSF, 10mM Na2VO4, and Complete Mini TM protease inhibitor cocktail (Roche). A protein assay was conducted to determine the protein concentration. Protein samples were then resolved by SDS-PAGE gel and transferred to the PVDF membrane. The membrane was then probed with different primary antibodies overnight at 4°C, followed by respective HRP-conjugated secondary antibodies for 1 hour. The membranes were soaked with “Chemiluminescent substrate SuperSignal ELISA Femto Substrate” (with peroxide and enhancer in 1:1 ratio) for visualizing protein bands as fluorescence signals. The fluorescence of protein bands was captured in ChemiDoc™ Touch Imager. ImageJ was used to analyze the strengths of fluorescence of the protein bands.
mRNA Extraction and Real-Time PCR
Trizol was used to extract endogenous mRNA from cells and the hippocampus of animals (Invitrogen). Reverse transcription was used to convert the mRNA into complementary DNA (cDNA). To determine the expression of various cytokines, real-time PCR with appropriate primers will be used. Primers used in the sample from Mus musculus 1) GFAP (F: AACCGCATCACCATTCCTGT, R: TGGCAGGGCTCCATTTTCAA); 2) TGF-β (F: AGGGCTACCATGCCAACTTC, R: CCACGTAGTAGACGATGGGC); 3) IL-1β (F: TGCCACCTTTTGACAGTGATG, R: TGATGTGCTGCTGCGAGATT); 4) IL-6 (F: CACTTCACAAGTCGGAGGCT, R: CTGCAAGTGCATCATCGTTGT); 5) TNF-α (F: ACAGAAAGCATGATCCGCGA, R: TTGCTACGACGTGGGCTAC); 6) APP (F: GAACCAGTCTCTGTCCCTGC, R: CAGAACCTGGTCGAGTGGTC); 7) BACE-1 (F: GCTGGGAGCTGGATTATGGT, R: CGTAGCTTTCGGGGTCTTCC); 8) PS1 (F: GAGACTGGAACACAACCATAGCC, R: AGAACACGAGCCCGAAGGTGAT). Primers used in the sample from Rattus norvegicus: 1) GFAP (F: CAACCTCCAGATCCGAGAAA, R: TCCTTAATGACCTCGCCATC); 2) TGF-β (F: GTCAACTGTGGAGCAACACG, R: CGTCAAAAGACAGCCACTCA); 3) IL-1β (F: CAGGAAGGCAGTGTCACTCA, R: AAAGAAGGTGCTTGGGTCCT); 4) IL-6 (F: ACCACCCACAACAGACCAGT, R: ACCACCCACAACAGACCAGT); 5) TNF-α (F: TGCCTCAGCCTCTTCTCATT, R: CCCATTTGGGAACTTCTCCT); 6) APP (F: CCAACCGTGGCATCCTTTTG, R: AGTGGTCAGTCCTCGGTCAG); 7) BACE-1 (F: CCTTCCGCATCACCATCCTT, R: GTCTTCCATGTCTGCCGTGA); 8) PS1 (F: GGTCCACTTCGTATGCTGG, R: GTTGTGTTCCAGTCCCCAC).
Statistical Analysis
Results were expressed as means ± standard error of the mean (SEM). Results were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s test, or t-test. P< 0.05 was considered statistically significant.
Results
MG Induces Spatial Learning Ability Impairment and Cognition Decline in vivo
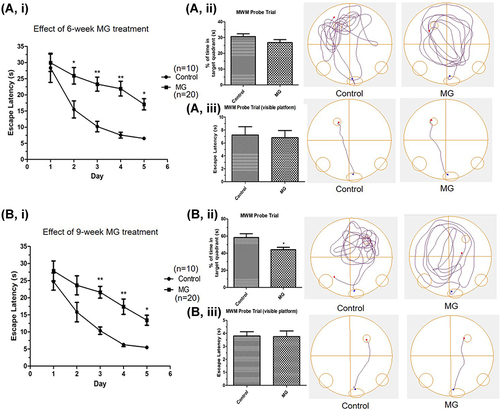
To examine the effect of MG on the spatial learning ability and cognition in vivo, the escape latency of the two groups of mice was examined by the MWM after 6 and 9 weeks of MG treatment. According to the results of 6-week-treatment (), MG-treated mice have a markedly longer escape latency than that of the control group. The difference in escape latency among the two groups is around 2–3 times on days 2–5. However, there was no major contrast shown in the probe trial on day 6. Similar results were observed in the reverse trial demonstrated after 9 weeks of treatment (), that the escape latency of the MG-treated group is markedly longer than that of the control group by over 2 to 3 folds starting from day 3. The probe trial result was more affirmative after 9 weeks of treatment showing a remarkable reduction of the percentage time spent in the target quadrant of the MG-treated mice.Citation28
Figure 2 Long term ip injection of MG induced spatial learning ability impairment and cognition decline in mice. MG solution (60 mg/kg) or vehicle solution were administrated into the mice respectively. Spatial learning ability and cognition of mice was examined by MWM after 6 and 9 weeks. (A) Significantly longer escape latency was observed in the MG-treated mice after 6 weeks treatment. (B) Significantly longer escape latency and significant reduction of the percentage time spent in the target quadrant of the MG-treated group were observed after 9 weeks treatment. i Graph indicated the escape latency of mice in experiment day 1 to 5. ii Representative track plots and the corresponding graph indicated the time spent in the target quadrant of mice in the probe trial on experiment day 6. iii Representative track plots and the corresponding graph indicated the escape latency of mice in the probe trial with visible platform on experiment day 6, showing mice in both groups have normal vision. Data correspond to the mean and SEM of 10 mice in control group and 20 mice in MG-treated group. *p < 0.05, **p < 0.01 versus the vehicle control.
MG Induces Astrogliosis, ERK Activation, Inflammatory Response, and Alteration of AD-Related Markers in the Hippocampus of C57BL/6 Mice
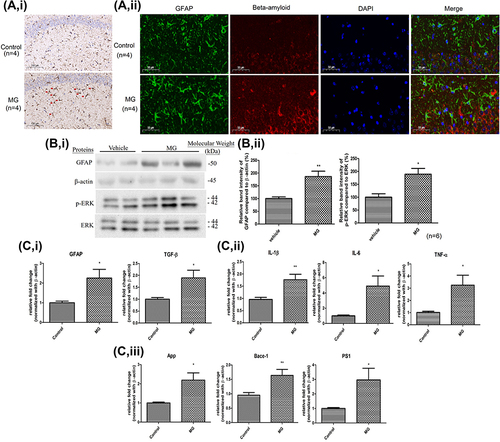
Hippocampi were extracted after an 11-week MG injection for analysis to assess the implications suggested from the in vitro results. IHC staining of visualization of GFAP was performed. Representative photomicrographs show that GFAP was strongly expressed by the reactive astrocytes in the MG-treat mice. While GFAP expression was less observable in the normal group (, i). IF staining for visualization of GFAP and beta-amyloid was also conducted. Representative photomicrographs show that the reactive astrocytes in the MG-treated group have a stronger GFAP and beta-amyloid immunoreactivity than that in the normal group (, ii). A significant increase in GFAP protein expression was found in the hippocampus of MG-treated mice (). The expressions of phosphorylated ERK were also markedly raised in the hippocampus of MG-treated mice (). The mRNA expression of an astrocytic marker GFAP, and TGF-β, pro-inflammatory cytokines, TNF-α, IL-1β, and IL-6, and AD-related markers, APP, BACE1, and PS1 in the hippocampus were significantly up-regulated as well ().
Figure 3 11 weeks ip injection of MG induced astrogliosis, ERK pathway activation, neuroinflammation response, and AD-related markers elevation in the hippocampus of C57BL/6 mice. MG solution (60 mg/kg) or vehicle solution were administrated into the mice for 11 weeks respectively. (A) Representative photomicrographs of i the immunohistochemistry staining for GFAP (indicated by red arrow) in the hippocampus of mice in each group. ii the immunofluorescence staining for GFAP (green) and beta-amyloid (red) in the hippocampus of mice in each group. One representative panel per group out of 4 mice is shown. (B) i Representative blots of GFAP and p-ERK expression in hippocampus of the two animal groups in relation to beta-actin and ERK respectively. ii Bar graph indicates quantified result in percentage. Data correspond to the mean and SEM of 6 mice in each group. *p< 0.05, **p< 0.01 versus the vehicle control. (C) Gene expressions of i astrocytic markers, ii inflammatory cytokines, and iii AD-related markers in the hippocampus of mice. Data correspond to the mean and SEM of 4 mice in each group. Beta-actin was used as mRNA internal control. *p< 0.05, **p< 0.01 versus the vehicle control.
MG Induces Cytotoxicity and Astrogliosis and Pro-Inflammatory Responses in DiTNC1 Cells
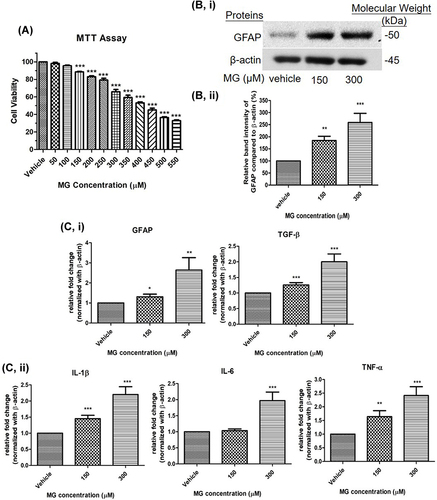
DITNC1 cells were treated with different dosages of MG, from 50 to 550μM to examine its cytotoxic effect. The result shows that the cytotoxicity of MG in DITNC1 cells was in a dose-dependent manner, while the median lethal dose (LD50) was observed at about 400μM (). After different durations of MG treatment, GFAP expression in DITNC1 cells was examined and were found significantly elevated after 24 hrs in a dose-dependent manner (). Further, mRNA expression of GFAP, transforming growth factor (TGF)-β, tumor necrosis factors (TNF)-α, interleukin (IL)-1β, and IL-6 were also found significantly raised as MG treatment concentration increased to 300μM ().
Figure 4 MG reduced cellular viability, increased astrocytic marker and pro-inflammatory cytokines in DITNC1 cells. (A) Cells were treated the indicated concentrations of MG for 24 hours. The cellular viability was found decreased in a dose dependent manner. Results are presented as means ± SEM, ***p < 0.001 versus the vehicle control. (n = 5) (B) GFAP expression of DITNC1 was examined by Western blot and was significantly increased after MG treatment. i Representative blots of GFAP expression at different concentrations of MG in relation to beta-actin. ii Bar graph indicates quantified result in percentage. Results are presented as means ± SEM, **p < 0.01, ***p < 0.001 versus the vehicle control. (n = 10) (C) The mRNA expression of DITNC1 were assessed by RT-PCR. i astrocytic markers, ii pro-inflammatory cytokines. All tested markers were found significantly up-regulated after 24 hours MG treatment. Beta-actin was used as mRNA internal control. Results are presented as means ± SEM, **p < 0.01, ***p < 0.001 versus the vehicle control. (n = 10).
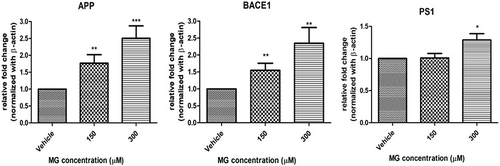
MG Altered AD-Related Markers APP, BACE-1, and PS1 Expression in DITNC1 Cells
The gene expression of AD-related markers, amyloid precursor protein (APP), β-site APP cleaving enzyme 1 (BACE1), and presenilin-1 (PS1) in DITNC1 cells after 24 hrs MG treatment were examined through real-time PCR analysis. Results indicate that APP, BACE-1, and PS1 show a remarkable elevation in mRNA expression as MG concentration increases ().
Figure 5 MG altered AD-related markers in DITNC1 cells. Cells were treated with the indicated concentrations of MG for 24 hours and the mRNA expressions were assessed by RT-PCR. APP, BACE-1, PS1 mRNA expression in DITNC1 cells were found significantly elevated. Beta-actin was used as mRNA internal control. Results are presented as means ± SEM, *p< 0.05, **p < 0.01, ***p < 0.001 versus the vehicle control. (n = 6).
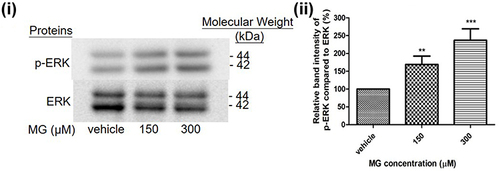
MG Increases Phosphorylation of ERK Kinase in DITNC1 Cells
Protein expressions were examined by Western blot to examine the influence of MG on the function of astrocytes through cellular pathways. In particular, phosphorylation of ERK was evaluated, with ERK as the endogenous control. Based on the MTT assay results, 150 and 300μM of MG were used to treat astrocytes cells for 24 hrs. According to the result, a marked increase in phosphorylation of ERK molecules was observed ().
Figure 6 MG induced ERK pathway activation in DITNC1 cells. Cells were treated the indicated concentrations of MG for 24 hours. p-ERK expression of DITNC1 were then assessed by Western blot. i Representative blots of p-ERK expression at different concentrations of MG in relation to ERK. ii Bar graph indicates quantified result in percentage. Results are presented as means ± SEM, **p < 0.01, ***p < 0.001 versus the vehicle control. (n = 10).
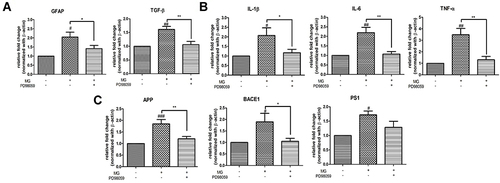
ERK Inhibition Attenuated mRNA Expression of Astrocytic Markers, Inflammatory Cytokines, and AD-Related Markers in DITNC1 Cells
To further examine the importance of the ERK pathway to astrocytic markers, pro-inflammatory responses, and AD-related markers in astrocytes, ERK upstream kinase MEK inhibitor PD98059 was applied to DITNC1 cells 2hrs before MG treatment. Trend of normalization of GFAP, TGF-β, TNF-α, IL-1β, IL-6, APP, BACE1, and PS1 mRNA expressions were observed after inhibitor pre-treatment ().
Figure 7 ERK inhibition attenuated mRNA expression of (A) astrocytic marker and TGF-β (B) inflammatory cytokines (C) AD-related markers in DITNC1 cells. Cells were incubated with 20μM of PD98059 for 2 hours prior to 300μM MG treatment. Cells were then harvested after 24 hours MG treatment. mRNA expression of all tested were markedly increased after MG treatment alone while trend of down-regulation of the markers were observed after PD98059 treatment. Results are presented as means ± SEM, *p< 0.05, **p< 0.01 versus MG. #p< 0.05, ##p< 0.01, ###p < 0.001 versus the vehicle control. (n = 6).
Discussion
The contribution of MG to a variety of diseases, including DM and neurodegenerative disorders, has been increasingly studied and recognized. Abnormal accumulation of MG is found in DM patients.Citation16 Fragmentation of MG is greatly increased as elevated production of dihydroxyacetone phosphate and glyceraldehyde-3-phosphate were promoted by an enhanced rate of glycolysis due to diabetes-induced hyperglycaemic conditions.Citation31 It is observed that the levels of MG in diabetes patients’ erythrocytes are 15- to 25-fold higher than that of healthy individuals. Even in the absence of hyperglycemia, the accumulation of high concentrations of MG was considered to be sufficient for inducing diabetic complications.Citation32 As an AGEs precursor, MG can turn into AGE inducing oxidative stress and contributing to neurodegeneration.Citation18,Citation33 In AD, accumulation of AGEs was observed in brain cells, including, astrocytes and neurons, and was found to correlated with the existence of amyloid plaques and NFTs.Citation34 Increased formation of AGEs and enhanced MG levels in the cerebrospinal fluid were also observed in AD and diabetes patients.Citation19 In addition, increased cognitive decline is associated with up-regulation of serum MG levels in the elderly.Citation35 These studies suggested that MG is a key factor in the progression of diabetes-related neurodegeneration.
Studies are now indicating the possible effect of MG on the modulation of memory, cognition, and executive function. For example, spatial memory impairment was observed in 3-month-old male rats after a single intravenous MG injection.Citation20 One study showed that alteration in cognitive function was not significant in rats after 12 months of 0.5% MG drinking water treatment,Citation21 and another study showed that memory decline was observed in mice after 4 weeks of 1% MG drinking water treatment.Citation22 A study demonstrated that 6-day intracerebroventricular MG injections (3µmol/µL/day) would induce cognitive decline,Citation23 and that memory impairment was observed in 3-month-old mice after acute (90 min), 7 and 10 days ip MG injection (10–200mg/kg).Citation24 The conditions reported were highly variable and the results were still inconclusive and require further examination.
Therefore, our current study proposes a more clinically comparable approach using in vivo model to demonstrate the effect of MG.Citation29 The MG dosage used in our study is correlated strongly with the clinical situations as briefly described previously.Citation25 To eliminate any possible additional effect due to aging and highlight the effect of MG, 5-week-old young-aged mice, corresponding to humans aged before 25,Citation36 were used in our setup. It is known that an increase in the intracellular accumulation of glucose and its metabolites, including MG, is promoted by the rise in blood glucose levels.Citation37 Long-term exposure to MG, for 11 weeks, was designed to imitate the accumulation of MG in DM patients due to chronic hyperglycemia-induced metabolic dysfunction. According to our MWM experiment results, MG-treated mice have a markedly longer escape latency than that of the control group (). A more affirmative probe trial result showing a remarkable reduction of the percentage time spent in the target quadrant of the MG-treated mice was observed after a 9-week treatment (). These results highlighted the long-term effect of MG, implying chronic hyperglycemia-induced MG accumulation could induce spatial learning ability impairment and cognition decline leading to Alzheimer’s disease.
Astrocytes are specialized glial cells that can be found throughout the CNS regions and makeup about a third of the total CNS cell population.Citation38 Astrocytes undertake several roles in retaining an optimal environment for neuronal survival and function. However, functional impairments of astrocytes due to stress promote the pathogenesis of neurodegeneration diseases. Astrogliosis, characterized by an increase in GFAP, is a universally acknowledged feature of AD. It is found in posthumous tissues from mouse and AD patients models. It is also found that the severity of astrogliosis is associated with cognitive deterioration.Citation10 Furthermore, neuroinflammation takes an essential part in the development of AD. AD patients have an increased level of pro-inflammatory cytokines. Astrocytes are closely engaged in neuroinflammatory events arising in the CNS because of their capability of secreting and responding to a wide range of inflammatory cytokines, including IL-1β, IL-6, TNF-α.Citation39 Meanwhile, TGF-β is typically upregulated in chronic neurodegeneration and neuroinflammation.Citation40 Being the major cause of the astroglial scar formation, fibrosis and sclerosis, TGF-β directly increases GFAP transcription in astrocytes. Extensive astrogliosis promoted by the presence of elevated TGF-β would hasten the pathogenesis of neurodegenerative disorder.Citation41
The effect of MG on the JNK pathway and the time frame of pro-inflammatory cytokines mRNA expression after MG treatment to astrocytes cells have been demonstrated previously.Citation25 Results of the dose-dependent effect of MG on astrocytes cells shown in this report, that MG induces astrogliosis and up-regulates pro-inflammatory responses in astrocytes cells (), were in line with our previous findings. Consistent in vivo results were also demonstrated. IHC visualization of increased GFAP expression of reactive astrocytes in the hippocampus of MG-treated mice supports that MG could aggravate astrogliosis (, i). Our results reinforce the implication that MG would induce neurodegeneration by causing astrocytic dysfunction and promoting neuroinflammation.
Other than neuroinflammation, AD is marked by the existence of extracellular amyloid plaques. Amyloid plaques are abnormal extracellular accumulation and deposition of β-amyloid (Aβ). While Aβ is formed by cleavage of APP by the enzymes BACE1 then by γ-secretase, where BACE1 only participates in the pathological condition leading to AD. It has been thought that neurons were the only cells that express BACE-1 and were capable to produce Aβ. However, studies have confirmed the contribution of astrocytes to Aβ formation that both APP and BACE1 also express in high levels in reactive astrocytes.Citation11 Moreover, different pro-inflammatory cytokines have been shown to upregulate APP and BACE-1. It is suggested that APP expression may be induced by IL-1β and TGF-β.Citation42,Citation43 While upregulation of BACE-1 may be promoted by TNF-α and other inflammatory mediators and pathways, including NF-kB,Citation44 JAK2 and ERK1/2 signaling.Citation45 In addition to Aβ production, mutations in the PSEN1 gene, encoding presenilin-1 (PS1), also take a part. Being the catalytic subunit of the γ-secretase complex, PS1 plays a role in the cleavage of APP. It has been suggested that PSEN1 mutations increase the Aβ42/Aβ40 ratio thus leading to neurodegeneration and dementia.Citation46 Considering the contribution of astrocytes in Aβ formation and the involvement in inflammatory events, together with our finding that MG-challenged astrocytes would become activated and promote inflammation, we further examine the effect of MG on APP, BACE1, and PS1 expression in astrocytes. According to our results, mRNA expression of the three markers in astrocytes cells was significantly increased after MG treatment (). The three markers were also found up-regulated in the hippocampus of mice after long-term MG treatment. Further, IF visualization of stronger GFAP and Aβ immunoreactivity in reactive astrocytes in the hippocampus of MG-treated group were observed (, ii). Taken together, these results implied that MG could contribute to the development of AD directly through promoting Aβ formation in astrocytes.
As the JNK pathway was found only contribute to a part of MG-induced neurodegeneration, we further examine the involvement of the ERK pathway. All members of the mitogen-activated protein kinases (MAPK) family have been implicated in the development of AD.Citation47 However, only the ERK/MAPK pathway takes the major role in Aβ generation by regulating APP and BACE1 genes.Citation27 Also, it was suggested that ERK1/2 was the only MAPK member with increased protein expression and positive associations with neuropathological grade.Citation28 Apart from regulation of the inflammatory response, ERK regulates neuronal function, development of the CNS, synaptic plasticity, learning, and memory formation by its crucial function in key cellular activities, including cell proliferation, differentiation, survival, and death. Growing evidence indicates that ERK involves in the pathogenesis of neurodegenerative disorders by several mechanisms. ERK1/2 cascades activation is found to contribute to neuronal apoptosis induction.Citation48 It is demonstrated in in vitro experiments that ERK would cause neuroinflammation by regulating inflammatory cytokine production in activated astrocytes.Citation49 Other than inflammation, ERK contributes to NFTs formation by phosphorylating tau.Citation50 ERK is also associated with senile plaques by mediating β- and γ-secretases and up-regulating APP.Citation43,Citation51–53 Moreover, elevated ERK levels have been observed in the brains of Alzheimer’s patients.Citation54 Abnormal ERK activation in the hippocampus may lead to memory impairments in Alzheimer’s patients by impairing hippocampal function.Citation49 The link between ERK and AD illustrates the significance of investigating the effect of MG on this pathway in astrocytes cells. We demonstrated that MG would activate ERK in astrocytes (). A significant increase in phosphorylation of ERK molecules was also found in the hippocampus of the MG-treated group. Together with the results mentioned, these findings suggested that MG could involve in the pathogenesis of diabetes-related neurodegeneration by promoting neuroinflammation and contributing to Aβ through activating the ERK pathway in astrocytes.
Regarding the link between the ERK signaling pathway and a variety of neurological conditions, including neurodegenerative illnesses of neurological disorders, including neurodegenerative diseases, it is suggested that ERK signaling inhibitors were likely to be developed for the therapeutic drugs of the diseases.Citation49 The main and most studied mechanism of inhibitor’s action to block ERK pathways is by preventing ERK1 and 2 phosphorylation through MEK1 and 2, the upstream kinases. The inhibitor was therefore introduced in this report to further evaluate the significance of ERK in the control of inflammatory cytokines. Our preliminary results show the trend of normalization in astrocytic marker and TGF-β, inflammatory cytokines TNF-α, IL-1β, IL-6, and AD-related markers APP, BACE1, PS1 mRNA expression after ERK inhibition (). According to the results, we propose that ERK pathway could take a part in regulating pro-inflammatory responses and contributing to neuroinflammation in reactive astrocytes leading to Aβ formation. In order to have a more comprehensive view of the mechanism of MG-induced neuroinflammation and neurodegeneration through the ERK pathway, we suggest that the time course of the events shall further be clarified. Also, other possible signaling pathways that are involved in regulating inflammation and promoting neurodegeneration, like NF-kB, p-38,Citation55 and JAK2,Citation45 should be investigated to evaluate their responsibility in regulating the markers.
Conclusion
To conclude, our data demonstrated for the first time that chronic MG accumulation could induce spatial learning ability impairment and cognition decline leading to AD in a more clinically comparable in vivo model. Our in vitro and in vivo results are in agreement that MG would induce astrogliosis, inflammatory response, up-regulation of AD-related markers and ERK activation. While ERK could participate in the promotion of neuroinflammation and Aβ formation causing neurodegeneration in MG-induced reactive astrocytes. The time cause of the events and other possible regulators of the makers should further be examined and clarified. Our findings provide significant evidence on the possible underlying mechanism of diabetes-related neurodegeneration that ERK may be one of the key pathways regulating Aβ formation, which is worth deeper investigation in future research. In addition, inhibition of the ERK pathway could be one of the potential therapeutic targets of diabetes-induced Alzheimer’s disease.
Abbreviations
AD, Alzheimer’s disease; AGEs, advanced glycation end products; APP, amyloid precursor protein; Aβ, beta amyloid; BACE1, beta site APP cleaving enzyme 1; CNS, central nervous system; DAPI, 4-, 6-diamidino-2-phenylindole; DM, diabetes mellitus; ERK, extracellular-signal-regulated kinase; GFAP, glial fibrillary acidic protein; IL-1β, interleukin 1 beta; IL-6, interleukin-6; JNK, C-Jun N-terminal kinases; MAPK, mitogen-activated protein kinases; MG, methylglyoxal; MTT, 3-[4, 5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide; MWM, Morris water maze; NFTs, neurofibrillary tangles; PS1, presenilin-1; ROS, reactive oxygen species, TGF-β, transforming growth factor beta; TNF-α, tumor necrosis factors-α.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
The authors thank the staff at the Hong Kong Baptist University for helpful technical assistance.
Additional information
Funding
References
- Biessels GJ, Despa F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol. 2018;14(10):591–604. doi:10.1038/s41574-018-0048-7
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189–a006189. doi:10.1101/cshperspect.a006189
- Vagelatos NT, Eslick GD. Type 2 diabetes as a risk factor for Alzheimer’s disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol Rev. 2013;35:152–160. doi:10.1093/epirev/mxs012
- Li K, Li J, Zheng J, Qin S. Reactive astrocytes in neurodegenerative diseases. Aging Dis. 2019;10(3):664–675. doi:10.14336/AD.2018.0720
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7–35. doi:10.1007/s00401-009-0619-8
- Phillips EC, Croft CL, Kurbatskaya K, et al. Astrocytes and neuroinflammation in Alzheimer’s disease. Biochem Soc Trans. 2014;42(5):1321–1325. doi:10.1042/BST20140155
- Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957–967. doi:10.1016/j.immuni.2017.06.006
- Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. doi:10.1038/nature21029
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28(3):138–145. doi:10.1016/j.it.2007.01.005
- Kashon ML, Ross GW, O’Callaghan JP, et al. Associations of cortical astrogliosis with cognitive performance and dementia status. J Alzheimers Dis. 2004;6(6):595–604. doi:10.3233/JAD-2004-6604
- Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017;7(12):170228. doi:10.1098/rsob.170228
- Wong DP, Chu JM, Hung VK, et al. Modulation of endoplasmic reticulum chaperone GRP78 by high glucose in hippocampus of streptozotocin-induced diabetic mice and C6 astrocytic cells. Neurochem Int. 2013;63(6):551–560. doi:10.1016/j.neuint.2013.09.010
- Steele ML, Robinson SR. Reactive astrocytes give neurons less support: implications for Alzheimer’s disease. Neurobiol Aging. 2012;33(2):423e1–13. doi:10.1016/j.neurobiolaging.2010.09.018
- Singh R, Barden A, Mori T, Beilin L. Advanced glycation end-products A review. Diabetologia. 2001;44(2):129–146. doi:10.1007/s001250051591
- Srikanth V, Westcott B, Forbes J, et al. Methylglyoxal, cognitive function and cerebral atrophy in older people. J Gerontol a Biol Sci Med Sci. 2013;68(1):68–73. doi:10.1093/gerona/gls100
- Kong X, Ma MZ, Huang K, et al. Increased plasma levels of the methylglyoxal in patients with newly diagnosed type 2 diabetes 2. J Diabetes. 2014;6(6):535–540. doi:10.1111/1753-0407.12160
- Matafome P, Sena C, Seica R. Methylglyoxal, obesity, and diabetes. Endocrine. 2013;43(3):472–484. doi:10.1007/s12020-012-9795-8
- Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. 2005;15(7):16R–28R. doi:10.1093/glycob/cwi053
- Kuhla B, Luth HJ, Haferburg D, Boeck K, Arendt T, Munch G. Methylglyoxal, glyoxal, and their detoxification in Alzheimer’s disease. Ann N Y Acad Sci. 2005;1043:211–216. doi:10.1196/annals.1333.026
- Huang X, Wang F, Chen W, Chen Y, Wang N, von Maltzan K. Possible link between the cognitive dysfunction associated with diabetes mellitus and the neurotoxicity of methylglyoxal. Brain Res. 2012;1469:82–91. doi:10.1016/j.brainres.2012.06.011
- Watanabe K, Okada K, Fukabori R, et al. Methylglyoxal (MG) and cerebro-renal interaction: does long-term orally administered MG cause cognitive impairment in normal Sprague-Dawley rats? Toxins. 2014;6(1):254–269. doi:10.3390/toxins6010254
- Chun HJ, Lee Y, Kim AH, Lee J. Methylglyoxal causes cell death in neural progenitor cells and impairs adult hippocampal neurogenesis. Neurotox Res. 2016;29(3):419–431. doi:10.1007/s12640-015-9588-y
- Hansen F, Pandolfo P, Galland F, et al. Methylglyoxal can mediate behavioral and neurochemical alterations in rat brain. Physiol Behav. 2016;164(Pt A):93–101. doi:10.1016/j.physbeh.2016.05.046
- Szczepanik JC, de Almeida GRL, Cunha MP, Dafre AL. Repeated methylglyoxal treatment depletes dopamine in the prefrontal cortex, and causes memory impairment and depressive-like behavior in mice. Neurochem Res. 2020;45(2):354–370. doi:10.1007/s11064-019-02921-2
- Chu JM, Lee DK, Wong DP, Wong GT, Yue KK. Methylglyoxal-induced neuroinflammatory response in in vitro astrocytic cultures and hippocampus of experimental animals. Metab Brain Dis. 2016;31(5):1055–1064. doi:10.1007/s11011-016-9849-3
- Kim HK. Role of ERK/MAPK signalling pathway in anti-inflammatory effects of Ecklonia cava in activated human mast cell line-1 cells. Asian Pac J Trop Med. 2014;7(9):703–708.
- Du Y, Du Y, Zhang Y, et al. MKP-1 reduces Abeta generation and alleviates cognitive impairments in Alzheimer’s disease models. Signal Transduct Target Ther. 2019;4:58. doi:10.1038/s41392-019-0091-4
- Chen MJ, Ramesha S, Weinstock LD, et al. Microglial ERK signaling is a critical regulator of pro-inflammatory immune responses in Alzheimer’s disease. bioRxiv. 2019:798215. doi:10.1101/798215
- Li WY, Wu SL, Fcy L, Yue K. 417-P: AGE precursor methylglyoxal leads to behavioral pattern changes characteristics of Alzheimer’s Disease in mice. Diabetes. 2021;70(Supplement_1). doi:10.2337/db21-417-P
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1(2):848–858. doi:10.1038/nprot.2006.116
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(December):813–820. doi:10.1038/414813a
- Fleming T, Cuny J, Nawroth G, et al. Is diabetes an acquired disorder of reactive glucose metabolites and their intermediates? Diabetologia. 2012;55(4):1151–1155. doi:10.1007/s00125-012-2452-1
- Sena CM, Matafome P, Crisostomo J, et al. Methylglyoxal promotes oxidative stress and endothelial dysfunction. Pharmacol Res. 2012;65(5):497–506. doi:10.1016/j.phrs.2012.03.004
- Luth HJ, Ogunlade V, Kuhla B, et al. Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer’s disease brains. Cereb Cortex. 2005;15(2):211–220. doi:10.1093/cercor/bhh123
- Beeri MS, Silverman J, Moshier E, et al. Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Alzheimers Dement. 2011;7:S144–S145. doi:10.1016/j.jalz.2011.05.388
- Kevin Flurkey JMC, Harrison DE. Mouse models in aging research. In: The Mouse in Biomedical Research. 2nd ed. Elsevier; 2007:637–672.
- Schalkwijk CG, Stehouwer CDA. Methylglyoxal, a highly reactive dicarbonyl compound, in diabetes, its vascular complications, and other age-related diseases. Physiol Rev. 2020;100(1):407–461. doi:10.1152/physrev.00001.2019
- Carson MJ, Thrash JC, Walter B. The cellular response in neuroinflammation: the role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin Neurosci Res. 2006;6(5):237–245. doi:10.1016/j.cnr.2006.09.004
- Heneka MT, Carson MJ, Khoury JE, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. doi:10.1016/s1474-4422(15)70016-5
- Doyle KP, Cekanaviciute E, Mamer LE, Buckwalter MS. TGFβ signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflammation. 2010;7:1–13. doi:10.1186/1742-2094-7-62
- Reilly JF, Maher PA, Kumari VG. Regulation of astrocyte GFAP expression by TGF-beta1 and FGF-2. Glia. 1998;22(2):202–210. doi:10.1002/(SICI)1098-1136(199802)22:2<202::AID-GLIA11>3.0.CO;2-1
- Burton T, Liang B, Amara F, Amara F. Transcriptional activation and increase in expression of Alzheimer’s β-amyloid precursor protein gene is mediated by TGF-β in normal human astrocytes. Biochem Biophys Res Commun. 2002;295(3):702–712. doi:10.1016/S0006-291X(02)00724-6
- Blasko I, Veerhuis R, Stampfer-Kountchev M, Saurwein-Teissl M, Eikelenboom P, Grubeck-Loebenstein B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol Dis. 2000;7(6Pt B):682–689. doi:10.1006/nbdi.2000.0321
- Bourne KZ, Ferrari DC, Lange-Dohna C, Rossner S, Wood TG, Perez-Polo JR. Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and glia upon exposure to beta-amyloid peptides. J Neurosci Res. 2007;85(6):1194–1204. doi:10.1002/jnr.21252
- Cho HJ, Kim SK, Jin SM, et al. IFN-gamma-induced BACE1 expression is mediated by activation of JAK2 and ERK1/2 signaling pathways and direct binding of STAT1 to BACE1 promoter in astrocytes. Glia. 2007;55(3):253–262. doi:10.1002/glia.20451
- Sun L, Zhou R, Yang G, Shi Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc Natl Acad Sci USA. 2017;114(4):E476–E485. doi:10.1073/pnas.1618657114
- Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802(4):396–405. doi:10.1016/j.bbadis.2009.12.009
- Subramaniam S, Unsicker K. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 2010;277(1):22–29. doi:10.1111/j.1742-4658.2009.07367.x
- Sun J, Nan G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: a potential therapeutic target (Review). Int J Mol Med. 2017;39(6):1338–1346. doi:10.3892/ijmm.2017.2962
- Ferrer I, Blanco R, Carmona M, et al. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol. 2001;11(2):144–158. doi:10.1111/j.1750-3639.2001.tb00387.x
- Tamagno E, Parola M, Bardini P, et al. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem. 2005;92(3):628–636. doi:10.1111/j.1471-4159.2004.02895.x
- Kwak YD, Choumkina E, Sugaya K. Amyloid precursor protein is involved in staurosporine induced glial differentiation of neural progenitor cells. Biochem Biophys Res Commun. 2006;344(1):431–437. doi:10.1016/j.bbrc.2006.03.054
- Tran MD. P2 receptor stimulation induces amyloid precursor protein production and secretion in rat cortical astrocytes. Neurosci Lett. 2011;492(3):155–159. doi:10.1016/j.neulet.2011.01.078
- Pei JJ, Braak H, An WL, et al. Up-regulation of mitogen-activated protein kinases ERK12 and MEK12 is associated with the progression of neurofibril. Mol Brain Res. 2002;109(1–2):45–55. doi:10.1016/S0169-328X(02)00488-6
- Kheiri G, Dolatshahi M, Rahmani F, Rezaei N. Role of p38/MAPKs in Alzheimer’s disease: implications for amyloid beta toxicity targeted therapy. Rev Neurosci. 2018;30(1):9–30. doi:10.1515/revneuro-2018-0008