Abstract
Over the last decade, new medical treatment modalities have emerged based on increased insights into amyloid formation. With the increased possibilities for treatment of amyloidosis caused by transthyretin (TTR) amyloid deposits comes the need for diagnostic procedures for early diagnosis and better tools to follow disease progression. This is of particular importance in clinical trials evaluating the efficacy of new treatments. Until recently, the treatment of TTR amyloidosis (ATTR) was based solely on liver transplantation, a procedure that has halted disease progression in many patients. Liver transplantation has been especially effective in patients under the age of 50 years carrying the TTR V30M mutation, whereas the outcome of the procedure has been variable for others, particularly elderly male patients and those carrying a non-V30M mutation. This review concentrates on new insights derived from our center’s experience with liver transplantation, how to implement this experience in evaluation of new treatment modalities for ATTR, and how to facilitate early diagnosis of neuropathy with easily available diagnostic tools. Attention has focused on manifestations of the disease that involve the heart and the peripheral nervous system; change in peripheral nerve function has been the primary endpoint in two controlled clinical trials, one finished and one ongoing. New insights into the amyloid formation process and the lessons learned from liver transplantation give the opportunity to design potentially effective treatment modalities for ATTR. It appears reasonable to suspect that a combination of different treatment modalities may be required to treat the disease, and that different treatment regimes will be designed according to the phenotype of the disease. For the patients and their relatives there is now a solid foundation for optimism, with prospects of several effective medical treatment possibilities within the coming decade.
Introduction
Transthyretin (TTR) amyloid contains misfolded TTR that has assembled into beta-pleated sheets: amyloid fibrils. It is generally believed that mutations in the TTR gene alter the stability of the TTR tetramer, leading to formation of misfolded monomers that assemble into amyloid fibrils.Citation1,Citation2 More than 100 amyloidogenic mutations have been identified,Citation3,Citation4 with the most common probably being the V122I, which has a prevalence of close to 4% in the African-American population.Citation5 The V122I mutation gives rise to cardiomyopathy caused by amyloid deposition in the myocardium, whereas another common mutation, V30M, is primarily associated with a peripheral neuropathy. However, it should be noted that wild-type TTR can assemble into amy-loid, giving rise to cardiomyopathy that generally occurs after the seventh decade of life and with a male predominance.Citation6 Interestingly, in the vervet monkey the V122I mutation has been found in aged animals that developed cardiomyopathy and heart failure; thus, the V122I mutation is naturally occurring in humans and monkeys, and it causes cardiomyopathy in both species.Citation7
The clinical presentation of TTR amyloidosis (ATTR) is extremely varied, not only between mutations but also between different geographic populations carrying the same mutation.Citation8–Citation10 In addition, variation in phenotype within a geographic area has been described for the V30M mutation.Citation11,Citation12 The disease is relentlessly progressive and the survival rate is generally between 10 and 15 years.Citation13–Citation16
Since liver transplantation was introduced as a treatment for the disease, knowledge has expanded further. It has become clear that no improvement has been noted for certain mutations after liver transplantation, especially in patients with symptoms from the central nervous system (CNS) and those whose foremost complications involve the heart, whereas for other mutations, particularly the V30M mutation in younger patients, the progress of the disease has been halted.Citation17–Citation26 In patients with TTR amyloid cardiomyopathy, combined liver and heart transplantations have been performed with encouraging results.Citation27,Citation28 Considering the risk that the patient takes by undergoing a liver transplantation, the failure to halt disease progression in many cases, the shortage of organs available for transplantation, and the need for transplant patients to be on lifelong immune suppression therapy, the need for effective medical treatments is conspicuous.
Evaluation of the efficacy of a treatment must rely on outcome measurements. To achieve reliable outcome measurements in patients with ATTR has proven to be difficult: the disease has a very variable phenotype, and improved survival – the ultimate measurement of efficacy – takes considerable time to validate in a disease with an expected survival rate of 10 years or more. To overcome this lack of validated outcome measures in ATTR, recent double-blind studies of a TTR-stabilizing agent, tafamidis meglumine, have employed rating scales developed for scoring of neuropathy in diabetes mellitus as markers of disease progression.Citation29,Citation30 However, a neurological scale is probably not a useful tool for patients with dominant cardiomyopathy.
This review will concentrate on new insights into amyloid fibril composition and its impact on the disease phenotype, derived from our center’s experience with ATTR amyloidosis. In addition, medical treatment and tools for early recognition of neurological impairment will be discussed.
The authors performed a search for published papers within the area of ATTR and its complications, including cardiomyopathy, autonomic and peripheral neuropathy, and liver transplantation: the search tools used were PubMed and Web of Science.
New insights into amyloid fibril composition
Histopathological examination of amyloid deposits has revealed the existence of two different forms of TTR deposits: one with a strong congophilic reaction and the other with a weaker affinity for Congo red.Citation31,Citation32 The weak staining has been noted for patients with wild-type ATTR but has also been noted for some V30M patients. Further analysis by Western blot showed that the differences in affinity for Congo red was associated with differences in fibril composition: full-length TTR amyloid fibrils were associated with a strong affinity for Congo red, whereas weak affinity was noted in deposits containing a mixture of full-length and truncated TTR amyloid fibrils.Citation31,Citation33 Examination of the fibril composition from different organs showed that the composition was similar in all tissues from the same individual. Therefore, fibril composition in material obtained from subcutaneous fat was similar to that found in the heart.Citation33 Importantly, the difference could be linked to the phenotype of the disease, where the mixed fibril type was found in late-onset V30M patients as well as in senile systemic amyloidosis patients, and the full-length TTR fibrils were found in early-onset V30M patients. In addition, the mixed fibril type was associated with cardiomyopathy, whereas full-length fibrils were found in patients where neuropathy dominated the clinical picture.Citation33
Since cardiomyopathy has been a problem after liver transplantation in late-onset V30M patients, especially males, who have a phenotype resembling that of senile systemic amyloidosis, the possibility that amyloid fibril composition may have an effect on posttransplant cardiomyopathy has been explored. Analysis of amyloid deposits in transplant patients showed that wild-type TTR was rapidly incorporated in TTR amyloid deposits in patients with mixed fibrils, whereas this process was significantly slower in patients with full-length fibrils.Citation34 Deterioration in heart function after transplantation was observed for patients with mixed fibrils, whereas no such deterioration was noted for patients with full-length fibrils.Citation21 In addition, mixed fibrils were found in T60A amyloidosis patients and liver transplantation has not been successful for this mutation, with a rapid development of cardiomyopathy after the procedure.Citation35 Thus, amyloid fibril composition appears to be related to outcome following liver transplantation for ATTR. The individual’s type of amyloid fibril composition remains the same over time and is not changed by transplantation.Citation34
The differences in fibril composition raise some questions: is the amyloid formation process in ATTR the same for the two different forms of fibrils? Are differences in outcome of treatment modalities related to fibril composition? These questions should be taken into consideration in future evaluations of treatment modalities.
Clinical expressions of ATTR
Variations in phenotype
ATTR is often divided into two phenotypes, characterized by the main targeted organ: (1) the heart leading to a hypertrophic/restrictive cardiomyopathy or (2) the peripheral nervous system leading to neuropathy. However, mutations with oculoleptomeningeal amyloidosis with symptoms from the CNS have also been described.Citation36–Citation39 The vast variation in ATTR is intriguing, and currently there is no explanation why different organs are targeted in different mutations.
V30M, one of the best-studied mutations, has displayed an unexpected variation in presenting symptoms and complications as well as variation in age of disease onset.Citation8,Citation10,Citation11,Citation40 For Swedish patients, the median age at onset is 56 years,Citation41 whereas for Portuguese, Brazilian, and Japanese patients, the onset in the endemic areas has been reported to be in the third decade of life.Citation13,Citation14 It was originally suspected that all V30M patients share a common Portuguese founder, but haplotype analysis has clearly shown that all Swedish patients share a common Swedish founder.Citation42 Reports from Japan, Portugal, and France have described cases outside the endemic areas with a phenotype characterized by late onset (over 50 years of age) and often suffering from cardiomyopathy in addition to neuropathy.Citation10,Citation43 Even differences in nerve fiber losses have been noted between early- and late-onset cases: a predominance of small-fiber loss has been noted in early-onset cases while late-onset cases have been characterized by relative preservation of unmyelinated fibers.Citation44 A study of Portuguese early- and late-onset cases has suggested that differences in genes coupled to proteins are associated with amyloid deposits, and an analysis of mitochondrial haplogroups has shown differences between early- and late-onset Swedish patients.Citation43,Citation45 Differences in maternally inherited mitochondrial DNA were also shown to be a possible explanation for the maternal anticipation noted for Portuguese families with the V30M mutation, but it could not solely explain the maternal anticipation observed in Swedish families with the V30M mutation.Citation46 So far, no genetic factor has been identified that can with certainty be linked to the variation in disease phenotype for V30M patients, or to the variation of phenotypes observed for the different TTR mutations.
Medical treatment for ATTR has been concentrated on V30M patients, with encouraging outcomes found in the tafamidis trial.Citation29 Considering the low prevalence of non-V30M patients, it will probably be difficult to conduct studies targeting other ATTR variants; however, the differences in phenotype even for V30M amyloidosis should be taken into consideration when treatment studies are designed.
Neuropathy
Somatic neuropathy: early screening
Amyloidosis is often associated with peripheral neuropathy, and evaluations of peripheral nerve function bring important contributions to a comprehensive diagnosis of the disease. The most important means for objective assessment of the state of the peripheral nervous system are electroneurography (ENeG) and needle electromyography (EMG), which evaluate the functional state of the large-diameter myelinated nerve fibers. However, the neuropathy found in V30M patients is initially predominantly characterized by affection of small-diameter nerve fibers, with clinical observations of disturbed temperature and pain perception, or signs of organ dysfunctions secondary to impaired autonomic innervation (see below). As electrophysiological testing primarily addresses the function of large myelinated fibers, patients may thus report marked symptoms of sensory impairment without showing any detectable abnormalities in ENeG or EMG assessment, occasionally even for periods of several years. This may critically delay the diagnosis and may lead to pronounced nerve damage before the patient receives adequate therapy.
The evaluation of peripheral small-diameter myelinated or unmyelinated fibers is more cumbersome, as there are no methods for direct recording from these fibers readily available for clinical routine use. Quantitative evaluation has to be achieved indirectly by studies of the effects of thin-diameter fiber on the target organsCitation47,Citation48 that require advanced equipment and which, because of this requirement, are not suitable for widespread screening of patients with minor symptoms. Likewise, the elaborate routines needed may not be mastered at nonspecialized centers and would be difficult to utilize for follow-up purposes at most centers. Skin biopsy for nerve density quantification has been utilized in ATTR patients with advanced neuropathy, and has been found to correlate with disease severity.Citation49 However, the method has not been tested for early diagnosis of ATTR neuropathy and it requires neuropathological expertise not readily available at most centers. In addition, decreased blood flow in peripheral tissues of ATTR patients with ensuing impaired wound healing makes the method less attractive.Citation50
Another well-established rationale for early identification of small-diameter fiber polyneuropathy is thermal quantitative sensory testing (QST), which is based on the patient’s thermal detection capacity – as determined by psychophysical assessments of the thermal perception thresholds (TPTs).Citation51–Citation59 Such testing is routinely performed at specialist clinics, but a more general use of thermal QST in patients with possible amyloidosis has been hampered by the fact that a TPT is dependent on a series of compound underlying somatosensory mechanisms, ranging from the peripheral sensors over the ascending pathways to the central processes contributing to thermal perception.Citation60,Citation61 Several alternatives have been suggested for thermal QST, essentially divided into reaction-time-inclusive and reaction-time-exclusive methods.Citation52,Citation62 For practical reasons, the reaction-time-inclusive “method of limits” (MLI) can be recommended for clinical use, as it comprises a quick, reliable, and easily adopted rationale well understood by both examiners and patients.Citation62–Citation69
When tested with the MLI, a group of Swedish ATTR patients with genetically and biopsy-verified disease, having clinical symptoms of polyneuropathy but lacking electro-physiological signs of nerve fiber abnormalities, showed significantly increased perception thresholds for cold and warmth compared with controls. The patients also showed more abnormal thresholds at distal test sites than the controls (see ).Citation70 These findings are concordant with the notion of a small-diameter fiber involvement early in the course of the disease. However, it is important to note that the QST must be performed bilaterally in the legs and arms, and preferably with both distal and proximal test locations, at least in the legs. Such a testing strategy is crucial for a firm interpretation of the test results, as asymmetric alterations in peripheral sensory detection capacity may also be caused by other nerve affections, such as impingements of peripheral nerves or spinal roots,Citation71,Citation72 or by referred sensations originating from altered segmental sensory processing.Citation73 Hence, to confirm a putative small-diameter fiber neuropathy, the QST should indicate bilateral threshold elevations with distal predominance, paralleling the clinically characteristic “stocking and glove” distribution of the sensory disturbances. Such alterations in TPTs of the extremities therefore strongly corroborate the presence of small-diameter peripheral neuropathy or a debuting generalized peripheral neuropathy.
Figure 1 Box plots of thermal thresholds in a group of transthyretin V30M amyloidosis patients with no sign of electrophysiological abnormalities (familial amyloid polyneuropathy [FAP]; n = 23) and a reference group (controls; n = 43), at the dorsum of the foot (DF), medial (ML) and lateral (LL) parts of the lower leg, and ventral part of the thigh (VT). Thermal thresholds are expressed as change in degrees Celsius (absolute values) from an adapted starting temperature of 32°C.
Figure reproduced from Heldestad and Nordh,Citation70 with permission from the publisher.
Abbreviations: CT, cold threshold; NS, not significant; WT, warm threshold. *P < 0.05; **P < 0.01; ***P < 0.001
![Figure 1 Box plots of thermal thresholds in a group of transthyretin V30M amyloidosis patients with no sign of electrophysiological abnormalities (familial amyloid polyneuropathy [FAP]; n = 23) and a reference group (controls; n = 43), at the dorsum of the foot (DF), medial (ML) and lateral (LL) parts of the lower leg, and ventral part of the thigh (VT). Thermal thresholds are expressed as change in degrees Celsius (absolute values) from an adapted starting temperature of 32°C.Figure reproduced from Heldestad and Nordh,Citation70 with permission from the publisher.](/cms/asset/6bb6fb17-67f1-43f9-9ac5-c2d1d5f08c76/dnnd_a_24652_f0001_b.jpg)
In a general outpatient or nonspecialist center setting where screening of unselected patient materials is available, a more basic approach must be adopted. To this end, testing of temperature discrimination with two traditional metal cylinders, one cold and one warm, rolled over the skin bilaterally at distal and proximal test sites is suggested. The cylinders should have suggested suprathreshold stimulus temperatures of 25°C (cold) and 40°C (warm), and the patient should be blinded to the presentation and repetition order.
QST utilizing the method of MLI appears to be attractive for the early diagnosis of neuropathy in ATTR, since it detects abnormalities before ENeG and EMG. It should also be suited for follow-up purposes in clinical trials. So far, no improvement of neurological impairment has been noted in transplant patients, either by QST or by electrophysiological testing.
Autonomic neuropathy
Autonomic disturbances are commonly encountered in ATTR and may be the presenting manifestation of the disease. However, measurement of the autonomic nervous system is challenging, and few methods are available that offer the possibility for early detection of autonomic neuropathy or of grading the disturbances. The intricate direct intraneural micrographic recordings of autonomic nerve fiber activity,Citation74 laboratory evaluation of adrenergic “spillover,”Citation75 and detailed quantitative evaluations of sweatingCitation47,Citation76,Citation77 are not readily available for clinical use but have shown promising outcomes for diagnosis of amyloidosis-related autonomic neuropathy in a study by the Mayo Clinic.Citation78 However, the widely used galvanic skin response (or somatosensory evoked response) test is less reliable, as it cannot be used for the grading of autonomic activity – it can only inform whether or not a galvanic skin response is present.
Analysis of heart rate variability (HRV) has proven to be a valuable method for detecting and measuring autonomic impairment in ATTR patients.Citation79–Citation82 Two different method ologies have been used for evaluating cardiac autonomic function before and after liver transplantation for ATTR: (1) as a short-term HRV recording (up to 30 minutes) in a controlled laboratory environment with the patient placed on a manually operated tilt tableCitation83 or (2) as a long-term HRV recording using 24-hour electrocardiographic monitoring (Holter monitoring).Citation79,Citation80 Studies of Swedish patients have not shown any improvement in cardiac autonomic function after liver transplantation, but studies have also shown that the development of cardiac arrhythmias continues after liver transplantation.Citation79,Citation80 The latter is a major confounder when HRV recordings are evaluated in ATTR patients, where it is presumed that the heart rate fluctuations originate from activity in the autonomous nervous system. The present authors have found that many patients present subtle atrial arrhythmias that lead to increased HRV, falsely indicating normal autonomic function.Citation82,Citation84,Citation85 Therefore, all HRV recordings in ATTR patients must be carefully investigated to detect subtle atrial arrhythmias.
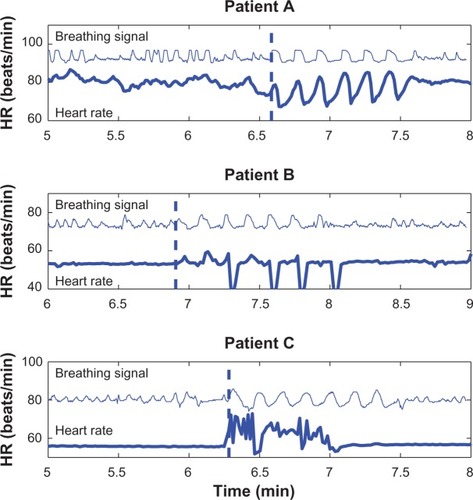
In the tafamidis study of V30M amyloidosis patients, the deep breathing test was selected as an integrated part of a scoring system for nerve function. The test is performed as a short sequence with controlled deep breathing at a rate of six breaths per minute, but since no previous study has presented any data from this test in ATTR patients, the reliability of the test is questionable. shows three examples of heart rate responses during the deep breathing test taken from the authors’ database of recordings in Swedish patients. Patient A showed the expected response during deep breathing with the same fluctuations in heart rate as in the breathing signal (six cycles per minute), although there were only small respiratory-related fluctuations in heart rate during spontaneous breathing. Patient B had even more reduced HRV during spontaneous breathing but presented supraventricular escape beats during deep breathing, resulting in large fluctuations in heart rate that at first glance appeared to be related to the breathing pattern. Finally, patient C also lacked HRV during spontaneous breathing but presented an irregular atrial arrhythmia during deep breathing. The authors have observed this pattern in many ATTR patients – the deep breathing test triggers arrhythmias. Therefore, interpretation of HRV recordings and the subsequent analysis require good knowledge of the methods and its limitations, particularly in patients presenting high indices of HRV.
Figure 2 Recordings of heart rate (HR) variability patterns in three transthyretin amyloidosis patients during spontaneous and deep breathing, where the recordings were performed in the supine position. Patient A showed the normal response in HR during deep breathing, whereas patients B and C presented marked increases in HR variability because of cardiac arrhythmia during the test.
Recently, a marked overshoot in blood pressure was observed after tilt reversal in ATTR patients.Citation86 The study was based on 15 ATTR patients and 14 healthy subjects, showing a significant inverse relationship between blood pressure overshoot and HRV, as well as an overshoot in four of the five patients who had arrhythmia. Thus, this investigation could provide an alternative method for the assessment of autonomic neuropathy in patients with cardiac arrhythmia.Citation86 Finally, in addition to autonomic dysregulation, HRV abnormalities can also be found in patients with cardiac amyloidosis.Citation87
The heart
Heart complications are important in ATTR diseases. They consist primarily of a restrictive-hypertrophic cardiomyopathy caused by amyloid deposits in the myocardium and/or conduction disturbances caused by amyloid infiltrations in the conductive system of the heart. In addition, atrial and even ventricular arrhythmias may develop.
Cardiomyopathy
It is not unusual that patients under evaluation for ATTR present with increased wall thickness on echocardiographic examination but do not show any symptoms consistent with heart failure.Citation88 Cardiomyopathy is more often encountered in elderly patients, particularly in males, or in patients with non-V30M mutations.Citation12,Citation89
It is difficult to use two-dimensional (2-D) echocardiography to differentiate amyloid disease from other causes of left ventricular hypertrophy, although it may detect signs of amyloid infiltration of the heart. Increased septal and posterior wall thickness, including in the right heart, with abnormal systolic and diastolic function are common findings in amyloid heart disease. Atrial involvement as well as thickened papillary muscles, valve leaflets, and small to moderate increased pericardial effusion should raise the suspicion of amyloid heart disease ().Citation90,Citation91
Figure 3 Typical patterns found in echocardiographic examinations in transthyretin amyloidosis patients.
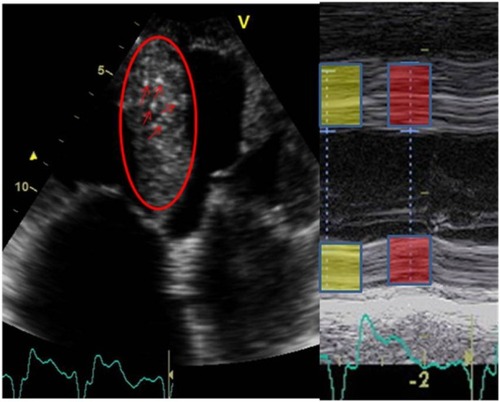
The differentiation between cardiac amyloidosis and hypertrophic cardiomyopathy has clinical implications, since heart transplantation and new medical treatments are options for patients with TTR amyloid heart disease. St John Sutton et alCitation92 found reduced radial systolic wall motion and reduced wall thickening during systole were highly specific for amyloid heart disease. Amyloid heart disease is also characterized by a high degree of highly reflective echoes that corresponds to nodules containing amyloid or a mixture of collagen and amyloid.Citation90,Citation91,Citation93,Citation94
Examination of the heart function by 2-D strain measurements disclosed impaired heart function in V30M patients, even before increased wall thickness or any restrictive filling pattern were present,Citation95 but 2-D strain measurements was unable to differentiate between hypertrophic or amyloid cardiomyopathy.Citation96
Global strain measurement of the heart function, derived from speckle tracking, was used to compare the heart function of patients with full-length TTR amyloid fibrils and patients with mixed fibril type after liver transplantation, and deterioration in heart function was able to be identified in the latter group.Citation21 In addition, since the method is largely based on a computerized imaging program, it is not influenced by intraindividual variations to the same extent as manual measurements of interventricular septal thickness, which makes it attractive for follow-up studies of treatment modalities.
Three-dimensional (3-D) echocardiography is a technique that provides assessment of volumes, dyssynchrony, and 3-D strain. Migrino et alCitation97 used 3-D echocardiography to compare patients with light-chain (AL) amyloidosis and healthy controls, and this showed left ventricular dyssynchrony in the AL amyloidosis patients. This technique still has the limitation of being highly dependent on good image quality, but it is under continual development and could become an important diagnostic tool in the future.
Even though echocardiography with the addition of new techniques should raise the suspicion of amyloid cardio-myopathy and although 2-D speckle tracking measurements appear to be suited for follow-up examinations, additional noninvasive tools are needed.
One very promising technique is scintigraphic examination, either by 99 mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (DPD)Citation98 or by 99 mTc-pyrophosphate.Citation99,Citation100 This technique has proven to have high sensitivity and specificity in the detection of TTR amyloid disease and is an important tool for differentiating between ATTR and other types of cardiomyopathy. However, scintigraphic examinations have not yet been developed for follow-up purposes.
Magnetic resonance investigation of the heart with gadolinium enhancement has been reported to disclose amyloid deposition within the myocardium and to give accurate estimation of heart dimensions. However, it does not appear to be as sensitive as DPD scintigraphyCitation101 for the diagnosis of TTR amyloid deposits, and since it is more costly and cannot be used in patients with pacemakers, its use for diagnosis and follow-up purposes is limited.
Serum N-terminal pro-brain natriuretic peptide has proven to be a reliable indicator of heart involvement in AL amyloidosis, and to be useful for follow-up evaluation of disease treatment.Citation102,Citation103 The analysis could also be well suited for diagnosis and follow-up purposes in ATTR, since it correlates with echocardiographic measurements of heart function. Troponin examination appears to be less useful in TTR amyloid cardiomyopathy than in AL amyloidosis,Citation103 but high-sensitivity troponin may prove to be more useful.Citation104
For diagnosis, the suspicion of amyloid heart disease should arise from echocardiographic examination, and a DPD scintigraphic examination should be able to differentiate between TTR amyloid cardiomyopathy and other forms of cardiomyopathy. Echocardiography with 2-D strain measurements in combination with serum markers such as pro-brain natriuretic peptide are useful for follow-up examinations. Since heart complications are common in ATTR, removal of amyloid deposits for treatment of cardiomyopathy or substantially decreasing TTR levels in the plasma to diminish amyloid formation are attractive options. The benefit of tafamidis or other TTR stabilizers for heart function has not been proven in controlled trials; an open-label study is completed but the outcome is not yet published.
Rhythm disturbances
Amyloid deposits in the heart are the main cause of electro-cardiographic disturbances. Findings include heavy amyloid infiltrations in the sinoatrial node, the atrial musculature, and the atrioventricular conduction system.Citation105,Citation106 Moreover, the heart rhythm can also be affected by the autonomic dysfunction commonly found in ATTR patients.Citation107,Citation108
Different cross-sectional studies, mainly based on conventional electrocardiograms (ECGs), have revealed a high prevalence of conduction disturbances, intermittent atrial and ventricular arrhythmias, and signs of low voltages in the ECGs.Citation109–Citation112 Development of severe arrhythmia is common in V30M patients. A longitudinal study of 12-lead ECG changes in non-transplant ATTR patients over a mean follow-up period of 8 years showed that 19 (31%) of 61 patients required pacemaker implantation during the observation period.Citation109 A study of 24-hour Holter ECG recordings showed a similar development of severe arrhythmia in transplant patients: pacemaker and intracardiac defibrillator implantation was performed in approximately 25% of patients over a median follow-up period of 11 years.Citation113 Therefore, patients who have undergone liver transplantation should be regularly reexamined by Holter monitoring for arrhythmia necessitating pacemaker implantation. It should be noted that the recommendation for pacemaker implantation in ATTR patients deviates from recommendations for other patient groups, since the presence of an autonomic neuropathy diminishes the patient’s ability to react to bradycardia. Even severe conduction disturbances may be asymptomatic, since episodes of bradycardia and/or ventricular tachyarrhythmia are often noted at night when the patient is asleep, increasing the risk of nocturnal sudden death.
The development of conduction disturbances is not coupled with the development of cardiomyopathy,Citation12 probably because the amyloid deposition in the conduction system does not follow deposition in the myocardium.Citation101 The aforementioned subtle arrhythmias may be induced by amyloid deposition in the atrium and be related to cardiomyopathy; nonetheless, supraventricular arrhythmia is predominantly found in elderly patients.Citation12,Citation114
Other organs commonly affected in ATTR
Since ATTR is a systemic disease and amyloid deposits can be found in all organs, a variety of expressions of the disease are described for ATTR, and the outcome for different organ systems after liver transplantation varies, as may the response of these organ systems to medical treatment.
Gastrointestinal
Impaired gastrointestinal function is a common complication in ATTR. The mechanism is not fully understood, but autonomic and enteric nervous system impairments are probably involved.Citation107,Citation115,Citation116 Unintentional weight loss, often before the patient complains of gastrointestinal disturbances, is frequently reported.Citation14 The decline in the patient’s nutritional status, measured by the modified body mass index (mBMI) (body mass index multiplied by serum albumin in grams per liter to compensate for edema) has been utilized.Citation15,Citation117 The mBMI correlates with survival and is also used as a predictor of mortality and morbidity after liver transplantation. It also serves as a secondary endpoint in the tafamidis and diflunisal clinical trials.Citation29,Citation118
No improvement in gastrointestinal function has been noted after liver transplantation,Citation119 but a significantly improved mBMI was noted for patients treated with tafamidis compared with those receiving placebo.Citation29 Thus, this index appears to be well suited for following patients in clinical studies and for evaluating the impact of treatment modalities.
Kidney
Both kidney and urinary bladder dysfunctions are common complications in ATTR. Impaired bladder function was coupled with an increased mortality after liver transplantation in a French study.Citation17 Combined liver and kidney transplantation may be considered in patients with kidney failure.Citation120,Citation121 The kidney function appears to remain stable after liver transplantation.Citation122
An increase in urinary tract infections was noted for patients treated with tafamidis compared with those receiving placebo.Citation29
Eyes
A variety of eye complications are noted in ATTR, of which vitreous opacities and glaucoma are the most important.Citation123 Neither of these two complications subsides after transplantation, since the retina synthesizes TTR.Citation124,Citation125 The development of glaucoma is troublesome, since the damage to the patient’s vision is irreversible, and the impairment of vision can be substantial before the patient notes it.
The impact of tafamidis on eye complications has not yet been examined. However, the efficacy of the treatment depends on the compound’s ability to travel into the eye and exert an effect in the intraocular environment. Again, this has not yet been examined for tafamidis, and neither antisense nor silencing RNA should penetrate from the circulation into the eye and affect TTR production.
Carpal tunnel syndrome
A peculiar feature of ATTR is the high incidence of carpal tunnel syndrome – this may occur before other symptoms of amyloid disease.Citation3,Citation126 It is often bilateral but, considering the high incidence of carpal tunnel syndrome in the general population, an active search for systemic amyloidosis, in the absence of other symptoms, is not indicated. However, for a patient with peripheral neuropathy or cardiomyopathy of unknown etiology, a history of carpal tunnel syndrome should raise the suspicion of ATTR. In addition, carpal tunnel syndrome is often found in patients with the rare oculoleptomeningeal form of ATTR.Citation127
CNS symptoms
CNS symptoms are rarely found in ATTR. However, they dominate in a few mutations and often resemble the symptoms of a minor stroke or transient ischemic attack.Citation5,Citation35 Dementia, seizures, subarachnoid hemorrhage, ataxia, myelopathy, and deafness may also present as symptoms. Vitreous opacities are often an accompanying complication and are often referred to as the oculoleptomeningeal form of ATTR. On cerebral magnetic resonance examination, the amyloid deposits can be found in the meningeal space.Citation3,Citation36
The effect of liver transplantation has generally been disappointing for mutations with CNS engagement and so the procedure is not recommended. No patients with CNS manifestations have been included in clinical trials.
Treatments for ATTR currently approved or on the horizon
TTR stabilizers
The suggestion that TTR amyloid was formed from misfolded monomers raised the question whether stabilization of the TTR tetramer could prevent amyloid formation, and in vitro studies showed that this appeared to be the case.Citation1 Nonsteroidal anti-inflammatory drugs appeared to be effective, but only one of the tested compounds, diflunisal, had a strong affinity for TTR – diflunisal is currently being evaluated in a controlled clinical trial.Citation1 Tafamidis, a new compound with strong binding affinity and stabilizing properties for TTR, has recently been approved in the European Union for the treatment of ATTR with neuropathy at an early stage of the disease (stage I: patient can walk without support by a stick or crutch). Tafamidis showed disease-modifying properties in an 18-month double-blind clinical trial, followed by an open-label 12-month extension study, through a diminished progression rate of neuropathy in the lower limb (as measured by the Neuropathy Impairment Score in the Lower Limbs), lower deterioration in quality of life for the patient (as measured by the Norfolk Questionnaire), and significantly improved nutritional status of the patient (as measured by the mBMI).Citation29 The patients included in the trial were predominantly early-onset cases, a group of patients where full-length TTR amyloid fibrils are most commonly found; therefore, it is not yet known if the response is different for late-onset patients or for patients with other mutations, as has been the case for liver transplantation.
Treatment targeting TTR synthesis
Since TTR amyloid can be formed from wild-type TTR, as shown to be the case after liver transplantation, treatments aimed to decrease the amount of amyloidogenic protein (ie, variant and wild-type TTR) appear attractive. From transgenic mice experiments, TTR in the cerebral spinal fluid appears to be necessary for normal brain function,Citation128 but outside the CNS, TTR is not required to sustain a normal function of vitamin A or thyroid hormone.Citation129
To effectively decrease TTR synthesis, two different approaches have been taken: (1) silencing RNA, which interferes with TTR synthesis in the ribosome, and (2) antisense nucleotide, which prevents transcription of messenger RNA in the nucleus. Both methods have been utilized in phase I trials and have shown the ability to significantly decrease TTR synthesis.Citation130,Citation131
Treatment aimed at removing amyloid deposits
Serum amyloid P component (SAP) is found in all amyloid deposits and it appears to protect amyloid from degradation. Recently, an antibody to SAP was shown to be able to degrade amyloid deposits in an amyloid mouse model, and a compound that effectively depressed SAP synthesis in patients with fibrinogen amyloidosis has been developed – CPHPC (R-1-[6-[R-2-carboxy-pyrrolidin-1-yl]-6-oxohexanoyl] pyrrolidine-2-carboxylic acid).Citation132,Citation133 Treatment with a combination of these two compounds may be effective for all types of amyloid diseases.
Doxycycline is another compound with the ability to dissolve amyloid deposits.Citation134 The European Committee for Orphan Medicinal Products has recently designated doxycy-cline for the treatment of hereditary amyloid polyneuropathy (http://www.emea.europa.eu/docs/en_GB/document_library/Orphan_designation/2012/05/WC500127736.pdf). The ability of doxycycline to treat ATTR is currently being evaluated in a clinical trial (ClinicalTrials.gov identifier NCT01171859), where doxycycline is combined with tauroursodeoxycholic acid. This combination has proven to be effective in a transgenic mouse model of V30M amyloidosis.Citation135
Conclusion
New insights into the amyloid formation process and the lessons learned from liver transplantation give the opportunity to design potentially effective treatment modalities for ATTR.
Early treatment is required – before the patient suffers from severe, irreversible organ damage – and increased awareness of the disease is an important means of enabling this. Improved and easily accessible diagnostic tools provide the possibility of diagnosing the disease soon after onset by less invasive methods. Noninvasive, readily available tests such as thermal threshold assessment and QST are well suited for peripheral neuropathy. Autonomic neuropathy can be assessed by analysis of beat-to-beat fluctuations in R-R intervals. However, for ATTR patients in whom atrial arrhythmias are common, the risk of misinterpretation of the outcome should be taken into consideration, especially in poorly validated deep breathing tests. This is of special importance, since the test is often incorporated into scoring of nerve function in clinical trials.
Heart complications of ATTR have achieved increased recognition as a major problem. DPD scintigraphy together with new echocardiographic methods such as global strain measurements are generally easily available and should be employed for diagnosis and assessment of heart function.
The TTR-stabilizing agent tafamidis appeared to be unable to halt the progress of the disease, even though it markedly diminished the rate of disease progression. So far the approval of tafamidis is restricted to patients with polyneuropathy in an early stage of the disease. For V30M patients with an early onset of the disease, liver transplantation is still a valid treatment option and should be offered to the patients.
The finding of a continuous exchange of amyloid, where variant TTR is rapidly replaced by wild-type TTR in the amyloid deposits after liver transplantation, is encouraging, since it demonstrates that amyloid deposits constantly undergo formation and degradation. If the concentration of the amyloidogenic protein is diminished, the amyloid deposits may subside; this should provide hope for the improvement of patients, especially for those suffering from cardiomyopathy.
It appears reasonable to suspect that a combination of different treatment modalities may be required for the treatment of the disease, and that different treatment regimes will be designed according to the phenotype of the disease.
For ATTR patients and their relatives there is now a solid foundation for optimism, with prospects of several effective medical treatment possibilities within the coming decade.
Acknowledgments
The authors wish to acknowledge the support received from the following: the Swedish Heart and Lung Foundation (OBS and PL), Central and Clinical ALF grants (OBS and RH), Spearhead grant from Västerbotten County (OBS), the Research Foundation of the Department of Pharmacology and Clinical Neuroscience, Umeå University (EN and VH), and FAMY, FAMY Norrbotten, and the Amyl Foundation (OBS, RH, UW, and EN). The award of a visiting professorship at the Rehabilitation Research Chair, King Saud University, Riyadh, Saudi Arabia, to author EN is gratefully acknowledged.
Disclosure
OBS has served as a consultant for Pfizer, Alnylam Pharmaceuticals, and Isis Pharmaceuticals companies. OBS has participated as an investigator in clinical trials with tafamidis, diflunisal, and small interfering RNA. EN received lecture honoraria from Ipsen and Pfizer. The authors have no other conflicts of interest to report.
References
- HammarströmPWisemanRLPowersETKellyJWPrevention of transthyretin amyloid disease by changing protein misfolding energeticsScience2003299560771371612560553
- KellyJWColonWLaiZTransthyretin quaternary and tertiary structural changes facilitate misassembly into amyloidAdv Protein Chem1997501611819338081
- BensonMDKincaidJCThe molecular biology and clinical features of amyloid neuropathyMuscle Nerve200736441142317554795
- ConnorsLHLimAProkaevaTRoskensVACostelloCETabulation of human transthyretin (TTR) variants, 2003Amyloid200310316018414640030
- JacobsonDRPastoreRDYaghoubianRVariant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black AmericansN Engl J Med199733674664739017939
- WestermarkPSlettenKJohanssonBCornwellGG3rdFibril in senile systemic amyloidosis is derived from normal transthyretinProc Natl Acad Sci U S A1990877284328452320592
- UedaMAgeyamaNNakamuraSAged vervet monkeys developing transthyretin amyloidosis with the human disease-causing Ile122 allele: a valid pathological model of the human diseaseLab Invest201292347448422184092
- CoelhoTSousaALourençoERamalheiraJA study of 159 Portuguese patients with familial amyloidotic polyneuropathy (FAP) whose parents were both unaffectedJ Med Genet19943142932998071954
- KoikeHTanakaFHashimotoRNatural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late-onset cases from non-endemic areasJ Neurol Neurosurg Psychiatry201283215215822228785
- Planté-BordeneuveVLaluTMisrahiMGenotypic-phenotypic variations in a series of 65 patients with familial amyloid polyneuropathyNeurology19985137087149748014
- HellmanUAlarconFLundgrenHESuhrOBBonaiti-PelliéCPlanté-BordeneuveVHeterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish populationAmyloid200815318118618925456
- HörnstenRPennlertJWiklundULindqvistPJensenSMSuhrOBHeart complications in familial transthyretin amyloidosis: impact of age and genderAmyloid2010172636820462364
- ArakiSKuriharaTTawaraSKuribayashiTFamilial amyloidotic polyneuropathy in JapaneseGlennerGGPinho e CostaPde FreitasAFAmyloid and AmyloidosisAmsterdamExcerpta Medica19806777
- CoutinhoPda SilvaAMLimaJKBarbosaARForty years of experience with type I amyloid neuropathy: review of 483 casesGlennerGGPinho e CostaPde FreitasAFAmyloid and AmyloidosisAmsterdamExcerpta Medica19798898
- SuhrODanielssonAHolmgrenGSteenLMalnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathyJ Intern Med199423554794858182405
- TashimaKSuhrOBAndoYGastrointestinal dysfunction in familial amyloidotic polyneuropathy (ATTR Val30Met): comparison of Swedish and Japanese patientsAmyloid19996212412910439119
- AdamsDSamuelDGoulon-GoeauCThe course and prognostic factors of familial amyloid polyneuropathy after liver transplantationBrain2000123Pt 71495150410869060
- De CarvalhoMConceiçãoIBentesCLuísMLLong-term quantitative evaluation of liver transplantation in familial amyloid polyneuropathy (Portuguese V30M)Amyloid20029212613312440485
- DubreySWDavidoffRSkinnerMBergethonPLewisDFalkRHProgression of ventricular wall thickening after liver transplantation for familial amyloidosisTransplantation199764174809233704
- García-HerolaAPrietoMPascualSProgression of cardiomyopathy and neuropathy after liver transplantation in a patient with familial amyloidotic polyneuropathy caused by tyrosine-77 transthyretin variantLiver Transpl Surg19995324624810226117
- GustafssonSIhseEHeneinMYWestermarkPLindqvistPSuhrOBAmyloid fibril composition as a predictor of development of cardiomyopathy after liver transplantation for hereditary transthyretin amyloidosisTransplantation201293101017102322395298
- HörnstenRWiklundUOlofssonBOJensenSMSuhrOBLiver transplantation does not prevent the development of life-threatening arrhythmia in familial amyloidotic polyneuropathy, Portuguese-type (ATTR Val30Met) patientsTransplantation200478111211615257048
- OlofssonBOBackmanCKarpKSuhrOBProgression of cardiomyopathy after liver transplantation in patients with familial amyloidotic polyneuropathy, Portuguese typeTransplantation200273574575111907421
- PomfretEALewisWDJenkinsRLEffect of orthotopic liver transplantation on the progression of familial amyloidotic polyneuropathyTransplantation19986579189259565095
- StangouAJHawkinsPNHeatonNDProgressive cardiac amyloidosis following liver transplantation for familial amyloid polyneuropathy: implications for amyloid fibrillogenesisTransplantation19986622292339701270
- SuhrOBHerleniusGFrimanSEriczonBGLiver transplantation for hereditary transthyretin amyloidosisLiver Transpl20006326327610827225
- GraziGLCesconMSalviFCombined heart and liver transplantation for familial amyloidotic neuropathy: considerations from the hepatic point of viewLiver Transpl20039998699212942463
- PilatoEDell’AmoreABottaLArpesellaGCombined heart and liver transplantation for familial amyloidotic neuropathyEur J Cardiothorac Surg200732118018217449267
- CoelhoTMaiaLFMartins da SilvaATafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trialNeurologyIn press2012
- DyckPJMeltonLJ3rdO’BrienPCServiceFJApproaches to improve epidemiological studies of diabetic neuropathy: insights from the Rochester Diabetic Neuropathy StudyDiabetes199746Suppl 2S5S89285491
- BergströmJGustavssonAHellmanUAmyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphologyJ Pathol2005206222423215810051
- KoikeHAndoYUedaMDistinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathyJ Neurol Sci20092871–217818419709674
- IhseEYboASuhrOLindqvistPBackmanCWestermarkPAmyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosisJ Pathol2008216225326118729067
- IhseESuhrOBHellmanUWestermarkPVariation in amount of wild-type transthyretin in different fibril and tissue types in ATTR amyloidosisJ Mol Med (Berl)201189217118021107516
- IhseEStangouAJHeatonNDProportion between wild-type and mutant protein in truncated compared to full-length ATTR: an analysis on transplanted transthyretin T60A amyloidosis patientsBiochem Biophys Res Commun2009379484685019118530
- BlevinsGMacaulayRHarderSOculoleptomeningeal amyloidosis in a large kindred with a new transthyretin variant Tyr69HisNeurology200360101625163012771253
- PetersenRBGorenHCohenMTransthyretin amyloidosis: a new mutation associated with dementiaAnn Neurol19974133073139066351
- SchweitzerKEhmannDGarciaRAlportEOculoleptomeningeal amyloidosis in 3 individuals with the transthyretin variant Tyr69HisCan J Ophthalmol200944331731919491989
- UemichiTUittiRJKoeppenAHDonatJRBensonMDOculoleptomeningeal amyloidosis associated with a new transthyretin variant Ser64Arch Neurol19995691152115510488818
- IkedaSNakazatoMAndoYSobueGFamilial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneityNeurology20025871001100711940682
- SousaAAnderssonRDruggeUHolmgrenGSandgrenOFamilial amyloidotic polyneuropathy in Sweden: geographical distribution, age of onset, and prevalenceHum Hered19934352882948406517
- ZarosCGeninEHellmanUOn the origin of the transthyretin Val30Met familial amyloid polyneuropathyAnn Hum Genet200872Pt 447848418460047
- KoikeHMisuKIkedaSType I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset formArch Neurol200259111771177612433265
- KoikeHMisuKSugiuraMPathology of early- vs late-onset TTR Met30 familial amyloid polyneuropathyNeurology200463112913815249622
- OlssonMHellmanUPlanté-BordeneuveVJonassonJLångKSuhrOBMitochondrial haplogroup is associated with the phenotype of familial amyloidosis with polyneuropathy in Swedish and French patientsClin Genet200975216316819018796
- BonaïtiBOlssonMHellmanUSuhrOBonaïti-PelliéCPlanté-BordeneuveVTTR familial amyloid polyneuropathy: does a mitochondrial polymorphism entirely explain the parent-of-origin difference in penetrance?Eur J Hum Genet201018894895220234390
- GibbonsCHIlligensBMCentiJFreemanRQDIRT: quantitative direct and indirect test of sudomotor functionNeurology200870242299230418541883
- NovakPQuantitative autonomic testingJ Vis Exp2011532502
- YangNCLeeMJChaoCCClinical presentations and skin denervation in amyloid neuropathy due to transthyretin Ala97SerNeurology201075653253820697105
- AndoYYamashitaTTanakaYRole of nitric oxide in the peripheral vessels of patients with familial amyloidotic polyneuropathy (FAP) type IJ Auton Nerv Syst199450179857844317
- DyckPJZimmermanIGillenDAJohnsonDKarnesJLO’BrienPCCool, warm, and heat-pain detection thresholds: testing methods and inferences about anatomic distribution of receptorsNeurology1993438150015088351002
- FruhstorferHLindblomUSchmidtWCMethod for quantitative estimation of thermal thresholds in patientsJ Neurol Neurosurg Psychiatry1976391110711075188989
- GruenerGDyckPJQuantitative sensory testing: methodology, applications, and future directionsJ Clin Neurophysiol19941165685837860720
- JamalGAHansenSWeirAIBallantyneJPAn improved automated method for the measurement of thermal thresholds: 1. Normal subjectsJ Neurol Neurosurg Psychiatry19854843543603998741
- LinYHHsiehSCChaoCCChangYCHsiehSTInfluence of aging on thermal and vibratory thresholds of quantitative sensory testingJ Peripher Nerv Syst200510326928116221286
- RolkeRBaronRMaierCQuantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): standardized protocol and reference valuesPain2006123323124316697110
- SelimMMWendelschafer-CrabbGHodgesJSVariation in quantitative sensory testing and epidermal nerve fiber density in repeated measurementsPain2010151357558120851518
- YarnitskyDQuantitative sensory testingMuscle Nerve19972021982049040659
- YarnitskyDSprecherEThermal testing: normative data and repeatability for various test algorithmsJ Neurol Sci1994125139457964887
- KandelERSchwartzJHJessellTMPrinciples of Neural Science4th edNew YorkMcGraw-Hill/Appleton and Lange2000
- SchmidtRFAltnerHFundamentals of Sensory PhysiologyNew York (NY)Springer-Verlag1978
- BecserNSandTZwartJAReliability of cephalic thermal thresholds in healthy subjectsCephalalgia19981885745829827251
- ClausDHilzMJHummerINeundörferBMethods of measurement of thermal thresholdsActa Neurol Scand19877642882963687380
- HeldestadVLinderJSellersjöLNordhEReproducibility and influence of test modality order on thermal perception and thermal pain thresholds in quantitative sensory testingClin Neurophysiol2010121111878188520478739
- HilzMJGloriusSBerićAThermal perception thresholds: influence of determination paradigm and reference temperatureJ Neurol Sci199512921351407608727
- LevyDAbrahamRReidGA comparison of two methods for measuring thermal thresholds in diabetic neuropathyJ Neurol Neurosurg Psychiatry1989529107210772795077
- LowensteinLJesseKKentonKComparison of perception threshold testing and thermal-vibratory testingMuscle Nerve200837451451718067137
- MeierPMBerdeCBDiCanzioJZurakowskiDSethnaNFQuantitative assessment of cutaneous thermal and vibration sensation and thermal pain detection thresholds in healthy children and adolescentsMuscle Nerve200124101339134511562914
- VerdugoROchoaJLQuantitative somatosensory thermotest: a key method for functional evaluation of small calibre afferent channelsBrain1992115Pt 38939131628207
- HeldestadVNordhEQuantified sensory abnormalities in early genetically verified transthyretin amyloid polyneuropathyMuscle Nerve200735218919517094098
- HsuWBettegowdaCJalloGIIntramedullary spinal cord tumor surgery: can we do it without intraoperative neurophysiological monitoring?Childs Nerv Syst201026224124519902217
- KortelainenPPuranenJKoivistoELähdeSSymptoms and signs of sciatica and their relation to the localization of the lumbar disc herniationSpine (Phila Pa 1976)198510188923983706
- TravellJGSimonsDGMyofascial Pain and DysfunctionPhiladelphia (PA)Lippincott Williams & Wilkins1992
- VallboABHagbarthKEWallinBGMicroneurography: how the technique developed and its role in the investigation of the sympathetic nervous systemJ Appl Physiol20049641262126915016790
- EslerMClinical application of noradrenaline spillover methodology: delineation of regional human sympathetic nervous responsesPharmacol Toxicol19937352432538115306
- LowPAClinical Autonomic Disorders: Evaluation and ManagementBoston (MA)Little Brown and Co1993
- LowPACaskeyPETuckRRFealeyRDDyckPJQuantitative sudomotor axon reflex test in normal and neuropathic subjectsAnn Neurol19831455735806316835
- WangAKFealeyRDGehrkingTLLowPAPatterns of neuropathy and autonomic failure in patients with amyloidosisMayo Clin Proc200883111226123018990321
- DelahayeNRouzetFSardaLImpact of liver transplantation on cardiac autonomic denervation in familial amyloid polyneuropathyMedicine (Baltimore)200685422923816862048
- HörnstenRSuhrOBJensenSMWiklundUOutcome of heart rate variability and ventricular late potentials after liver transplantation for familial amyloidotic polyneuropathyAmyloid200815318719518925457
- KinoshitaOHongoMSaikawaYHeart rate variability in patients with familial amyloid polyneuropathyPacing Clin Electrophysiol19972012 Pt 1294929539455756
- WiklundUHörnstenRKarlssonMSuhrOBJensenSMAbnormal heart rate variability and subtle atrial arrhythmia in patients with familial amyloidotic polyneuropathyAnn Noninvasive Electrocardiol200813324925618713325
- WiklundUHörnstenROlofssonBOSuhrOBCardiac autonomic function does not improve after liver transplantation for familial amyloidotic polyneuropathyAuton Neurosci20101561–212413020478749
- AbeMAndoYHigashiKKanoTNon-neurogenic periodic fluctuations in heart rate and vasomotion appearing in familial amyloid polyneuropathy (FAP) type I (Met30)J Auton Nerv Syst1996601–271758884698
- HörnstenRSuhrOBOlofssonBOWiklundUArrhythmia: a pitfall in tests of cardiac autonomic function after liver transplantation for familial amyloidotic polyneuropathy; a long-term follow-up of Swedish patientsAmyloid20121928186
- ObayashiKHörnstenRWiklundUBlood pressure overshoot after tilt reversal in patients with familial amyloidotic polyneuropathyHypertens Res201134113313820927118
- ZhaoYHörnstenRLindqvistPWiklundUSuhrOBHeneinMYLeft ventricular dyssynchrony is associated with reduced heart rate variability in familial amyloidotic polyneuropathyInt J Cardiol2012155227327821056485
- SuhrOBLindqvistPOlofssonBOWaldenströmABackmanCMyocardial hypertrophy and function are related to age at onset in familial amyloidotic polyneuropathyAmyloid200613315415917062381
- RapezziCRivaLQuartaCCGender-related risk of myocardial involvement in systemic amyloidosisAmyloid2008151404818266120
- BhandariAKNandaNCMyocardial texture characterization by two-dimensional echocardiographyAm J Cardiol19835158178256829442
- FalkRHPlehnJFDeeringTSensitivity and specificity of the echocardiographic features of cardiac amyloidosisAm J Cardiol19875954184222949593
- John SuttonMGStReichekNKastorJAGiulianiERComputerized M-mode echocardiographic analysis of left ventricular dysfunction in cardiac amyloidCirculation19826647907996214334
- ChandrasekaranKAylwardPEFleagleSRFeasibility of identifying amyloid and hypertrophic cardiomyopathy with the use of computerized quantitative texture analysis of clinical echocardiographic dataJ Am Coll Cardiol19891348328402926037
- ErikssonPBackmanCErikssonAErikssonSKarpKOlofssonBODifferentiation of cardiac amyloidosis and hypertrophic cardiomyopathy: a comparison of familial amyloidosis with polyneuropathy and hypertrophic cardiomyopathy by electrocardiography and echocardiographyActa Med Scand1987221139463565084
- LindqvistPOlofssonBOBackmanCSuhrOWaldenströmAPulsed tissue Doppler and strain imaging discloses early signs of infiltrative cardiac disease: a study on patients with familial amyloidotic polyneuropathyEur J Echocardiogr200671223015869906
- EngvallCHeneinMHolmgrenASuhrOBMörnerSLindqvistPCan myocardial strain differentiate hypertrophic from infiltrative etiology of a thickened septum?Echocardiography201128440841521323993
- MigrinoRQHarmannLWoodsTBrightMTruranSHariPIntraventricular dyssynchrony in light chain amyloidosis: a new mechanism of systolic dysfunction assessed by 3-dimensional echocardiographyCardiovasc Ultrasound200864018687125
- RapezziCQuartaCCGuidalottiPLUsefulness and limitations of 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in the aetiological diagnosis of amyloidotic cardiomyopathyEur J Nucl Med Mol Imaging201138347047821069320
- FalkRHLeeVWRubinowASkinnerMCohenASCardiac technetium-99m pyrophosphate scintigraphy in familial amyloidosisAm J Cardiol1984548115011516093493
- HongoMHirayamaJFujiiTEarly identification of amyloid heart disease by technetium-99m-pyrophosphate scintigraphy: a study with familial amyloid polyneuropathyAm Heart J198711336546623030086
- PeruginiERapezziCPivaTNon-invasive evaluation of the myocardial substrate of cardiac amyloidosis by gadolinium cardiac magnetic resonanceHeart200692334334915939726
- PalladiniGCampanaCKlersyCSerum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosisCirculation2003107192440244512719281
- SuhrOBAnanIBackmanCDo troponin and B-natriuretic peptide detect cardiomyopathy in transthyretin amyloidosis?J Intern Med2008263329430118069997
- PalladiniGBarassiAKlersyCThe combination of high-sensitivity cardiac troponin T (hs-cTnT) at presentation and changes in N-terminal natriuretic peptide type B (NT-proBNP) after chemotherapy best predicts survival in AL amyloidosisBlood2010116183426343020644111
- ErikssonAErikssonPOlofssonBOThornellLEThe cardiac atrioventricular conduction system in familial amyloidosis with polyneuropathy: a clinico-pathologic study of six cases from northern SwedenActa Pathol Microbiol Immunol Scand A19839153433496225293
- ErikssonAErikssonPOlofssonBOThornellLEThe sinoatrial node in familial amyloidosis with polyneuropathy: a clinico-pathological study of nine cases from northern SwedenVirchows Arch A Pathol Anat Histopathol198440232392466322411
- AndoYSuhrOBAutonomic dysfunction in familial amyloidotic polyneuropathy (FAP)Amyloid19985428830010036588
- NiklassonUOlofssonBOBjerlePAutonomic neuropathy in familial amyloidotic polyneuropathy: a clinical study based on heart rate variabilityActa Neurol Scand19897931821872718736
- BeckmanABjerlePOlofssonBElectrocardiographic findings in familial amyloidotic polyneuropathyAm J Noninvas Cardiol19926192196
- De FreitasAFThe heart in Portuguese amyloidosisPostgrad Med J1986627286016053774701
- De FreitasAFBarbedoAConduction disturbances in 190 patients with familial amyloidotic polyneuropathy (Andrade’s type)Adv Cardiol197821206209202146
- SawayamaTKuriharaTArakiSNoninvasive cardiovascular findings in familial amyloid polyneuropathyBr Heart J1978401112881292718770
- OkamotoSHörnstenRObayashiKWijayatungaPSuhrOBContinuous development of arrhythmia is observed in Swedish transplant patients with familial amyloidotic polyneuropathy (amyloidogenic transthyretin Val30Met variant)Liver Transpl201117212212821280184
- HörnstenRWiklundUSuhrOBJensenSMVentricular late potentials in familial amyloidotic polyneuropathyJ Electrocardiol2006391576216387053
- AnanIEl-SalhyMAndoYColonic enteric nervous system in patients with familial amyloidotic neuropathyActa Neuropathol1999981485410412800
- AnanIEl-salhyMAndoYTerazakiHSuhrOBComparison of amyloid deposits and infiltration of enteric nervous system in the upper with those in the lower gastrointestinal tract in patients with familial amyloidotic polyneuropathyActa Neuropathol2001102322723211585246
- SuhrOBEriczonBGFrimanSLong-term follow-up of survival of liver transplant recipients with familial amyloid polyneuropathy (Portuguese type)Liver Transpl20028978779412200779
- BerkJLDyckPJObiciLThe diflunisal trial: update on study drug tolerance and disease progressionAmyloid201118Suppl 119119222080762
- LångKWikströmLDanielssonATashimaKSuhrOBOutcome of gastrointestinal complications after liver transplantation for familial amyloidotic polyneuropathyScand J Gastroenterol200035998598911063162
- LobatoLBeirãoISilvaMEnd-stage renal disease and dialysis in hereditary amyloidosis TTR V30M: presentation, survival and prognostic factorsAmyloid2004111273715185496
- LobatoLVenturaABeirãoIEnd-stage renal disease in familial amyloidosis ATTR Val30Met: a definitive indication to combined liver-kidney transplantationTransplant Proc20033531116112012947881
- NowakGSuhrOBWikströmLWilczekHEriczonBGThe long-term impact of liver transplantation on kidney function in familial amyloidotic polyneuropathy patientsTranspl Int200518111111515612992
- AndoEAndoYOkamuraRUchinoMAndoMNegiAOcular manifestations of familial amyloidotic polyneuropathy type I: long-term follow upBr J Ophthalmol19978142952989215058
- AndoEAndoYHaraokaKOcular amyloid involvement after liver transplantation for polyneuropathyAnn Intern Med20011351093193211712896
- SandgrenOKjellgrenDSuhrOBOcular manifestations in liver transplant recipients with familial amyloid polyneuropathyActa Ophthalmol200886552052418435819
- SvendsenIHSteensgaard-HansenFNordvågBYA clinical, echocardiographic and genetic characterization of a Danish kindred with familial amyloid transthyretin methionine 111 linked cardiomyopathyEur Heart J19981957827899717013
- SuhrOBAndersenOAronssonTReport of five rare or previously unknown amyloidogenic transthyretin mutations disclosed in SwedenAmyloid200916420821419922332
- BuxbaumJNYeZReixachNTransthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Abeta toxicityProc Natl Acad Sci U S A200810572681268618272491
- EpiskopouVMaedaSNishiguchiSDisruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormoneProc Natl Acad Sci U S A1993906237523798384721
- BodinKEllmerichSKahanMCAntibodies to human serum amyloid P component eliminate visceral amyloid depositsNature20104687320939720962779
- CoelhoTSuhrOBAdamsDPhase I safety, pharmacokinetic and pharmacodynamics results for ALN-TTR01, a novel RNAi therapeutic for the treatment of transthyretin amyloidosis (abstract)VIIIth International Symposium on Familial Amyloidotic PolyneuropathyKumamoto, Japan2011P62
- MoniaBAckermanEGuoSBensonMDClinical development of an antisense therapy for the treatment of transthyretin-associated polyneuropathy (abstract)VIIIth International Symposium on Familial Amyloidotic PolyneuropathyKumamoto, Japan2011P64
- GillmoreJDTennentGAHutchinsonWLSustained pharmacological depletion of serum amyloid P component in patients with systemic amyloidosisBr J Haematol2010148576076720064157
- CardosoIMerliniGSaraivaMJ4′-Iodo-4′-deoxydoxorubicin and tetracyclines disrupt transthyretin amyloid fibrils in vitro producing noncytotoxic species: screening for TTR fibril disruptersFASEB J200317880380912724338
- CardosoIMartinsDRibeiroTMerliniGSaraivaMJSynergy of combined doxycycline/TUDCA treatment in lowering transthyretin deposition and associated biomarkers: studies in FAP mouse modelsJ Transl Med201087420673327