Abstract
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease. It is typically characterized by adult-onset degeneration of the upper and lower motor neurons, and is usually fatal within a few years of onset. A subset of ALS patients has an inherited form of the disease, and a few of the known mutant genes identified in familial cases have also been found in sporadic forms of ALS. Precisely how the diverse ALS-linked gene products dictate the course of the disease, resulting in compromised voluntary muscular ability, is not entirely known. This review addresses the major advances that are being made in our understanding of the molecular mechanisms giving rise to the disease, which may eventually translate into new treatment options.
Introduction
Amyotrophic lateral sclerosis (ALS), also known as Charcot’s disease or Lou Gehrig’s disease is the most widespread type of motor neuron disease. Striking later in life, the disease causes degeneration of motor neurons and consequently progressive atrophy of associated muscle tissues and supporting cells. Unlike similar motor neuron diseases that primarily affect only a single subgroup of neurons (eg, Primary Muscular Atrophy or Primary Lateral Sclerosis), ALS patients typically have both lower motor neuron (LMN) and upper motor neuron (UMN) involvement. The symptoms of ALS commonly are muscle weakness and wasting, especially in the limbs, cramps, twitching, and difficulties in speaking. The lifetime risk of acquiring ALS by age 70 is between 1 in 400 and 1 in 1000,Citation1 and in general, ALS individuals succumb to the disease within 2–3 years due to respiratory failure.
A growing number of ALS-causing genes have been identified recently and are now under investigation, providing promise for increased understanding of the etiology of the disease. SOD1, encoding the highly conserved, cytosolic antioxidant enzyme Cu,Zn-superoxide dismutase (Cu,ZnSOD), was the first such gene to be identified with ALS.Citation2,Citation3 SOD1 mutations are common in both familial ALS (FALS) and sporadic ALS (SALS), and have been studied in the most depth. Other genes such as OPTN Citation4 or TARDBP, FUS, and ANG (involved in RNA metabolism)Citation5 were later identified as causative factors in both FALS and SALS. Suggestive of proteolytic disfunction, UBQLN2 was recently implicated in ALS,Citation6,Citation7 and very recently, nucleotide repeat expansions in C9ORF72,Citation8–Citation10 were found to comprise the largest fraction of ALS-causing mutations known to date. The present era is an exciting time for ALS research with the major challenge of understanding how these distinct, underlying triggers lead to a common aberrant cellular dyshomeostasis phenotype, resulting in toxic protein aggregates, neuronal death, and subsequently muscle atrophy that ultimately paralyzes the ALS patient.
Only one drug, riluzole, has been approved to treat ALS, which typically provides a meager gain of a few months of survival.Citation11 With advances in diagnostics and personalized medicine, however, future ALS patients will hopefully find improved treatment regimes to follow for their specific ALS manifestations. In this review, we will focus on the recent breakthroughs that will likely provide new avenues to reach this outcome. These include increased understanding of the basic biology of ALS and progress toward upcoming therapeutics in development.
Diagnosis of ALS
Epidemiology
Worldwide, the incidence rate of ALS varies from approximately 0.3–2.5 cases per year per 100,000 persons.Citation12 Five percent or greater of all cases run in families (FALS),Citation13 with a range from 2%–15% in different populations,Citation14 although regional and/or ethnic variations in incidenceCitation15,Citation16 and penetranceCitation17 complicate the estimation,Citation18 as do the organization of the studies themselves, being either population-or clinic-based.Citation19 Aside from family history, the clinical presentation of FALS and SALS can be very similar.Citation20 The onset for FALS is typically several years before that of SALS, although an exact age is difficult to estimate. In one study, for example, the mean FALS age was 48, as compared to 66 for a population-based group,Citation21 whereas in another larger study the discrepancy, although still present, was not as large (52 versus 56, respectively).Citation22 Typically, in SALS cases, but not always in FALS,Citation21,Citation22 males appear to predominate,Citation23 but this may vary among ethnic backgrounds and may be trending toward equality with time.Citation24 The higher incidence of ALS among war veterans and smokers,Citation25–Citation27 potentially accounts for the increased male risk, in addition to factors such as male hormones.Citation28 Interestingly, a recent study suggested that a lower-than-average ratio of the index to ring finger is represented in ALS patients.Citation29 This measurement (termed the 2D:4D ratio) is thought to reflect androgen exposure in the wombCitation30,Citation31 and therefore postulates a role for prenatal developmental factors in the disease. Sports (soccer and football) and sport-specific effects (soccer, but not basketball or cycling)Citation32 have also been implicated in ALS disease development.Citation25,Citation33 Finally, higher body mass index (up to 30–35) was found to correlate to disease survival,Citation34 possibly due to the common weight loss phenotype from muscle wasting associated with disease progression. An improved awareness of risk factors and trends for ALS might eventually establish better preventative measures or treatments, especially for those with a family history of the disease.
Symptom presentation and examination
No single test for diagnosing ALS exists; most cases are established based on symptom presentation, progression, and tests to eliminate overlapping conditions.Citation35 ALS is typically characterized by combined symptoms of the UMNs and LMNs. The UMNs of the central nervous systems originate in the motor cortex or brainstem and relay motor information to the LMNs. The LMNs are located in the brainstem and spinal cord and relay impulses from the UMNs to the muscles at neuromuscular synapses to innervate skeletal muscles controlling the arms and legs. UMN symptoms include weakness, speech problems, overactive reflexes, spasticity, and inappropriate emotionality; LMN symptoms also include weakness, as well as decreased reflexes, cramps, twitching and muscle wasting.Citation36,Citation37 Disease onset usually begins in the limbs (termed spinal onset), although about a quarter of ALS patients have “bulbar” onset,Citation38 the term describing the facial, mouth/jaw, and tongue muscles controlled by the “bulb,” an early name for the lower brainstem. Associated with poorer prognosis, bulbar onset is more common in elderly patients and women.Citation39,Citation40 A hallmark of ALS is rapid progression, and over time most patients will display both spinal and bulbar features (including emotionality, yawning, jaw jerking, tongue twitching, wasting, drooling, and difficulties swallowing). The El Escorial Criteria are a set of guidelines for ALS diagnosis, frequently used to gauge clinical trial participation and clinical practice. In some cases, though, these criteria may be overly stringent when used in diagnosis.Citation41
Diagnosis may be seen as a process of elimination, although family history can also be useful. The battery of tests performed, ie, blood tests, electromyography, magnetic resonance imaging, and nerve conduction studies, can aid in ruling out other conditions.Citation42 For example, in some patients, creatine kinase activity may be slightly elevated.Citation43 Cerebrospinal fluid (CSF) examination, on the other hand, is typically normal but can aid in diagnosing conditions such as multiple sclerosis. Furthermore, muscle biopsy can rule out inclusion body myositis.Citation44 Indeed, a central challenge in ALS diagnosis is distinguishing the many mimics. These include injuries (eg, herniated disk, spinal compression, or heavy metal poisoning), cervical spondylosis, metabolic problems such as enzyme/vitamin deficiency (B-12 etc), copper deficiency or thyroid problems, stroke, myopathies or neuropathies, inclusion body myositis, infections such as Lyme or HIV, or diseases such as myasthenia gravis, syringomyelia, cancer, Kennedy’s disease, Tay-Sachs diseases, or multiple sclerosis, among others.Citation12,Citation20,Citation36,Citation37,Citation44–Citation46 Misdiagnoses are in fact very common,Citation20,Citation47 about 10% of patients with other disorders are diagnosed erroneously with ALS.Citation48,Citation49 These findings may result in incorrect (potentially harmful) treatments, and delays in obtaining the necessary therapies and support and in seizing clinical trial opportunities.
Attempts to identify ALS-specific biomarkers may prove useful. For example, a study examining blood plasma found statistically significant distinctions in a panel of several hundred metabolites among ALS patients, allowing the authors to cleanly separate control patients from diseased patients (on taking or not taking riluzole), and even to sub-classify LMN-affected patients.Citation50 Such efforts may eventually aid the clinician in more specifically diagnosing motor neuron disease.
Pathophysiology
Protein inclusions and cellular dyshomeostasis
Typical hallmarks of ALS revealed from post-mortem examinations of patient brain and spinal cord sections are neuronal atrophy and the presence of cellular inclusions. Inclusions typical of affected cells include the small, cystatin-C and transferrin-immunoreactive Bunina bodies.Citation51 Also very common are ubiquitinated cellular inclusions, most often skein-like or of the round Lewy-body hyaline variety.Citation52 The presence of ubiquitin-reactive inclusions is consistent with a very recent study demonstrating that defects in the ubiquitin proteasome system may be a more generalized feature of ALS.Citation6 Degenerative cellular abnormalities can afflict the motor cortex, the brainstem, the anterior horn of the spinal cord, the lateral and/or anterior corticospinal tracts. Distinct cellular inclusions, suggested by differential protein composition, are observed in ALS arising from different genetic backgrounds (discussed below).
Another common facet of ALS pathophysiology is irregular glutamate metabolism, targeted by riluzole, the only drug approved to treat ALS.Citation53 Elevated synaptic glutamate can lead to excessive stimulation of glutamate receptors (eg, AMPA and NMDA) on the postsynaptic neuron, resulting in nerve damage and death through excitotoxicity. Interestingly, the above-described features may also occur in the supporting glia, including astrocytes in which inclusions and downregulation of GLT-1 (also known as EAAT2) glutamate transporter were observed.Citation54 Other relevant cellular abnormalities in ALS include an increase of p53-mediated apoptosis, impaired axonal transport, and cytoskeletal and mitochondrial dysfunction.Citation55–Citation58 Additionally, as disease symptoms appear at mid-to-late life, cumulative damage occurring through increased levels of oxidative stress may be a significant contributor to the disease.Citation59 A recent study analyzing the CSF of ALS patients suggested distinct metabolic signatures discernible between SALS patients and those with SOD1 and non-SOD1 FALS. The metabolomes of SOD1 FALS patients were observed to be more homogeneous than those of non-SOD1 FALS patients, which were more homogeneous than those of SALS patients.Citation60 These observations suggest that genetic contributions to the disease may influence ALS physiology.
FALS and SALS genes
Despite the identification of some ALS-causing genetic defects in individual families, ALS is not a single-pathway, single-gene condition. Therefore in recent years, high throughput, genome wide association studies have become a favored tactic for filling in the significant remaining space of unknown FALS-causing genes.Citation61 Nonetheless, consistency in reproducing candidate genes had been a problemCitation62 until the recent, notable exception of the C9ORF72 gene in the 9p21 locus,Citation8,Citation9,Citation63 a major ALS breakthrough. The disease subtypes associated with FALS mutations have been assigned designations of ALS1-ALS15 (). However, several known FALS mutations have now been documented in SALS cases, suggesting a broader role for these gene products in ALS pathogenesis. Although a variety of genes have been implicated in ALS (), we will focus on this subset of genes, in which genetic lesions can cause and contribute to both FALS and SALS.
Table 1 Common genes involved in ALS
SOD1
The SOD1 gene encodes the cytosolic enzyme Cu,ZnSOD, which is conserved from bacteria to humans. Cu,ZnSOD catalyzes the dismutation of the superoxide (O2−) radical anion, a toxic byproduct of cellular respiration, to produce molecular oxygen and hydrogen peroxide,Citation64 with the toxicity of the latter being removed by conversion through a peroxidase or catalase. Over 150 SOD1 mutations () account for a significant fraction of FALS, and are typically present in about 20% of such cases (ranging from 2.5%–23.5%), as well as in 0.44% to 7% of SALS cases.Citation19,Citation65 The majority of inherited SOD1 mutations are dominant, and individuals with two copies of a mutation may have much earlier onset.Citation66,Citation67 The common D90A SOD1 mutation is an exception that can be inherited in either a dominant or recessive fashion, as well as appearing sporadically.Citation68,Citation69
Figure 1 Known mutations in FALS and SALS-associated proteins.
Abbreviations: FALS, familial amyotrophic lateral sclerosis; SALS, sporadic amyotrophic lateral sclerosis; NLS, nuclear localization sequence; NES, nuclear export sequence; Sec, cleaved signal sequence; RRM, RNA regnition motif; X rich, X (amino acid residue) rich motifs; UBD, ubiquitin binding domain; ZnF, zinc finger; UBL, ubiquitin like domain; STI1, heat-shock-chaperonin-binding motifs; UBA, ubiquitin associated domain.
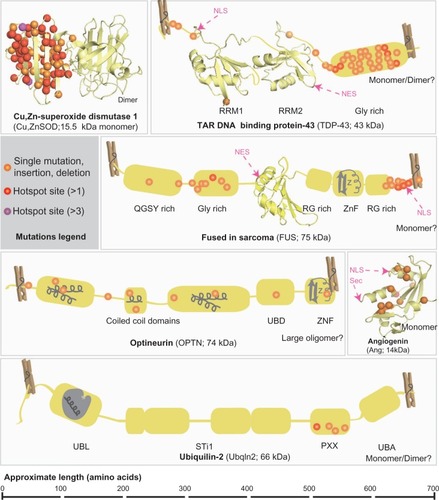
SOD1 mutations do not appear to cause disease by a loss of function. For example, transgenic expression of SOD1 mutants in mice is pathogenic without altering enzyme activity.Citation70 This is also evidenced by the fact that Cu,ZnSOD deficient mice do not develop motor neuron diseaseCitation71 and that mutations are not restricted to the active site of the enzyme.Citation2 Instead, mutant Cu,ZnSODs form toxic, misfolded species within neuronal and glial Lewy-body like inclusionsCitation72,Citation73 that usually appear before symptom presentation.Citation73,Citation74 Within these aggregates, mutant Cu,ZnSOD can be associated with heat shock protein Hsc70Citation75–Citation77 or 14-3-3 proteins, suggesting in the latter case that sequestration of anti-apoptotic proteins could contribute to cell death.Citation78 In a recent report, strong mutant Cu,ZnSOD immuno-reactivity was observed in small, granular non-ubiquitin reactive inclusions that localize to the cytosol and/or lysosomes of FALS (SOD1 and non-SOD1) and non-SOD1 SALS patients.Citation79 Also, Cu,ZnSOD-positive nuclear inclusions have been observed in spinal-cord derived glia from FALS and SALS patients.Citation80 Therefore, Cu,ZnSOD aggregates, found in tissues from distinct ALS patients, may be a component of diverse cellular inclusions in affected motor neurons and their supporting cells.
Detailed analyses of Cu,ZnSOD structures and enzymatic mechanismsCitation81,Citation82 including comparisons to bacterial Cu,ZnSODCitation83 and the human mitochondrial MnSODCitation84–Citation86 provided an informed foundation to evaluate the diverse mutations.Citation2,Citation87 To explain the complex effects of Cu,ZnSOD mutations in ALS pathogenesis, we and others have proposed a framework destabilization hypothesis.Citation87–Citation89 In this hypothesis each of the diverse set of mutations can cause local unfolding events that contribute to a globally defective, self-aggregating protein, which can deleteriously co-aggregate with other cellular proteins.Citation88 Such framework-destabilizing mutations are associated with other neurodegenerative and cancer prone diseases as typified by mutants of the XPD helicase.Citation90 Several studies have attempted to characterize the aggregation propensity of mutant forms of Cu,ZnSOD in vitro and in cultured cells, but a direct correlation between mutant protein stability and clinical phenotype has been elusive.Citation91–Citation94 This lack of correlation could be due to a multitude of contributing factors, ranging from important roles for metals in architectural stability,Citation95 to aberrant oxidative modifications of the free cysteines,Citation96,Citation97 to anomalous interactions of mutant Cu,ZnSOD with other cellular components. These components likely include proteins involved in stress responses (eg, Derlin-1, Rac-1)Citation98,Citation99 folding/maturation (eg, Hsc70 and the Cu,ZnSOD copper chaperone)Citation77,Citation100 and vesicular transport associated proteins (eg, chromogranin, dynein heavy chain).Citation101–Citation103
TARDBP
The TARDBP gene encodes TAR DNA binding protein 43 (TDP-43), a modular DNA/RNA binding protein (), localized to the cytosol and the nucleus, which is involved in splicing and transcriptional regulation.Citation104 In vivo, TDP-43 depletion in mice resulted in mRNA reduction and splicing errors in many mRNA transcripts and a few non-coding RNAs, particularly long intron-containing transcripts. This suggests a broad role for TDP-43 in alternative splicing and prevention of nonsense-mediated decay of transcripts expressed in neurons.Citation105 The nearly 40 mutations identified in the TARDBP gene encoding TDP-43 () may contribute to up to 6.5% of dominantly-inherited FALS cases,Citation106,Citation107 in addition to 0%–5% of sporadic cases.Citation107–Citation110 A reduced nuclear pool of TDP-43 is associated with some mutations, and cytoplasmic, ubiquitin-reactive hyperphosphorylated TDP-43 inclusions are observed in tissues from frontotemporal dementia (FTD) patientsCitation111,Citation112 and in neuronal and glial tissues samples from SALS and Guam ALS patients.Citation113 The inclusions commonly co-localize with ubiquitin and the protein p62.Citation113 However, TDP-43 inclusions are not present in SOD1 FALS individualsCitation114 (with the exception of one caseCitation113) or FUS mutant patients.Citation115
FUS
FUS encodes fused in sarcoma (FUS, also known as Translated in Liposarcoma, TLS), a modular nucleic acid-associated protein with many similarities to TDP-43, including conservation of protein domains (), a role in RNA processingCitation115 and localization in both the cytosol and nucleus in many cells. About 30 known FUS mutations account for approximately 3%–5% of FALS and ∼1% of SALS casesCitation116,Citation117 and all but the one known recessive variant, H517QCitation118 cause a dominant phenotype. As with some TARDBP mutations, certain FUS mutations located near the nuclear localization sequence may shift the nuclear/cytoplasmic balance towards cytosolic. This imbalance occurs by impairing the transportin-mediated import of FUS into the nucleus.Citation119 FUS-reactive inclusions have been found in tissues from FUS mutant FALS patients but not in SOD1 mutant patients.Citation115,Citation117 Furthermore, although earlier studies failed to see FUS-immunoreactivity in SALS cases Citation115 a more recent study did report FUS staining in inclusions from SALS patients.Citation117 FUS inclusions are commonly seen in FTD patients,Citation115,Citation118,Citation120 in addition to ALS patients, and these FUS-proteinopathy phenotypes might be distinguished through co-localization of other FUS family member proteins in FTD, but not in ALS.Citation121 Furthermore, FUS and TDP-43 inclusion phenotypes are thought to be mutually exclusive in FTD,Citation122,Citation123 but this may not be the case in ALS; although TDP-43 reactivity was not observed in FUS ALS mutant tissues,Citation115 FUS-reactivity was later reported in TDP-43 ALS mutant tissues.Citation117
OPTN
A recent Italian study indicated that approximately 3.5% of SALS patients, in addition to 1.2% of FALS patients, had mutations in the OPTN gene,Citation4 which encodes Optineurin. About a dozen mutations in OPTN can lead to ALS, with gain of function mutations dominant and loss of function mutations recessive.Citation124,Citation125 Optineurin is a multifunctional cytosolic and Golgi-associated coiled-coil domain-containing, ubiquitin-binding phosphoprotein (). It is involved in vesicular trafficking and Golgi maintenance, signaling in the tumor-necrosis factor α/NF-κB pathway,Citation126 mGluR signalingCitation127,Citation128 and autophagy.Citation129 Optineurin has been shown to form homo-complexes and heteromultimerize with Rab8, myosin VI, and transferrin receptor proteins. In both FALS- and SALS-affected cells, Optineurin can co-localize in inclusion bodies with FUSCitation130 and TDP-43,Citation124 although the frequency of such inclusions was shown to be low in another study.Citation131 Furthermore, Optineurin localization has been observed in basophilic inclusions from SOD1 FALS patient tissues,Citation124 although conflictingly this co-localization was not observed in another study in patient-derived or mouse model tissues.Citation132
ANG
Angiogenin (Ang, encoded by the ANG gene), a small, hypoxia- and ischemia-inducibleCitation133 ribonuclease A () involved in angiogenesis, is mutated in a smaller number of FALS and SALS cases.Citation134 Expressed in many tissues, including motor neurons,Citation135 where it promotes cell survival,Citation136 Ang is required for the VEGF-mediated stimulation of angiogenesis.Citation137 Ang is secreted and taken up by effector cells via endocytosis, then translocated to the nucleus, to stimulate transcription of rRNA, among other roles.Citation135 Due to loss of ribonuclease and/or nuclear translocation activity,Citation135 ANG mutations appear to attenuate angiogenesis although the protein stability is not compromised.Citation138 Eighteen ANG mutations, therefore, can cause a loss-of-function phenotype, with most ANG ALS patients presenting with bulbar onset (discussed above).Citation134
UBQLN2
UBQLN2, a gene on the X-chromosome, was recently found to be causative for X-linked dominant FALS.Citation6,Citation139 In affected families, incomplete penetrance was noted in females, presumably due to X-inactivation. The encoded ubiquilin-2 protein () normally performs effector functions in the ubiquitin proteasome pathway by tethering degradation-targeted proteins (through its C-terminal ubiquitin-associated domain) to the proteasome (through association with its N-terminal ubiquitin-like domain). The intervening regions within the protein are less well characterized, and include a PXX (proline-rich) domain, where five distinct mutations were found. In tissues derived from UBQLN2-mutant patients, ubiquitin-positive skein-like inclusions were also reactive for ubiquilin 2. This phenotype was particularly notable in the spinal cord and hippocampus, correlating with the appearance of dementia in 20% of the X-linked ALS patients. Furthermore, these inclusions were also positive for TDP-43, FUS and OPTN, but not Cu,ZnSOD. Notably, ubiquilin-2 inclusion staining was present in all samples from a wide panel of genetically-distinct ALS patient tissues (sporadic, SOD1-mutant, TARDBP mutant, and non-FUS/non-TARDBP/non-SOD1 FALS, and ALS with dementia) but not in non-ALS controls.Citation6 Expression of mutant ubiquilin-2 protein significantly slowed down proteosomal degradation of a reporter substrate in Neuro-2a cells,Citation6 suggesting a mechanistic contribution for these mutants. Unlike the other mutations described, those in the UBQLN2 gene have not yet been implicated in SALS. However, these findings suggest ubiquilin-2 could be generally relevant to ALS pathogenesis.
C9ORF72
Very recently, two independent research groups flagged C9ORF72 as the gene at locus 9p21 that was linked to dominant cases of ALS/FTDCitation8,Citation9 in previous genome-wide association studies. Strikingly, a substantial hexanucleotide repeat (GGGGCC) within an intron of this gene was identified in 24%–46% of FALS cases and 4%–21% of SALS cases, making this the most commonly mutated ALS gene. The expansion appeared to result in nuclear foci and directed preferential splicing of an alternatively spliced transcript.Citation8 However, precisely how the aberrant RNA metabolism of C9ORF72 causes ALS is not yet known, and the protein, aside from nuclear localization,Citation9 has no ascribed function. Interestingly, postmortem examination of several patients with the C9ORF72 hexanucleotide repeat, who exhibited ALS and FTD-like symptoms, also revealed neuronal TDP-43 inclusions.Citation8
Commonalities and crosstalk
One puzzle for understanding ALS is that the known ALS-causing gene products have diverse physiological functions. However, some common themes in pathogenesis are beginning to emerge. For example, RNA processing defects are visible in mutants of TARDBP, FUS, and ANG (as well as a FALS gene called SETX).Citation5 Nucleotide repeat expansions have also now been identified in C9ORF72 (and an ALS-susceptibility protein called Ataxin-2).Citation140 Proteinacious cellular inclusions are also a common denominator in ALS patient-derived tissues; these can involve ubiquilin-2, as well as SOD, FUS, TDP-43, and/or optineurin. Interestingly however, different disease subtypes appear to reveal aggregates with distinct protein composition. Due to their roles in both ALS and FTD, TDP-43, FUS, OPTN, and ubiquilin-2 have been proposed to function in the context of a unified pathway.Citation141 Thus, interactions among these components should be a focus for future research. Along these lines, a recent study in zebrafish found that the expression of human FUS could rescue the motor neuron phenotype associated with knockdown of TARDBP expression, whereas, conversely, TARDBP could not rescue FUS knockdown, suggesting that TARDBP is genetically upstream of FUS.Citation142 These results are consistent with a study showing that TDP-43 regulates the mRNA processing of FUS transcripts as well as its own.Citation105
Genetic overlap between ALS and other diseases
Gene products whose mutations cause ALS have been implicated in other diseases. For example, FUS, TDP-43, ubiquilin-2, and/or optineurin-positive inclusions are found in many FTD patients,Citation131,Citation143 and C9ORF72 is implicated also in ALS/FTD.Citation8,Citation9 TDP-43-immunoreactivity is sometimes seen in hippocampal sclerosis, Pick’s disease, and Alzheimer’s disease (AD), and ubiquitin staining can occur in the latter disease.Citation109 Likewise, optineurin has recently been implicated in AD due to its inclusion body staining in neurofibrillary tangles.Citation144 Furthermore, optineurin interacts with the protein huntingtin, suggesting some role in Huntington’s disease,Citation145 and mutations in optineurin are associated with glaucomaCitation146 and Paget’s disease of the bone.Citation147 The ubiquilin-1 paralog, with a domain structure similar to ubiquilin-2, is associated with AD.Citation6 The 14-3-3 protein isoforms co-localized in Cu,ZnSOD inclusions have also been found in a Parkinson’s disease model, suggesting some commonalities in inclusion formation.Citation148 Angiogenin has been implicated in a gamut of diseases, from cancers to diabetes, asthma, and heart disease.Citation149 Finally, nucleotide repeats (as in C9ORF72) are known to cause a variety of neurodegerative diseases such as Huntington’s disease, Fragile X-syndrome, Kennedy’s disease and others.Citation150 These observations underscore the need for meaningful synergistic collaborations among researchers studying these different complex diseases that often involve protein aggregation, allowing new insights to be compounded.
Treatment of ALS
The primary goal of ALS treatment is the inhibition of disease progression, although an important secondary consideration is the treatment of damage already done. Palliative care (eg, home care and hospice) remains a significant focus of the treatment program for the ALS patient. Non-invasive ventilation, for example, can improve the quality of life and extend survival in non-bulbar patients.Citation151 A support team, and hospice care toward the end of life can help the ALS patient to prepare nutritive food that is easy to swallow, provide medications for muscle spasticity, weariness, sleep and depression, and adjust ventilators, enabling the patient to adjust to lifestyle limitations.
Although domestic alterations can provide significant relief to current patients, biochemical and pharmacological advances will drive forward better therapeutics. A panel of ALS biomarkers from non-invasive analyses would be a major gain not only in diagnosis and monitoring progression, but also in identifying affected biological pathways in ALS to target therapeutically.Citation152 Multiple studies have sought to identify protein biomarkers for ALS, including increased blood or CSF levels of TDP-43, or the cysteine protease inhibitor cystatin C, or a skewed CSF ratio of phospho-neurofilament heavy chain to complement C3.Citation153–Citation156 Furthermore, the combined efforts of GC/MS (gas chromatography coupled to mass spectrometry), LC/MS (liquid chromatography coupled to mass spectrometry), and NMR (nuclear magnetic resonance) could potentially span the whole metabolome in identifying biomarker signatures.Citation50,Citation60 Better disease markers could reduce the long duration, averaging 14 months, between initial symptom presentation and diagnosis,Citation47 helping to improve the disease trajectory.Citation157 Such endeavors would also provide a platform for personalized medicine for ALS patients. At present, at least one clinical trial (NCT00677768) is being organized to analyze the blood and CSF of ALS patients for biological markers.
Pharmacological interventions
The only approved medicine to treat the general symptoms of ALS is the anti-excitotoxicity drug riluzole.Citation158 The drug is thought to preserve motor neuron function by decreasing toxic glutamate levels at glutamatergic nerve terminals by (a) inactivating sodium channels, (b) inhibiting glutamate release, and (c) blocking postsynaptic actions of NMDA receptors.Citation159 The safety and efficacy profiles for riluzole are better than those for other excitotoxicity drugs, but riluzole only increases the chance of an additional year of survival by about 9%, typically prolonging survival for about 2–3 months.Citation11 The drug serves to slightly preserve limb and bulbar function but actual muscle strength is typically not improved.Citation11 Recently approved for treating purely the pseudobulbar affect symptoms less commonly observed in ALS patients is dual-acting dextromethorphan/quinine (sold as Neudexta®; Avanir Pharmaceuticals, Aliso Viejo, CA).Citation160 Like riluzole, dextromethorphan also inhibits glutamatergic signaling, and quinine helps to increase its bioavailability, providing modest benefit to a subset of patients.Citation160
Promising new therapeutic developments, several of which are in late-phase clinical trials, may provide strides forward in treating ALS. One such drug in phase III clinical trials (NCT00349622) is the antibiotic ceftriaxone, used to treat pneumonia and bacterial meningitis. In ALS patients, ceftriaxone appears to upregulate the GLT-1 (EAAT2) glutamate transporter, potentially correcting cellular glutamate levels.Citation161 Another potential treatment option is high-dose methylcobalamin (vitamine B-12), currently in phase II/III studies (NCT00444613 and NCT00445172) to determine safety and efficacy for long-term use in ALS.Citation162 This compound was recently shown to reduce homocysteine (another excitatory amino acid)-mediated toxicity in NSC-34 cells.Citation163 Finally, an antioxidant targeting the mitochondria is currently in phase III trials (NCT01281189), sponsored by Biogen Idec (Westin, MA) and Knopp Biosciences LLC (Pittsburgh, PA). This drug, dexpramipexole,Citation164 is the R(+)-isomer of the amino-benzothiazole drug pramipexole (currently approved to treat Parkinson’s disease and restless legs syndrome). Dexpramipexole was well tolerated in phase II clinical trials, revealing positive trends in slowing function decline and improving survivability.
SOD1-targeting therapies
The establishment of mutant SOD1 transgenic mice in the late 1990s was a major breakthrough in the field, providing the first disease models for ALS.Citation70 Now, about a dozen such SOD1 ALS mouse models exist.Citation165 Other distinctive ALS models have been developed,Citation166,Citation167 including the newer TARDBP mouse models that similarly display ALS-like symptoms such as gait abnormalities, weight loss, and spasticity.Citation104 However, the use of SOD1 mouse models has predominated much of the therapeutic progress, in part because SOD1 represents a major disease target. For example, because the SOD1 gene is predominately dispensible,Citation71 reducing its expression and perturbing aggregation are favored strategies for treatment of ALS. These transgenic animals are appropriate models in many cases, and guidelines have been suggested for standardizing studies in SOD1 mice.Citation168
Both small molecules and siRNAs are being explored to downregulate and diminish SOD levels. The hydroxylamine drug arimoclomol (Orphazyme) is currently in stage II/III clinical trials (NCT00706147). This compound induces a heat shock response that resulted in a decrease in ubiquitin-positive aggregates in G93A SOD1 mouse models,Citation169 and is now being tested in SOD1 FALS patients. A free radical scavenger, edaravone (Mitsubishi Tanabe Pharma Corporation, Osaka, Japan) was recently found to ameliorate ALS symptoms and diminish SOD aggregate deposition in interior horn cells. Phase III clinical trials were recently completed (NCT00330681; NCT00424463; NCT00415519), with results pending publication, so the future success of the drug remains to be seen. Studies aimed at silencing SOD1 using siRNA-based strategies in mice have met with some success,Citation170,Citation171 although the inability of siRNA to pass the blood–brain barrier makes delivery a problem. Accordingly, Isis Pharmaceuticals Inc (Carlsbad, CA) has developed a CSF-infused delivery method for Isis-SOD1RX antisense oligos that recently were successful in animal models,Citation172 and are now being examined in phase I clinical trials (NCT01041222). Finally, an approach aimed at prevention, which is in its infancy, is immunization against mutant Cu,ZnSOD through vaccination with mutant Cu,ZnSOD or metal-free Cu,ZnSOD (exhibiting some similar pathogenic properties).Citation173 As stable Cu,ZnSOD polymers expected to break tolerance exist,Citation174 and as antibodies favor reactions with more flexible regions,Citation175,Citation176 such antibody experiments may be promising.
A recent study used patient-derived progenitor cells to derive cultured astrocyte cell lines, and these were found to be toxic to motor neurons, via a mechanism involving secretion of uncharacterized factors. Interestingly, both FALS (mutant SOD1) and SALS-derived cells, but not non-ALS derived astrocyte cells, had common pathway changes (namely NF-κB, MAPK, JNK, and AKT), and knockdown of SOD1 rescued the motor neuron killing phenotype in four of six cell lines examined.Citation177 This study interestingly reaffirms the use of SOD1-targeted therapeutics in the context of SALS (although the effects on other FALS genetic backgrounds were not tested) and also suggests that such cell cultures could prove useful for therapeutic screening in the absence of an all-encompassing ALS disease model. Indeed, a few years ago, astrocyte replenishment by injection of glial precursor cells in SOD1 model rats was found to prolong life and improve motor performance.Citation178 Similarly, a phase I clinical trial (NCT01348451) aimed at spinal implantation of spinal cord-derived stem cells is being sponsored by Neuralstem Inc (Rockville, MD). This treatment previously extended the life of SOD1 transgenic rats by 10 days,Citation179 and provides the first regenerative medicine strategy for ALS.
Future directions
Where do we go from here? ALS was first described about 150 years agoCitation180 and recent biotechnological advances have allowed researchers to begin pinpointing the precise genetics and pathological mechanisms behind the disease. Yet, many questions still remain: How do the distinct pathways involved in the disease overlap and converge to cause similar phenotypes? Can diagnostics improve to the point of early screening and detection? Arguably most importantly, how can we best treat individual patients? Fortunately, the complex nature of the disease also allows for many potential targets and means for therapeutic intervention.
The discovery of the role of SOD1 in ALS was a triggering event that significantly advanced our current understanding of the disease aided by the basic science of SOD structure and biochemistry.Citation87,Citation181 Although we now know that the mutant proteins aggregate, we are only starting to appreciate the key architectural features of the proteins involved in triggering this aggregation and its consequences. More recently, we have realized the significant contributions of TDP-43 and FUS in ALS and other degenerative diseases.Citation182 Indeed, RNA metabolism appears to be a common thread. The recent identification of ubiquilin-2 as a co-immunolocalized component of ALS inclusions in a wide variety of ALS cell types has also been a major breakthrough in the field.Citation6 Thus, follow-up work is now needed in order to determine the mechanism of this ubiquilin-mediated pathology, as well as its potential contributions to other ALS-linked pathways. Finally, determining the pathogenic mechanism of action of newly identified C9ORF72 repeats may prove extremely useful in understanding a significant majority of ALS cases, both sporadic and inherited. Newer disease models will undoubtedly play a significant role in facilitating these studies.
A critical element of progress in the ALS field will be the dissemination of genetic, epidemiologic, and therapeutic information. Fortunately, several helpful online databases and resource are now available, including the ALS online genetics database,Citation183 the Genetic Association studies website,Citation184 the ALS forum,Citation185 and the Northeast ALS Consortium (NEALS).Citation186 Outreach and social networking is provided by sites such as the Twitter-based ALS Untangled,Citation187 which hosts a forum for patient conversations. These assets will increase awareness and discourse among ALS patients and drive future research collaborations.
Conclusions
Currently, ALS is an unrelenting and incurable neuromuscular disease that paralyzes its victims, eventually leaving them incapable of breathing. Gradually, thanks in part due to strides in molecular genetics, the mechanisms leading to aberrant cellular physiology and toxic inclusions are being sewn together. At present, therapeutic strategies aim to slow down the pace of the disease. Ultimately, however, future efforts will work to block the initial events leading to neuronal death. This will prevent damage to the patient’s motor ability before it happens, stemming from earlier diagnosis and leading to better prognosis.
Acknowledgments
This work was supported by NIH grant R03 AR059968 (to JJP) and NIH grant R01 GM39345 (to EDG). AJP is a predoctoral fellow of the National Science Foundation and the Skaggs Institute for Chemical Biology at the Scripps Research Institute.
Disclosure
The authors report no conflicts of interest in this work.
References
- WijesekeraLCLeighPNAmyotrophic lateral sclerosisOrphanet J Rare Dis20094319192301
- DengHXHentatiATainerJAAmyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutaseScience19932615124104710518351519
- RosenDRSiddiqueTPattersonDMutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosisNature1993362641559628446170
- Del BoRTilocaCPensatoVNovel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosisJ Neurol Neurosurg Psychiatry201182111239124321613650
- StrongMJThe evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS)J Neurol Sci20102881–211219840884
- DengHXChenWHongSTMutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementiaNature2011477736321121521857683
- DaoudHRouleauGAA role for ubiquilin 2 mutations in neurodegenerationNat Rev Neurol201171159960021989241
- DeJesus-HernandezMMackenzieIRBoeveBFExpanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALSNeuron201172224525621944778
- RentonAEMajounieEWaiteAA hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-Linked ALS-FTDNeuron201172224526821944778
- WoodHA hexanucleotide repeat expansion in C9ORF72 links amyotrophic lateral sclerosis and frontotemporal dementiaNat Rev Neurol201171159522009283
- MillerRGMitchellJDLyonMMooreDHRiluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND)Cochrane Database Syst Rev20071CD00144717253460
- SathasivamSMotor neurondisease: clinical features, diagnosis, diagnostic pitfalls and prognostic markersSingapore Med J201051536737320593140
- ByrneSCHardimanORate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysisNeurology2010749A56
- ConwitRAPreventing familial ALS: a clinical trial may be feasible but is an efficacy trial warranted?J Neurol Sci20062511–21217070848
- HaberlandtWFGenetic aspects of amyotrophic lateral sclerosis and progressive bulbar paralysisActa Genet Med Gemellol (Roma)1959836937413830663
- MurrosKFogelholmRAmyotrophic lateral sclerosis in Middle-Finland: an epidemiological studyActa Neurol Scand198367141476601351
- WilliamsDBFloateDALeicesterJFamilial motor neuron disease: differing penetrance in large pedigreesJ Neurol Sci1988862–32152303221241
- AndersenPMAl-ChalabiAClinical genetics of amyotrophic lateral sclerosis: what do we really know?Nat Rev Neurol201171160361521989245
- AndersenPMAmyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase geneCurr Neurol Neurosci Rep200661374616469270
- TraynorBJCoddMBCorrBFordeCFrostEHardimanOAmyotrophic lateral sclerosis mimic syndromes: a population-based studyArch Neurol200057110911310634456
- MulderDWKurlandLTOffordKPBeardCMFamilial adult motor neuron disease: amyotrophic lateral sclerosisNeurology19863645115173960325
- LiTMAlbermanESwashMComparison of sporadic and familial disease amongst 580 cases of motor neuron diseaseJ Neurol Neurosurg Psychiatry19885167787843404186
- JohnstonCAStantonBRTurnerMRAmyotrophic lateral sclerosis in an urban setting: a population based study of inner city LondonJ Neurol2006253121642164317219036
- WormsPMThe epidemiology of motor neuron diseases: a review of recent studiesJ Neurol Sci20011911–23911676986
- ChioABenziGDossenaMMutaniRMoraGSeverely increased risk of amyotrophic lateral sclerosis among Italian professional football playersBrain2005128Pt 347247615634730
- HornerRDGrambowSCCoffmanCJAmyotrophic lateral sclerosis among 1991 Gulf War veterans: evidence for a time-limited outbreakNeuroepidemiology2008311283218535397
- WangHO’ReillyEJWeisskopfMGSmoking and risk of amyotrophic lateral sclerosis: a pooled analysis of 5 prospective cohortsArch Neurol201168220721321320987
- AbhinavKStantonBJohnstonCAmyotrophic lateral sclerosis in South-East England: a population-based study. The South-East England register for amyotrophic lateral sclerosis (SEALS Registry)Neuroepidemiology2007291–2444817898523
- VivekanandaUManjalayZRGanesalingamJLow index-to-ring finger length ratio in sporadic ALS supports prenatally defined motor neuronal vulnerabilityJ Neurol Neurosurg Psychiatry201182663563721551173
- ManningJTScuttDWilsonJLewis-JonesDIThe ratio of 2nd to 4th digit length: a predictor of sperm numbers and concentrations of testosterone, luteinizing hormone and oestrogenHum Reprod19981311300030049853845
- BerenbaumSABrykKKNowakNQuigleyCAMoffatSFingers as a marker of prenatal androgen exposureEndocrinology2009150115119512419819951
- ChioACalvoADossenaMGhiglionePMutaniRMoraGALS in Italian professional soccer players: the risk is still present and could be soccer-specificAmyotroph Lateral Scler200910420520919267274
- AbelELFootball increases the risk for Lou Gehrig’s disease, amyotrophic lateral sclerosisPercept Mot Skills20071043 Pt 21251125417879657
- PaganoniSDengJJaffaMCudkowiczMEWillisAMBody mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosisMuscle Nerve2011441202421607987
- HardimanOvan den BergLHKiernanMCClinical diagnosis and management of amyotrophic lateral sclerosisNat Rev Neurol201171163964921989247
- Lomen-HoerthCAmyotrophic lateral sclerosis from bench to bedsideSemin Neurol200828220521118351522
- CristiniJMisdiagnosis and missed diagnoses in patients with ALSJAAPA2006197293516869150
- KiernanMCVucicSCheahBCAmyotrophic lateral sclerosisLancet2011377976994295521296405
- ForbesRBColvilleSSwinglerRJThe epidemiology of amyotrophic lateral sclerosis (ALS/MND) in people aged 80 or overAge Ageing200433213113414960427
- McCombePAHendersonRDEffects of gender in amyotrophic lateral sclerosisGend Med20107655757021195356
- AndersenPMBorasioGDDenglerREFNS task force on management of amyotrophic lateral sclerosis: guidelines for diagnosing and clinical care of patients and relativesEur J Neurol2005121292193816324086
- de CarvalhoMDenglerREisenAElectrodiagnostic criteria for diagnosis of ALSClin Neurophysiol2008119349750318164242
- TalbotKMotor neuron disease: the bare essentialsPract Neurol20099530330919762894
- DabbyRLangeDJTrojaborgWInclusion body myositis mimicking motor neuron diseaseArch Neurol20015881253125611493165
- WeihlCCLopateGMotor neuron disease associated with copper deficiencyMuscle Nerve200634678979316929546
- SilaniVMessinaSPolettiBThe diagnosis of amyotrophic lateral sclerosis in 2010Arch Ital Biol2011149152721412713
- ChioAISIS Survey: an international study on the diagnostic process and its implications in amyotrophic lateral sclerosisJ Neurol1999246Suppl 3III1III510631652
- DavenportRJSwinglerRJChancellorAMWarlowCPAvoiding false positive diagnoses of motor neuron disease: lessons from the Scottish Motor Neuron Disease RegisterJ Neurol Neurosurg Psychiatry19966021471518708642
- LudolphACKnirschUProblems and pitfalls in the diagnosis of ALSJ Neurol Sci1999165Suppl 1S14S2010448976
- RozenSCudkowiczMEBogdanovMMetabolomic analysis and signatures in motor neuron diseaseMetabolomics20051210110818820733
- OkamotoKMizunoYFujitaYBunina bodies in amyotrophic lateral sclerosisNeuropathology200828210911518069968
- LeighPNWhitwellHGarofaloOUbiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificityBrain1991114Pt 27757881646064
- PlaitakisACaroscioJTAbnormal glutamate metabolism in amyotrophic lateral sclerosisAnn Neurol19872255755792892463
- BarbeitoLHPeharMCassinaPA role for astrocytes in motor neuron loss in amyotrophic lateral sclerosisBrain Res Brain Res Rev2004471–326327415572176
- BarbosaLFCerqueiraFMMacedoAFIncreased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALSBiochim Biophys Acta20101802546247120097285
- BilslandLGSahaiEKellyGGoldingMGreensmithLSchiavoGDeficits in axonal transport precede ALS symptoms in vivoProc Natl Acad Sci U S A201010747205232052821059924
- KingAEDicksonTCBlizzardCANeuron-glia interactions underlie ALS-like axonal cytoskeletal pathologyNeurobiol Aging201132345946919427060
- ZhuHNShiPWeiYMZhangJYGalJMitochondrial dysfunction is a converging point of multiple pathological pathways in amyotrophic lateral sclerosisJ Alzheimers Dis201020Suppl 2S311S32420463400
- Miana-MenaFJGonzalez-MingotCLarrodePMonitoring systemic oxidative stress in an animal model of amyotrophic lateral sclerosisJ Neurol2011258576276921108037
- WuolikainenAMoritzTMarklundSLAnttiHAndersenPMDisease-related changes in the cerebrospinal fluid metabolome in amyotrophic lateral sclerosis detected by GC/TOFMSPLoS One201164e1794721483737
- TalbotKDo twin studies still have anything to teach us about the genetics of amyotrophic lateral sclerosis?J Neurol Neurosurg Psychiatry201081121299130021087924
- GarberKGenetics. The elusive ALS genesScience200831958592018174402
- DaoudHBelzilVDionPARouleauGAChromosome 9p21 in amyotrophic lateral sclerosis: the plot thickensLancet Neurol201091094594720801719
- McCordJMFridovichISuperoxide dismutase. An enzymic function for erythrocuprein (hemocuprein)J Biol Chem196924422604960555389100
- van EsMADahlbergCBirveAVeldinkJHvan den BergLHAndersenPMLarge-scale SOD1 mutation screening provides evidence for genetic heterogeneity in amyotrophic lateral sclerosisJ Neurol Neurosurg Psychiatry201081556256619965850
- MarucciGMorandiLBartolomeiEAmyotrophic lateral sclerosis with mutation of the Cu/Zn superoxide dismutase gene (SOD1) in a patient with Down syndromeNeuromuscul Disord2007179–1067367617624778
- HaywardCBrockDJMinnsRASwinglerRJHomozygosity for Asn86Ser mutation in the CuZn superoxide dismutase gene produces a severe clinical phenotype in a juvenile onset case of familial amyotrophic lateral sclerosisJ Med Genet1998352174
- JonssonPABackstrandAAndersenPMCuZn-superoxide dismutase in D90A heterozygotes from recessive and dominant ALS pedigreesNeurobiol Dis200210332733312270693
- MancusoMFilostoMNainiAA screening for superoxide dismutase-1 D90A mutation in Italian patients with sporadic amyotrophic lateral sclerosisAmyotroph Lateral Scler Other Motor Neuron Disord20023421521812710511
- GurneyMEPuHFChiuAYMotor-neuron degeneration in mice that express a human Cu,Zn superoxide-dismutase mutationScience19942645166177217758209258
- ReaumeAGElliottJLHoffmanEKMotor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injuryNat Genet199613143478673102
- ShibataNHiranoAKobayashiMIntense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvementJ Neuropathol Exp Neurol19965544814908786408
- BruijnLIBecherMWLeeMKALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusionsNeuron19971823273389052802
- KatoSShimodaMWatanabeYNakashimaKTakahashiKOhamaEFamilial amyotrophic lateral sclerosis with a two base pair deletion in superoxide dismutase 1: gene multisystem degeneration with intracytoplasmic hyaline inclusions in astrocytesJ Neuropathol Exp Neurol19965510108911018858006
- ZetterstromPGraffmoKSAndersenPMBrannstromTMarklundSLProteins that bind to misfolded mutant superoxide dismutase-1 in spinal cords from transgenic amyotrophic lateral sclerosis (ALS) model miceJ Biol Chem201128623201302013621493711
- WatanabeMDykes-HobergMCulottaVCPriceDLWongPCRothsteinJDHistological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissuesNeurobiol Dis20018693394111741389
- WangJFarrGWZeissCJProgressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALSProc Natl Acad Sci U S A200910651392139719171884
- OkamotoYShirakashiYIharaMColocalization of 14-13-3 proteins with SOD1 in Lewy body-like hyaline inclusions in familial amyotrophic lateral sclerosis cases and the animal modelPLoS One201165e2042721655264
- ForsbergKJonssonPAAndersenPMNovel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patientsPLoS One201057e1155220644736
- ForsbergKAndersenPMMarklundSLBrannstromTGlial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosisActa Neuropathol2011121562363421287393
- TainerJAGetzoffEDRichardsonJSRichardsonDCStructure and mechanism of copper, zinc superoxide dismutaseNature198330659402842876316150
- PargeHEHallewellRATainerJAAtomic structures of wild-type and thermostable mutant recombinant human Cu,Zn superoxide-dismutaseP Natl Acad Sci U S A1992891361096113
- BourneYRedfordSMSteinmanHMLepockJRTainerJAGetzoffEDNovel dimeric interface and electrostatic recognition in bacterial Cu,Zn superoxide dismutaseP Natl Acad Sci U S A199693231277412779
- BorgstahlGEPargeHEHickeyMJBeyerWFJrHallewellRATainerJAThe structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundlesCell19927111071181394426
- GuanYHickeyMJBorgstahlGECrystal structure of Y34F mutant human mitochondrial manganese superoxide dismutase and the functional role of tyrosine 34Biochemistry19983714472247309537987
- PerryJJHearnASCabelliDENickHSTainerJASilvermanDNContribution of human manganese superoxide dismutase tyrosine 34 to structure and catalysisBiochemistry200948153417342419265433
- PerryJJShinDSGetzoffEDTainerJAThe structural biochemistry of the superoxide dismutasesBiochim Biophys Acta20101804224526219914407
- DiDonatoMCraigLHuffMEALS mutants of human superoxide dismutase form fibrous aggregates via framework destabilizationJ Mol Biol2003332360161512963370
- ShinDSDiDonatoMBarondeauDPSuperoxide dismutase from the eukaryotic thermophile alvinella pompejana: structures, stability, mechanism, and insights into amyotrophic lateral sclerosisJ Mol Biol200938551534155519063897
- FanLFussJOChengQJXPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutationsCell2008133578980018510924
- WangQJohnsonJLAgarNYAgarJNProtein aggregation and protein instability govern familial amyotrophic lateral sclerosis patient survivalPLoS Biol200867e17018666828
- VassallKAStubbsHRPrimmerHADecreased stability and increased formation of soluble aggregates by immature superoxide dismutase do not account for disease severity in ALSProc Natl Acad Sci U S A201110862210221521257910
- BystromRAndersenPMGrobnerGOlivebergMSOD1 mutations targeting surface hydrogen bonds promote amyotrophic lateral sclerosis without reducing apo-state stabilityJ Biol Chem201028525195441955220189984
- PrudencioMHartPJBorcheltDRAndersenPMVariation in aggregation propensities among ALS-associated variants of SOD1: correlation to human diseaseHum Mol Genet200918173217322619483195
- LindbergMJTibellLOlivebergMCommon denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo stateProc Natl Acad Sci U S A20029926166071661212482932
- BoscoDAMorfiniGKarabacakNMWild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALSNat Neurosci201013111396140320953194
- ProctorEADingFDokholyanNVStructural and thermodynamic effects of post-translational modifications in mutant and wild type Cu, Zn superoxide dismutaseJ Mol Biol2011408355556721396374
- NishitohHKadowakiHNagaiAALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1Genes Dev200822111451146418519638
- HarrazMMMardenJJZhouWSOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS modelJ Clin Invest2008118265967018219391
- KatoSSumi-AkamaruHFujimuraHCopper chaperone for superoxide dismutase co-aggregates with superoxide dismutase 1 (SOD1) in neuronal Lewy body-like hyaline inclusions: an immunohistochemical study on familial amyotrophic lateral sclerosis with SOD1 gene mutationActa Neuropathol2001102323323811585247
- UrushitaniMSikASakuraiTNukinaNTakahashiRJulienJPChromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosisNat Neurosci20069110811816369483
- KieranDHafezparastMBohnertSA mutation in dynein rescues axonal transport defects and extends the life span of ALS miceJ Cell Biol2005169456156715911875
- ZhangFStromALFukadaKLeeSHaywardLJZhuHInteraction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complexJ Biol Chem200728222166911669917403682
- Da CruzSClevelandDWUnderstanding the role of TDP-43 and FUS/TLS in ALS and beyondCurr Opin Neurobiol 20112011216904919
- PolymenidouMLagier-TourenneCHuttKRLong pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43Nat Neurosci201114445946821358643
- Van DeerlinVMLeverenzJBBekrisLMTARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysisLancet Neurol20087540941618396105
- KuhnleinPSperfeldADVanmassenhoveBTwo German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutationsArch Neurol20086591185118918779421
- SreedharanJBlairIPTripathiVBTDP-43 mutations in familial and sporadic amyotrophic lateral sclerosisScience200831958701668167218309045
- RutherfordNJZhangYJBakerMNovel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosisPLoS Genet200849e100019318802454
- KabashiEValdmanisPNDionPTARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosisNat Genet200840557257418372902
- NeumannMSampathuDMKwongLKUbiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosisScience2006314579613013317023659
- AraiTHasegawaMAkiyamaHTDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosisBiochem Biophys Res Commun2006351360261117084815
- MaekawaSLeighPNKingATDP-43 is consistently co-localized with ubiquitinated inclusions in sporadic and Guam amyotrophic lateral sclerosis but not in familial amyotrophic lateral sclerosis with and without SOD1 mutationsNeuropathology200929667268319496940
- MackenzieIRBigioEHIncePGPathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutationsAnn Neurol200761542743417469116
- VanceCRogeljBHortobagyiTMutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6Science200932359181208121119251628
- NeumannMMackenzieIRRademakersRTDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementiaLancet Neurol2010910995100720864052
- DengHXZhaiHBigioEHFUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosisAnn Neurol201067673974820517935
- KwiatkowskiTJJrBoscoDALeclercALMutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosisScience200932359181205120819251627
- DormannDRoddeREdbauerDALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear importEMBO J201029162841285720606625
- SnowdenJSHuQRollinsonSThe most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS geneActa Neuropathol201112219911021424531
- NeumannMBentmannEDormannDFET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutationsBrain2011134Pt 92595260921856723
- NeumannMRademakersRRoeberSBakerMKretzschmarHAMackenzieIRA new subtype of frontotemporal lobar degeneration with FUS pathologyBrain2009132Pt 112922293119674978
- WoulfeJGrayDAMackenzieIRFUS-immunoreactive intra-nuclear inclusions in neurodegenerative diseaseBrain Pathol201020358959719832837
- MaruyamaHMorinoHItoHMutations of optineurin in amyotrophic lateral sclerosisNature2010465729522322620428114
- SwarupGNagabhushanaAOptineurin, a multifunctional protein involved in glaucoma, amyotrophic lateral sclerosis and antiviral signallingJ Biosci201035450150521289430
- ZhuGWuCJZhaoYAshwellJDOptineurin negatively regulates TNFalpha- induced NF-kappaB activation by competing with NEMO for ubiquitinated RIPCurr Biol200717161438144317702576
- YingHShenXParkBYueBYPosttranslational modifications, localization, and protein interactions of optineurin, the product of a glaucoma genePLoS One201052e916820161783
- AnborghPHGodinCPampilloMInhibition of metabotropic glutamate receptor signaling by the huntingtin-binding protein optineurinJ Biol Chem200528041348403484816091361
- WildPFarhanHMcEwanDGPhosphorylation of the autophagy receptor optineurin restricts Salmonella growthScience2011333603922823321617041
- ItoHHFujitaKNakamuraMOptineurin is co-localized with FUS in basophilic inclusions of ALS with FUS mutation and in basophilic inclusion body diseaseActa Neuropathol2011121455555721327942
- HortobagyiTTroakesCNishimuraALOptineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disordersActa Neuropathol2011121451952721360076
- DengHXBigioEHZhaiHDifferential involvement of optineurin in amyotrophic lateral sclerosis with or without SOD1 mutationsArch Neurol20116881057106121825243
- HuangLHuangYGuoHDominant expression of angiogenin in NeuN positive cells in the focal ischemic rat brainJ Neurol Sci20092851–222022319666176
- GreenwayMJAndersenPMRussCANG mutations segregate with familial and sporadic amyotrophic lateral sclerosis. Nat GenetApr2006384411413
- WuDYuWHKishikawaHAngiogenin loss-of-function mutations in amyotrophic lateral sclerosisAnn Neurol200762660961717886298
- PrehnJHKieranDSebastiaJControl of motoneuron survival by angiogeninJ Neurosci20082852140561406119109488
- HuGFKishimotoKLiuSMTsujiTOlsonKAEndogenous angiogenin in endothelial cells is a general requirement for cell proliferation and angiogenesisOncogene200524344545615558023
- CrabtreeBThiyagarajanNPriorSHCharacterization of human angiogenin variants implicated in amyotrophic lateral sclerosisBiochemistry20074642118101181817900154
- HongSBSiddiqueTX-linked dominant locus for late-onset familial amyotrophic lateral sclerosisSoc Neurosci Abst199824478
- LeeTLiYRIngreCAtaxin-2 intermediate-length poly-glutamine expansions in European ALS patientsHum Mol Genet20112091697170021292779
- FectoFSiddiqueTMaking connections: pathology and genetics link amyotrophic lateral sclerosis with frontotemporal lobe dementiaJ Mol Neurosci201145366367521901496
- KabashiEBercierVLissoubaAFUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosisPLoS Genet201178e100221421829392
- MackenzieIRNeumannMRademakersRRoeberSBakerMKretzschmarHAA new subtype of frontotemporal lobar degeneration with FUS pathologyBrain20091322922293119674978
- LiuYHTianTHypothesis of optineurin as a new common risk factor in normal-tension glaucoma and Alzheimer’s diseaseMed Hypotheses201177459159221764520
- del ToroDAlberchJLazaro-DieguezFMutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineurin/Rab8 complex from the Golgi apparatusMol Biol Cell20092051478149219144827
- RezaieTChildAHitchingsRAdult-onset primary open-angle glaucoma caused by mutations in optineurinScience200229555571077107911834836
- AlbaghaOMViscontiMRAlonsoNGenome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of boneNat Genet201042652052420436471
- ShirakashiYKawamotoYTomimotoHTakahashiRIharaMAlpha-synuclein is colocalized with 14-13-3 and synphilin-1 in A53T transgenic miceActa Neuropathol2006112668168916957925
- van BlitterswijkMLandersJERNA processing pathways in amyotrophic lateral sclerosisNeurogenetics201011327529020349096
- GatchelJRZoghbiHYDiseases of unstable repeat expansion: mechanisms and common principlesNat Rev Genet200561074375516205714
- BourkeSCTomlinsonMWilliamsTLBullockREShawPJGibsonGJEffects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trialLancet Neurol20065214014716426990
- BowserRTurnerMRShefnerJBiomarkers in amyotrophic lateral sclerosis: opportunities and limitationsNat Rev Neurol201171163163821989244
- KasaiTTokudaTIshigamiNIncreased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosisActa Neuropathol20091171556218989684
- FouldsPMcAuleyEGibbonsLTDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer’s disease and frontotemporal lobar degenerationActa Neuropathol2008116214114618506455
- WilsonMEBoumazaILacomisDBowserRCystatin C: a candidate biomarker for amyotrophic lateral sclerosisPLoS One2010512e1513321151566
- GanesalingamJAnJShawCEShawGLacomisDBowserRCombination of neurofilament heavy chain and complement C3 as CSF biomarkers for ALSJ Neurochem2011117352853721418221
- SwashMEarly diagnosis of ALS/MNDJ Neurol Sci1998160Suppl 1S33S369851646
- BensimonGLacomblezLMeiningerVA controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study GroupN Engl J Med199433095855918302340
- DobleAThe pharmacology and mechanism of action of riluzoleNeurology1996476 Suppl 4S233S2418959995
- Garnock-JonesKPDextromethorphan/quinidine: in pseudobulbar affectCNS Drugs201125543544521476614
- RothsteinJDPatelSReganMRBeta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expressionNature20054337021737715635412
- IzumiYKajiRClinical trials of ultra-high-dose methylcobalamin in ALSBrain Nerve200759101141114717969354
- HemendingerRAArmstrongEJ3rdBrooksBRMethyl Vitamin B12 but not methylfolate rescues a motor neuron-like cell line from homocysteine-mediated cell deathToxicol Appl Pharmacol2011251321722521237187
- CheahBCKiernanMCDexpramipexole, the R(+) enantiomer of pramipexole, for the potential treatment of amyotrophic lateral sclerosisI Drugs2010131291192021154151
- TurnerBJTalbotKTransgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALSProg Neurobiol20088519413418282652
- SwarupVJulienJPALS pathogenesis: recent insights from genetics and mouse modelsProg Neuropsychopharmacol Biol Psychiatry201135236336920728492
- TudorELGaltreyCMPerkintonMSAmyotrophic lateral sclerosis mutant vesicle-associated membrane protein-associated protein-B transgenic mice develop TAR-DNA-binding protein-43 pathologyNeuroscience2010167377478520188146
- GordonPHMeiningerVHow can we improve clinical trials in amyotrophic lateral sclerosis?Nat Rev Neurol201171165065421947135
- KalmarBNovoselovSGrayACheethamMEMargulisBGreensmithLLate stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALSJ Neurochem2008107233935018673445
- RalphGSRadcliffePADayDMSilencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS modelNat Med200511442943315768029
- RaoulCAbbas-TerkiTBensadounJCLentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALSNat Med200511442342815768028
- MillerTMSmithRAYamanakaKAntisense oligo-nucleotide therapy for neurodegenerative diseaseJ Clin Invest200611682290229616878173
- TakeuchiSFujiwaraNIdoAInduction of protective immunity by vaccination with wild-type apo superoxide dismutase 1 in mutant SOD1 transgenic miceJ Neuropathol Exp Neurol201069101044105620838241
- HallewellRALariaITabriziAGenetically engineered polymers of human CuZn superoxide-dismutase. Biochemistry and serum half-livesJ Biol Chem19892649526052682647749
- TainerJAGetzoffEDAlexanderHThe reactivity of anti-peptide antibodies is a function of the atomic mobility of sites in a proteinNature198431259901271346209578
- TainerJAGetzoffEDPatersonYOlsonAJLernerRAThe atomic mobility component of protein antigenicityAnnu Rev Immunol198535015352415142
- Haidet-PhillipsAMHesterMEMirandaCJAstrocytes from familial and sporadic ALS patients are toxic to motor neuronsNat Biotechnol201129982482821832997
- MaragakisNJLeporeACRauckBFocal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron diseaseNature Neuroscience200811111294130118931666
- XuLYYanJChenDHuman neural stem cell grafts ameliorate motor neuron disease in SOD-1 transgenic ratsTransplantation200682786587517038899
- CharcotJMJofforyATwo cases of progressive muscular atrophy with lesions in the gray matter and in the anterolateral bundles of the spinal cord [Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux anterolateraux de la moelle epiniere]Arch Physiol Neurol Pathol 21869744754 French
- PerryJJFanLTainerJADeveloping master keys to brain pathology, cancer and aging from the structural biology of proteins controlling reactive oxygen species and DNA repairNeuroscience200714541280129917174478
- DormannDHaassCTDP-43 and FUS: a nuclear affairTrends Neurosci201134733934821700347
- ALS online genetics database Available from: http://alsod.iop.kcl.ac.uk/alsAccessed December 28, 2011
- Genetic Association studies Available from: http://www.alsgene.orgAccessed December 28, 2011
- ALS forum Available from: http://www.researchals.orgAccessed December 28, 2011
- Northeast ALS Consortium Available from: http://www.alsconsortium.orgAccessed December 28, 2011
- ALS untangled Available from: http://www.alsuntangled.comAccessed December 28, 2011