Abstract
Duchenne muscular dystrophy (DMD), an allelic X-linked progressive muscle-wasting disease, is one of the most common single-gene disorders in the developed world. Despite knowledge of the underlying genetic causation and resultant pathophysiology from lack of dystrophin protein at the muscle sarcolemma, clinical intervention is currently restricted to symptom management. In recent years, however, unprecedented advances in strategies devised to correct the primary defect through gene- and cell-based therapeutics hold particular promise for treating dystrophic muscle. Conventional gene replacement and endogenous modification strategies have greatly benefited from continued improvements in encapsidation capacity, transduction efficiency, and systemic delivery. In particular, RNA-based modifying approaches such as exon skipping enable expression of a shorter but functional dystrophin protein and rapid progress toward clinical application. Emerging combined gene- and cell-therapy strategies also illustrate particular promise in enabling ex vivo genetic correction and autologous transplantation to circumvent a number of immune challenges. These approaches are complemented by a vast array of pharmacological approaches, in particular the successful identification of molecules that enable functional replacement or ameliorate secondary DMD pathology. Animal models have been instrumental in providing proof of principle for many of these strategies, leading to several recent trials that have investigated their efficacy in DMD patients. Although none has reached the point of clinical use, rapid improvements in experimental technology and design draw this goal ever closer. Here, we review therapeutic approaches to DMD, with particular emphasis on recent progress in strategic development, preclinical evaluation and establishment of clinical efficacy. Further, we discuss the numerous challenges faced and synergistic approaches being devised to combat dystrophic pathology effectively.
Introduction
Duchenne muscular dystrophy (DMD) is the most common fatal genetic disorder diagnosed in childhood, with a sex-linked inheritance pattern of one in 3500 live male births.Citation1,Citation2 Affected individuals can be diagnosed at birth on the basis of elevated serum creatine kinase (CK), a biochemical marker of muscle necrosis,Citation3 prior to visible difficulty in walking between 1 and 3 years of age. The clinical course of DMD is progressive; muscle weakness by age 5 years eventually leads to loss of independent ambulation by the middle of the second decade and death during the third decade, primarily as a result of respiratory or cardiac complications.Citation2 The genetic causation of DMD was established by localization of candidate complementary DNAs (cDNAs) to the short arm of the X chromosome (Xp band 21.2),Citation4 which led to full characterization of the 2.5-Mb DMD locus and corresponding 427-kDa dystrophin protein.Citation5 The sheer size of the resulting 14-kb dystrophin messenger RNA transcript served to explain how one-third of DMD cases arise from spontaneous new mutations.Citation6 In terms of clinical manifestation, DMD results from failure to produce functional dystrophin protein as a result of nonsense or frame-shift DNA mutations,Citation6 whereas those retaining the amino acid reading frame result in partially functional dystrophin and the milder allelic variant, Becker muscular dystrophy (BMD).
The genetic link to dystrophic pathology was elucidated by localization of dystrophin protein to the sarcolemma of skeletal and cardiac muscle, which is absent in DMD patients.Citation5 Structural and functional studies illustrate that dystrophin is pivotal for maintaining structural integrity by linking the internal actin cytoskeleton of individual muscle fibers via F-actin binding of its N-terminusCitation7 and C-terminal binding to the dystrophin-associated protein complex (DAPC) through β-dystroglycan (β-DG).Citation8 The DAPC comprises several internal scaffold and transmembrane proteins, including α/β-DGs, sarcoglycans, sarcospan, and biglycan, by which linkage to collagen and laminin is achieved; while evidence suggests some of these links have functional sig-naling roles, their predominant purpose appears mechanical (reviewed in Davies and Nowak).Citation9 In addition, the C-terminus of dystrophin interacts with neuronal nitric oxide synthase,Citation10 dystrobrevin,Citation11 and the syntrophins.Citation12 At the molecular level, loss of dystrophin and consequential loss of the DAPC create sarcolemmal instability, enhancing susceptibility to mechanically induced damage and degeneration.Citation13 Although the muscle initially responds through enhanced regeneration,Citation14 successive rounds of necrosis eventually deplete the supply of muscle progenitor cells, which leads to infiltration of adipose and fibrotic connective tissue and exacerbates muscle wasting.Citation15
Fragility of the DAPC also results in stretch-induced membrane permeability, leading to disruption of cellular homeostasis.Citation16 The resultant elevation of intracellular calcium ([Ca2+]i) levels triggers increased production of reactive oxygen species (ROS) by the mitochondria,Citation17 which contributes to the self-perpetuating cycle of increased oxidative stress, sarcolemmal damage, and eventual myofiber death.Citation6 Chronic activation of signaling pathways involved in the inflammatory response further exacerbates dystrophic pathology by increasing myofiber expression of the major histocompatibility complexCitation18 and the secretion of chemokines and cytokines.Citation19 Although profibrotic signaling is initially activated in an attempt to repair compromised myofibers, it is susceptible to upregulation by the unremitting nature of muscle damage, which triggers fibrosis and perpetuates inflammation.Citation20 As the cellular mechanisms that govern these secondary responses are intimately linked, it is difficult to ascribe hierarchical order to the molecular events that exacerbate DMD pathogenesis.
Although standards of care are improving, with better quality of life and prolonged survival,Citation3 there is no cure for DMD. Clinical intervention is generally restricted to symptom management, such as ventilators for respiratory support and administration of glucocorticoids to stem progressive muscle damage. Long-term corticosteroid treatment purportedly extends functional ability for up to 2 yearsCitation21 by modifying cellular events, including inflammation and Ca2+ homeostasis; however, their relative nonspecificity also causes unfavorable effects such as weight gain and loss of bone density.Citation22 Nonetheless, established steroidal efficacy provides a basis for devising therapeutic strategies able specifically to target molecular defects underlying dystrophic pathology. Several promising approaches have emerged due to advances in experimental design, delivery, and efficacy for all three subgroups: gene therapy, cell therapy, and pharmacological therapy. In this review, we describe the current status of each approach, with particular emphasis on clinical application. Further, we discuss emerging combinatorial strategies that are most likely to provide future candidates for a definitive DMD therapy.
Mammalian models of Duchenne muscular dystrophy
Animal models have been an invaluable resource to elucidate the molecular basis of DMD pathogenesis and in assessing therapies that may carry substantial risk in humans (). As the dystrophin-deficient phenotype significantly differs between species, the suitability of each animal model is primarily based on phenotypical similarity to DMD, weighed against the extent of pathological characterization, scope for genetic manipulation, accessibility, and breeding costs.Citation23
Table 1 Animal models used in assessing therapeutic strategies for Duchenne muscular dystrophy (DMD)
Murine models of DMD
Mouse models are indispensable for developing therapeutic approaches for DMD, since they are easily and reliably reproduced. The widely used X-linked muscular dystrophy mouse mdx modelCitation24 arises from a spontaneous nonsense mutation in exon 23Citation25 and absence of dystrophin protein.Citation5 Although muscle necrosis and high CK levels are evident from 2 weeks, the mdx phenotype is most pronounced between 3 and 4 weeks, when, in contrast to DMD patients, successive cycles of extensive necrosis are countered by regeneration,Citation24 eventually decreasing to chronic low-level damage by 8 weeks, permitting a near-normal lifespan.Citation23 Further, deterioration of skeletal and cardiac muscle (including fibrosis and inflammatory cell infiltration at later stages) in mdx is comparatively mild, where only the diaphragm is considered to recapitulate the severity of human disease.Citation26 This phenotypic disparity extends to N-ethyl-N-nitrosourea (ENU)-induced genetic variant mdx strains (mdx2–5cv)Citation27 that are not commonly used for therapeutic studies. Despite issues involving body size, genetic background, and pathological features, mdx is the established model for in vivo efficacy due to its desired transgenic and breeding capacity. For example, gene-based skipping of exon 51 (a strategy that is theoretically applicable to the highest percentage of DMD patients with out-of frame deletion mutations)Citation28 can be assessed using exon 52 knockout mice (mdx52).Citation29 Further, the development of “humanized” (hDMD) transgenic mice containing full-length human dystrophin has recently enabled direct preclinical screening of human-specific exon-skipping approaches.Citation30
Although inaccurate as genetic models, several double knockouts, including the myogenic transcription factor MyoD,Citation31 the discriminant analysis of principal component (DAPC) α-DB,Citation32 parvalbumin,Citation33 α7 integrin,Citation34 cytidine monophosphate–sialic acid hydroxylase,Citation35 and the dystrophin autosomal paralogue utrophinCitation36 have been developed. The most clinically relevant and widely used are dystrophin/utrophin knockout mice (mdx; utrn–/–, commonly referred to as dko),Citation36 which illustrate similar pathology to mdx at 4–5 weeks, after which this model progressively recapitulates DMD disease pathogenesis, resulting in a dramatically reduced lifespan.Citation23 As decreased survival of dko mice potentially hampers experimental design, a haploinsufficiency model (mdx; utrn+/–) has been generated but is not widely used.Citation37
Canine X-linked muscular dystrophy
Spontaneous canine X-linked muscular dystrophy (CXMD) has been reportedCitation38 in golden retriever (GRMD),Citation39 beagle (CXMDJ),Citation40 rottweiler,Citation41 German shorthaired pointer,Citation42 Japanese spitz,Citation43 and Cavalier King Charles spaniel (CKCS-MD)Citation44 breeds. Of these, the phenotypic progression of GRMD, resulting from an intron 6 splice acceptor mutation (leading to skipping of exon 7 and truncated dystrophin protein)Citation39 has been the most extensively characterized.Citation23 GRMD represents the most accurate animal model for DMD in recapitulating phenotypic timing and severity, where muscle degeneration and generalized necrosis noted from birth onwards results in extensive fibrosis by 6 months and respiratory failure commensurate to human pediatric age.Citation39 Given the retriever’s suitability in respect of genetic background and body size, GRMD has been instrumental in predicting disease pathogenesis, severity, and treatment efficacy,Citation45 providing proof of concept for numerous cell- and gene-therapy approaches (see ). However, the use of GRMD is restricted by dramatic phenotypical variation between sibs (causing difficulties in preclinical standardization),Citation39 welfare implications, and high costs of maintenance and treatment.Citation23 These concerns have been partially addressed by interbreeding GRMD dogs with smaller beagle sires (canine X-linked muscular dystrophy in Japan [CXMDJ]), resulting in a near-identical phenotype to GRMD but with an improved survival rate.Citation46
Although GRMD and CXMDJCitation47 dogs have several advantages over mdx as an exon-skipping model, they also retain a similar disadvantage where the disease-causing mutation lies outside the region commonly affected in humans.Citation48 The recent characterization of severe CXMD in CKCS dogs is of particular clinical relevance given its genotypic causation (a donor splice acceptor mutation in exon 50 and predicted protein truncation).Citation44 Further, success in inducing exon 51 skipping in cultured CKCS-MD myoblastsCitation44 indicates the potential of CKCS-MD as a suitable in vivo model (see gene-therapy section).
Feline and porcine models
Hypertrophic feline muscular dystrophy (HFMD)Citation49 and the 238 tailored pig model (238-DMD)Citation50 represent two large animal models of DMD that substantially differ in their genetic derivation that are suitable candidates for therapeutic assessment. While HFMD represents spontaneous dystrophin deficiency as a result of an extensive promoter deletion,Citation51 it is not widely used to limited pathological similarity to DMD.Citation38 In contrast, the exon 52–deleted 238-DMD pig, similar to mdx52, was engineered to assess exon 51 skipping methodologies,Citation50 and appears to be a bona fide model, as ascertained by absence of dystrophin protein, elevated serum CK levels, and early degenerative changes on muscle histology.Citation50 Further, porcine models have a number of practical advantages, such as the ability to circumvent numerous issues that currently preclude experimental transition from mdx into larger models (such as transgenic manipulation and breeding considerations), while retaining a similar size and physiology to humans.
As there is no definitive DMD animal model, GRMD and mdx currently represent the most appropriate model for preclinical testing by consensus.Citation23 It is tempting to speculate reliance on the mdx mouse model has hampered therapeutic progress, as the mild clinical phenotype results in difficulty in assessing certain issues, such as devising gene or cell therapies for larger muscle.Citation52 Nonetheless, the genetic tractability, reproducibility, and convenient size make mouse models invaluable tools in DMD research, provided their physiological differences are acknowledged. This reliance on smaller animals is largely due to practical difficulties imposed by larger models such as GRMD, which more closely represent DMD patients in size and pathological expression, but are never likely to supersede mdx in high-throughput studies. However, the future use of dog (and possibly pig) models to hone mouse-developed technologies on a more comparable phenotype is unclear, given a number of proof-of-concept strategies developed in mdx and human DMD cell lines have circumvented their use to successfully progress to human safety trials.
Cell-based therapeutic approaches
Cell-based therapies involve transplantation or transduction of allogeneic (donor-derived) or autologous (patient-derived) cells to engraft with existing myofibers or repopulate the cellular niche to promote functional muscle regeneration.Citation53
Myoblast transplantation
Allogeneic myoblast transfer was the first cell-based strategy to be assessed in immunologically tolerant mice, providing evidence of host–donor fusion and stimulating myofiber development;Citation54 parameters that were subsequently recapitulated in mdx mice (reviewed in Mouly et al).Citation55 Although allogeneic cell transplantation can circumvent the need for genetic manipulation to reintroduce functional dystrophin, the risk of graft rejection remains.Citation57 In addition, several unfavorable characteristics of using donor myoblasts, including (1) poor intramuscular migration, (2) low efficiency of dystrophin production, (3) limited cell survival, and (4) immune complications in mdx, were mirrored in early clinical trials assessing allogeneic implantation of immunohistocompatible myoblasts in DMD patient muscle.Citation56 Further, this approach leads to massive central ischemic necrosis in nonhuman primates.Citation57
Satellite cells and muscle-derived stem cells
As a result of the various pitfalls encountered with myoblast transfer, stem cell transplantation was deemed a more attractive option due to their differentiation potential and self-renewal capacity.Citation58 Among the first to be assessed were satellite cells (SCs), a quiescent and committed population of myogenic precursors that actively divide and differentiate in response to myofiber growth or damage.Citation59 When SCs remain attached to single myofibers for transplantation, they illustrate self-renewal and self-sufficiency as a regenerative source.Citation60 At present, direct SC engraftment faces two major hurdles: (1) the rapid decline of their autologous isolation potential, especially in the later stages of muscle degeneration, and (2) their individual isolation, in particular as in vitro expansion drastically reduces their engraftment and regeneration capacity.Citation61 It also remains unclear whether SCs derive from precursors resident in muscle or from circulating progenitors.Citation60 A number of these parameters can be alleviated through the use of muscle-derived stem cells (MDSCs), which are commonly thought to represent a predecessor of the SC.Citation59 As MDSCs represent a multipotential cell population, they are considered distinct from the myogenically committed SCs. Further, MDSCs have a number of advantages over SCs, including (1) increased engraftment ability, (2) expression of specific stem cell markers that allow specific isolation, and (3) expansion and maintenance in an in vitro progenitor state.Citation20,Citation59,Citation62 Systemic delivery of allogeneic murine or human MDSCsCitation63,Citation64 can restore dystrophin expression and ameliorate dystrophic pathology in immunotolerant mdx/severe combined immunodeficiency (SCID) mice. Further, autologous transplantation of MDSCs in DMD patients during a Phase I clinical safety study did not result in local or systemic side effects.Citation62 Despite these encouraging results, the typically heterogeneous nature of MDSCs may affect their efficacy, depending on their isolation and culturing conditions.Citation59
Pluripotent and non-muscle-derived progenitor cells
Several non-muscle cell types such as embryonic stem (ES) cells can converted to myogenic precursors after coculturing with skeletal myoblasts or by myogenic induction.Citation65 To circumvent ethical and legal restrictions associated with deriving ES cells,Citation66 allogeneic pluripotent human cells have been successfully isolated from early-age amniotic fluid (human AF-amniotic fluid stem cells) and umbilical cord (human umbilical cord-derived mesenchymal stem cells [hUC-MSCs]). Both of these donor cell populations were able to fuse with host myofibers after intramuscular or intravascular delivery, respectively, in immunosuppressed mice, although not within a dystrophic (ie, mdx) genetic background.Citation67,Citation68 However, given that hUC-MSC engraftment demonstrates effective elevation of muscle proteins in dysferlin-deficient dystrophic mice (an animal model for limb-girdle muscular dystrophy type 2B and Miyoshi myopathy, both caused by mutations of the dysferlin gene),Citation68 a planned Phase I/II trial is currently recruiting to assess their safety and efficacy in DMD patients.Citation69
In recent years, a number of tissue-specific adult stem cells, which maintain, generate, and replace terminally differentiated cells within their resident organ, have demonstrated myogenic potential.Citation53 Among the most promising are adult MSCs, which can differentiate to form myogenic cells in situ.Citation20 In contrast to other DMD cell-based therapies, MSCs also possess distinct anti-inflammatory activities and represent an ethical alternative to ES cells.Citation70 For example, intramuscular injection of bone marrow-derived MSCs was successful in regenerating muscle cells and repairing muscle degeneration in mdx/SCID mice.Citation71 Intramuscular or interarterial injection of myogenically induced canine wild-type allogeneic (dog leukocyte antigen matched with an unaffected littermate donor) bone marrow MSCs was able to establish long-term, widespread muscle engraftment and differentiation in CXMDJ dogs without requiring immunosuppression.Citation72
Another promising MSC-based approach is the use of vessel-associated mesoangioblasts (MABs), multipotential progenitors with the ability to differentiate into many mesodermal phenotypes, including myotubes.Citation70 Interarterial delivery of donor wild-type MABs in GRMD dogs illustrates impressive engraftment capability, leading to extensive recovery of muscle morphology and function.Citation70 Encouragingly, similar parameters can be achieved in dko mice, where proliferating MABs illustrated the ability to form new myofibers and promote expression of dystro-phin and its autosomal paralogue – utrophin.Citation73 These results establish MABs as a feasible candidate for DMD stem cell therapy, and an interarterial Phase I/IIa DMD clinical trial using MABs from healthy donors has been initiated.Citation74
From myoblast transfer to the use of stem and progenitor cells, a common hurdle remains in the effective use of cell therapy in DMD patients. For cell transplantation to be successful, hurdles such as immune rejection must be overcome, or advances in developing methods to manipulate autologous cells to reexpress dystrophin must be made. The most promising cell-based approach is thus likely to involve ex-vivo expansion of pluripotent patient-derived myogenic precursors (such as MABs) with gene therapy to enable autologous, genetically corrected cell engraftment. Recent progress in the rapidly expanding combined cell–gene therapy field is outlined in the next section.
Gene-based therapeutic approaches
As DMD is caused by recessive and monogenic genetic mutations, therapeutic strategies can be devised to correct or improve muscle function by (1) exogenous delivery of functionally engineered dystrophin gene constructs or (2) repair/augmentation of the endogenous locus (). Encouragingly, both approaches benefit from either the rapid progress in RNA-based technology or by combination with cell-based therapies.
Figure 1 Current genetic and pharmacological targets of dystrophic pathology.
Abbreviations: PA, polyaxamer 188; TGF-β, transforming growth factor beta; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B (PKB); IKK, IκB kinase; NF-κB, nuclear factor kappa-light-chain enhancer of activated B cells; BMP, bone morphogenic protein; AngII, angiotensin II; RI/RII, receptor I/II; AT, angiotensin; ACE, angiotensin-converting enzyme; NBD, NEMO binding domain; IGF-1, insulin growth factor 1; LAM-III, laminin-111 protein; VPA, valproic acid; ALK-4, activin receptor-like kinase; ActRII, activin receptor type II; BCL/ABL, breakpoint cluster region/Abelson murine leukemia viral oncogene homologue 1; Akt, acutely transforming retrovirus AKT8 in rodent T-cell lymphoma; [Ca2+]i, intracellular calcium; L-Arg, L-arginine; NO, nitric oxide; nNOS, neural nitric oxide synthase; cGMP, cyclic guanosine monophosphate; GMP, guanosine monophosphate; GC, guanylate cyclase; BGP-15, O-(3-piperidino-2-hydroxy-1-propyl) nicotinic amidoxime; PDE5, cGMP-specific phosphodiesterase type 5; TNF-α, tumour necrosis factor alpha; CsA, cyclosporine A; ROS, reactive oxygen species; MPTP, mitochondrial permeability transition pore; cycD, cyclophilin D; HSP72, heat shock protein 72; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; SG, sarcoglycans; Src, sarcospan; syn, sytrophin; db, dystrobrevin; RDO, RNA/DNA oligonucleotide; AON, antisense oligonucleotide; 2′OMePS, 2′-O-methyl oligoribonucleotide; PMO, phosphorodiamidate morpholino oligomer; AICAR, 5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide; UTRN/DMD/MSTN, utrophin/DMD/myostatin gene; DAPC, dystrophin-associated protein complex; LAM, laminin; Utr, utrophin minigene construct; cDNA, complementary DNA.
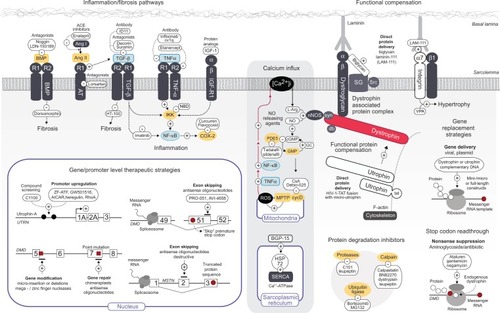
Gene-replacement therapy
Delivery of exogenous functional dystrophin is an attractive prospect to benefit all DMD patients (given the inconsequential nature of the endogenous mutation), and gene replacement is traditionally divided into viral and naked (nonviral) categories. The major challenge common to viral and nonviral approaches involves developing suitable delivery vectors and gene cassettes while avoiding a destructive immune response. Further, the large size of the dystrophin gene, coupled with the limited carrying capacity of vectors such as recombinant adeno-associated virus (rAAV) prompted construction of internally deleted but highly functional “mini”-dystrophin (mDYS)Citation75 and “micro”-dystrophin (µDYS)Citation76 constructs to facilitate gene transfer.
Historically, studies using systemic and intramuscular rAAV-mediated delivery of mDYS and µDYS in mdx demonstrated promising efficacy in a number of parameters, including successful formation of sarcolemmal mDYS−/µDYS-associated protein complexes and improved muscle function while reducing fibrosis (reviewed in Bowles et al).Citation77 Although in vivo rAAV-mediated gene transfer has been effective in reducing dystrophic pathology in both GRMD and dko animal models (reviewed in Seto et al),Citation78 the immune reaction against rAAV particles and dystrophin protein itself has been readily apparent in mdx and is particularly severe in GRMD.Citation79 Recent efforts to improve immune tolerance and transduction efficiency have led to increasing use of rAAV8-and AAV9-modified serotypes. For example, intramuscular rAAV2/9-µDYSCitation80 and rAAV9-mDYSCitation81 gene transfer in mdx and systemic injection of rAAV8-µDYS in CXMDJCitation82 and rAAV8-mDYS in GRMDCitation81 not only illustrate widespread transgene expression but also increase tropism in cardiac and skeletal muscle. However, lingering immune concerns continue to limit clinical assessment of rAAV-mediated gene transfer. This was evident in a 2010 Phase I dose-escalation study using intramuscular rAAV2.5-mDYS injection, which elicited failure in long-term transgene expression and severe T-cell reaction in a small cohort of DMD patients.Citation83 However, it is likely that AAV-mediated gene replacement will be subject to future trials, given recent improvements in translation optimization of the rAAV2.5 capsid has led to vastly improved immune tolerance in a Phase I follow-up safety study.Citation77
Nonviral gene-transfer methodologies have been explored by intramuscular injection of naked full-length and mDYS plasmid into mdx hindlimb muscle,Citation84,Citation85 mDYS in mdx4cv diaphragm,Citation86 and more recently by electrotransfer of full-length canine dystrophin into GRMD hind limb.Citation87 These studies indicate that plasmid-based gene transfer has greater potential for long-term expression compared to rAAV-mediated approaches but is hampered by lower comparative efficacy. Due perhaps to the latter, an initial clinical report detailing success of intramuscular delivery of full-length/mDYS plasmids in DMD patients to express functional protein within the injection siteCitation88 has not been repeated. Further, commercial development of a plasmid-based therapy (Myodys; Transgene) was assessed in a Phase I trial in 2008; however, no data has subsequently been released.Citation89
An increasingly promising alternative strategy to deliver functional dystrophin involves the ex vivo combination of cell and gene therapies. This approach involves the use of genetically modified cells as autologous delivery vehicles to circumvent immune challenges and reduce the risk of implant rejection.Citation90 Systemic efficacy of combined cell–gene therapy was originally established by successful interarterial delivery of MDSC cells transduced with lentivirus to regenerating mdx5cv muscle.Citation90 This study was extended by lentiviral-mediated transduction of canine mDYS in human and GRMD MDSCs prior to transplantation into mdx and GRMD by either intramuscular injection or electrotransfer.Citation91 Lentiviral vectors have also been used to demonstrate that µDYS-transduced autologous mdx4cv SCCitation92 and GRMD MABsCitation93 can regenerate dystrophin-positive myofibers in vivo. However, it is important to note that although the level of µDYS-expressing fibers was sufficient in treated GRMD dogs to ameliorate dystrophic morphology (5%–50%), their clinical performance remained poor, in direct contrast to the phenotypical improvement observed using systemic delivery of unmodified donor cell MABsCitation93 (see section on cell-based therapeutics.).
Alternative viral delivery vehicles such as retrovirus also demonstrate transduction efficacy, although the risk of immunogenic graft rejection is increased.Citation94,Citation96 Nonetheless, interarterial administration of isogenic MSCs containing retroviral-induced µDYS enabled persistent, long-term (12-week) dystrophin restoration in mdx muscle fibers and satellite cells.Citation94 Although plasmid-based mDYS transduction in MDSCCitation95 and mdx/DMD myoblastsCitation96,Citation97 induces high in situ expression, mini-gene approaches have been superseded by development of a human artificial chromosome (HAC) containing the entire dystrophin gene (DYS-HAC).Citation98 DYS-HAC has a number of distinct advantages over plasmid-based approaches, such as stable episomal maintenance and ability to carry large genomic regions (including regulatory elements). DYS-HAC transduction via microcell-mediated chromosome transfer enables complete genetic correction of engraftable induced pluripotent stem (iPS) cellsCitation99 from mdx and DMD patients.Citation100 Further, correct tissue expression of human dystrophin isoforms was evidenced in chimeric mice generated from DYS-HAC ES cell transfer.Citation100 Efficacy has recently been established in vivo using systemic delivery of DYS-HAC–transduced MABs, which illustrated morphological and functional amelioration of dystrophic pathology for up to 8 months posttransplantation.Citation101 These approaches have led to planned trials of DYS-HAC–transduced MDSCs for autologous transplantation in patients.Citation74
Gene editing
An alternative approach to exogenous delivery of functional dystrophin by gene replacement or cell-based therapies is to induce de novo dystrophin production. Gene editing aims to repair or modify the underlying genetic defect (gene repair) or to modulate RNA processing (by selectively “skipping” exons of the dystrophin gene) to ameliorate effects of the underlying gene mutation.
Gene repair
Initial approaches to gene editing were aimed at correcting point mutations in the dystrophin gene using synthetic RNA/DNA “chimeraplasts” (RDOs), which enter the cell and attach to the target gene. The DNA strands of the chimeraplast and the gene complement each other with the exception of the nucleotides that require editing, which are then targeted by DNA repair enzymes, allowing the permanent replacement of the DNA target sequence with that of the chimeraplast.Citation102 Although direct injection of RDOs into mdx muscle resulted in sarcolemmal localization of full-length dystrophin, myofiber conversion rates were poor (ranging from 1% to 15%).Citation102,Citation103 Similarly, direct intramuscular injection of RDOs illustrated sustained (over 48 weeks) in vivo repair of the GRMD point mutation and production of full-length dystrophin; resulting levels of dystrophin protein were similarly low, and almost exclusively restricted to the injection site.Citation104
Consequently, RDO-mediated editing has been superseded by antisense oligodeoxynucleotide (AON) approaches due to considerations such as modification ability, cost, scale, and experimental variability.Citation105 Although RDO-induced point mutations can successfully enable mdx exon-skipping in vitro,Citation106 it is notable that AON-mediated gene editing in vitro and in vivo using the mdx5cv mouse modelCitation107,Citation108 was originally devised to prevent not encourage exon skipping. The emergence of the latter as one of the most promising therapeutic approaches for DMD has led to a decline in using traditional gene-editing methodologies.
Exon skipping
Exon skipping is the most frequent alternative splicing mechanism known in mammals, and as such is a major contributor to protein diversity.Citation109 AONs aim to mimic exclusion (or “skipping”) of specific exons by hybridizing and thus blocking targeted pre-mRNA motifs involved in normal splicing to synthesize an internally truncated, semi-functional dystrophin protein.Citation48 Exon skipping has immense clinical potential, as 60% of DMD patients harbor deletions in exons 45–55 and sole targeting of exon 51 can address the majority of patients by addressing deletions of exon 50, exon 52, exons 48–50, or exons 49 and 50.Citation48 Further, a small-molecule “cocktail” approach enabling multiple exon skipping can feasibly be marketed as a single drug (reviewed in Benchaouir and Goyenvalle).Citation110
Preclinical in vitro proof of concept for AON-mediated exon skipping was established in primary cultured mdx myoblasts using targeted 2′-O-methyl oligoribonucleotides to exclude exon 23 and restore the dystrophin reading frame.Citation111 This result was recapitulated in vivo by intramuscular injection in mdx, which showed efficient AO nuclear uptake and sarcolemmal localization of dystrophin in treated muscle fibers.Citation112 As a result, two different AON chemistries have been under extensive study for clinical application: 2′-O-methyl-phosphorothioate (2′OMePS) and phosphorodiamidate morpholino oligomers (PMOs). Both 2′OMePS and PMO-induced exon-skipping approaches have been evaluated in cultured muscle cells from DMD patients, GRMD/CXMDJ dogs, and H2K-mdx and human explants (reviewed in Arechavala-Gomeza et al).Citation113 Further, systemic delivery of exon 23–skipping antisense compounds in mdx has been successful in restoring up to 50% of dystrophin expression in various muscle groups, improved muscle force, and reduced CK levels without tissue toxicity.Citation114 Specific exon 51 skipping was established using intramuscular injection of PMOs in mdx52Citation115 and for human exons 44, 46, and 49 by 2′OMePS in hDMD mice,Citation30 which restored dystrophin expression in whole-body skeletal muscles in addition to improving muscle function.Citation30,Citation115 Use of multiexon-skipping “cocktails” in vivo was first achieved by systemic PMO delivery in CXMDJ,Citation47 and subsequently validated using 2′OMePS and PMO combinations in CXMDJCitation116 and mdx4cv.Citation117 This approach appears more successful in increasing dystrophin expression in CXMDJ dogs (26% of normal levels)Citation116 compared to mdx4cv (5%–7%)Citation117 and trigger improvement in whole-body canine skeletal muscle (with the exception of heart) without adverse effects.Citation47 Further, PMOs generally illustrate in vivo superiority to 2′OMePS in the consistent and sustained induction of dystrophin protein.Citation118
Encouragingly, 2′OMePS and PMO exon 51–skipping technologies have progressed to clinical trials, with early indications of success at the biochemical and clinical level. Proof of concept for PRO-051, a 2′OMePS AON developed by Prosensa,Citation119 was established by intramuscular injection into DMD patients, which restored local sarcolemmal dystrophin in 64%–97% dystrophin-positive fibers and expression between 17% and 35% with no adverse effects.Citation120 This result is impressive, considering that a dystrophin expression level of 30% is postulated to avoid human pathogenesis,Citation121 although the precise level required to induce clinical and functional improvement remains unclear.Citation75 A follow-up Phase I/IIa clinical trial using systemic administration of PRO-051 was also well tolerated, with dose-dependent molecular efficacy (60%–100% positive fibers and up to 15.6% expression) accompanied by modest clinical improvement after 12 weeks’ extended treatment.Citation122 PRO-051 is currently licensed by GlaxoSmithKline (GSK2402968), who have initiated three clinical trials, including a large international Phase III study.Citation119 Prosensa have also opened a Phase I/IIa clinical study of PRO-044 (targeting exon 44) after assessment in DMD cultured cells, with preclinical trials of PRO-045 and PRO-053 (targeting exons 45 and 53, respectively) planned.Citation119
AVI-4658 (Eteplirsen) is a splice-switching PMO developed by Sarepta Therapeutics,Citation123 identified by exon 51–specific AON screening in two different chemical forms in cultured human muscle cells and explants (wild type and DMD) and by local in vivo administration in hDMD mice.Citation124 AVI-4658 has also been tested in cultured myoblasts of the CKCS-MD dog, which restored the reading frame and protein.Citation44 A single-blind, placebo-controlled, dose-escalation study illustrated encouraging local dystrophin induction,Citation125 leading to a systemic intravenous Phase IIb dose-escalation study to assess further the safety, tolerability, and pharmacokinetic properties of AVI-4658.Citation126 The initial 12-week study proved disappointing compared to PRO051, with 0%–55% positive fibers and up to 18% expression, with no significant improvement in clinical outcomes (even at higher doses), despite restoration of both components of the DAPC and neural nitric oxide synthase (nNOS) to the sar-colemma.Citation126 Although a subsequently longer clinical regime (24 weeks) has recently purported to improve dystrophin induction (averaging 22.5% dystrophin-positive fibers),Citation123 data on other parameters are currently unavailable. It is likely that additional trials to reoptimize delivery and dosage of AVI-4658 are planned by Sarepta Therapeutics, who have several other exon-skipping candidates, in particular AVI-5038 (targeting exon 50), which is currently in preclinical development.Citation123
An alternative exon-skipping methodology involves masking splicing sites using the endogenous targeting capacity of modified small nuclear RNAs (snRNAs), in particular U7 snRNA, to shuttle coupled AONs after rAAV vectorization (reviewed in Benchaouir and Goyenvalle).Citation110 Proof of principle was established in exons 48–50 deleted DMD patient cell lines, where U1/U7 snRNA successfully altered dystrophin pre-mRNA splicing to rescue synthesis,Citation127 confirmed by exon 23 skipping in mouse C2C12 cells.Citation128 In vivo systemic rescue of mdx dystrophic muscle by single intravenous (IV) administration of exon 23–targeted rAAV-U7 constructs induced sustained muscle expression and correction of dystrophic pathology;Citation129 parameters confirmed in rAAV-U1 and -U7 transduced mdx muscle after local injection.Citation130 The remarkable potential of systemic IV rAAV-U7–mediated therapeutics follows recent, single treatment of self-complementary rAAV-U7–mediated exon skipping in dko, which restored near-normal dystrophin levels and improved function in all muscles examined, including heart.Citation131 Human-specific multiexon skipping has also been achieved using rAAV-U7 in DMD cell lines and hDMD mice.Citation132 Combined with the recent success of using rAAV-U7–mediated exon 7 skipping in long-term restoration of dystrophin expression in GRMD cardiac muscle,Citation133,Citation134 this approach illustrates significant potential in effectively targeting DMD cardiomyopathy.
Increasing emergence of proof-of-principle studies in gene-based dystrophin replacement and endogenous augmentation provide significant promise for treatment of DMD pathology, including the recent use of meganucleases and zinc-finger nucleases to induce endogenous microdeletion or -insertions in the endogenous gene.Citation135 Despite the lack of long-term toxicology studies, multiple AON-mediated exon skipping potentially provides an applied therapeutic strategy for up to 83% of DMD patients.Citation136 However, difficulties in establishing long-term correction and circumventing immune challenges remain problematic, especially the inability of gene-replacement and PMO/2′OMePS-mediated exon skipping to effectively target cardiac tissue in mdx at doses corresponding to those required for clinical application.Citation137,Citation138 Several other issues, such as the timing of repeated administration, optimization of systemic delivery, and addressing poor cellular uptake, represent major hurdles in alleviating numerous chemical, clinical, and ethical issues. Moreover, further studies are required to clarify the mechanism through which AONs interfere with RNA splicing to optimize target sequences in humans.Citation139,Citation140 Recent studies to address these issues link inhibition of cell-cycle progression to enhance exon skipping,Citation141 exonic sequences as better exon-skipping targets, and enchanced efficacy by repeated intraperitoneal delivery over intramuscular or IV injection.Citation137 Encouragingly, significant progress has been made in improving systemic delivery (especially in cardiac muscle) and lowering dosage of AONs in a number of animal models (including mdx, dko, and GRMD) by conjugation to nanoparticles, cell-penetrating peptides, or enhanced delivery using artificial vesicles (reviewed in Arechavala-Gomeza et alCitation113 and Moulton).Citation142 It is therefore likely the first definitive DMD therapy will result from combining optimized multiexon-skipping methodologies with developing cell and pharmacological approaches.
Pharmacological approaches
DMD pharmacotherapy strategies involve the systemic delivery of small compounds that aim to (1) provide sarcolemmal-based compensation to directly address loss of the DAPC or (2) modify dysfunctional signaling pathways implicated in secondary pathology (). A number of pharmacological strategies show efficacy in circumventing immunological and delivery hurdles that currently hamper gene- and cell-based therapies.Citation143 However, as pharmaceuticals frequently target molecules involved in complex signaling pathways, their development is far from simple. Here, we summarize pharmacological approaches according to their intended molecular targets and discuss their progress, pitfalls and promise as treatment strategies for DMD.
Targeting primary DMD pathology by functional compensation or restoration of the DAPC
In conjunction with medicinal chemistry, a number of pharmacological approaches have been devised to specifically address the primary defect in DMD. These include (1) specific restoration of the DAPC by suppressing nonsense mutations in the dystrophin gene; (2) upregulation of its autosomal paralogue, utrophin, to provide a scaffold on which components of the DAPC can be restored to the sarcolemma; or (3) compensatory formation of integrin–laminin complexes, which have mechanosignaling similarities to the DAPC.
Aminoglycoside-mediated suppression of nonsense mutations
Approximately 10%–15% of DMD mutations convert an amino acid into a premature stop codon, while the rest of the mRNA is unaffected.Citation144 Nonsense mutation read-through strategies use aminoglycosides or small molecules to modify ribosomes to produce full-length functional protein by specifically targeting premature stop codons through contextual recognition of surrounding nucleotide sequences that differ between nonsense mutations and regular stop codons.Citation145 The most studied aminoglycoside for DMD therapeutic use is gentamicin, which acts via the 40S ribosomal subunit.Citation146 Proof-of-concept studies of gentamicin in mdx have been promising: a 2-week course of subcutaneous injection successfully enabled full-length dystrophin production with correct localization properties in both skeletal muscleCitation147 and the vascular system.Citation148 Further, these studies indicated improvement in a number of physiological parameters, including protection against contractile injury, normalization of blood flow and increased cardiac response to sheer stress.Citation147,Citation148 However, following Phase I DMD/BMD clinical outcomes were highly variable, from promisingCitation143,Citation149 to disappointing.Citation150 Potency issues due to batch consistencyCitation151 or dosage regimes may contribute to such conflicting outcomes, although the latter is less likely, given follow-up mdx studies could not replicate benefits described in one clinical trial.Citation152 In an attempt to combat gentamicin toxicity and increase target specificity, a drug-delivery system using hybrid liposomes has been developed,Citation153 and it will be of interest if this approach results in future clinical assessment.
The small-molecule ataluren (PTC124), which also acts via the 60S ribosomal subunit,Citation154 exhibits similar efficiency in mdx to gentamicin at a lower concentration.Citation155 Although PTC124 was well tolerated in patients, three Phase II DMD/BMD clinical studies were halted as predetermined primary outcomes were not met.Citation69 Therefore, despite the favorable pharmacodynamic response of both gentamicin and ataluren, their clinical development remains problematic, making their path to regulatory approval for DMD therapy a difficult one.Citation143 To circumvent toxicity concerns, an alternative approach is use of less toxic antibiotic peptides, such as negamycin, which inhibits eukaryotic RNA decoding.Citation156 Encouragingly, prolonged (4-week) intraperitoneal delivery in mdx enabled restoration of cardiac and skeletal muscle dystrophin levels comparable to those achieved with gentamicin,Citation156 making negamycin a promising therapeutic candidate.
Utrophin upregulation
A compensatory approach aimed at restoring components of the DAPC involves increasing levels of utrophin, the autosomal paralogue of dystrophin.Citation157 Although spatially restricted in adult myofibers to neuromuscular and myotendinous junctions,Citation158 extrajunctional utrophin is upregulated during embryonic development in mdx and DMD patients.Citation159 Utrophin-based upregulation therapy has a number of favorable attributes, notably (1) the ability to circumvent immunological challenges that accompany introduction of functional dystrophin protein; (2) in principle, effectiveness for all DMD patients, regardless of gene defect; and (3) amenability to systemic administration, given whole-body overexpression in mdx appears nontoxic.Citation160 Extensive proof-of-principle studies in mdx establish that a three- to fourfold increase in utrophin expression can enable functional restoration by formation of an alternative to the DAPC: the utrophin-associated protein complex (UAPC) complex (reviewed in Moorwood et al).Citation161 Historically, a number of endogenous transcriptional/posttranscriptional effectors of utrophin have been evaluated in mdx, including direct injection of the active Ras homologue gene family, member A (RhoA), heregulin, NO, and L-arginine, but none of these approaches has been able to reproducibly increase utrophin levels (extensively reviewed in Fairclough et al).Citation162
The observation that endogenous utrophin is elevated in slow-twitch muscleCitation163 led to investigation of how key regulators of muscle oxidative metabolism can be augmented to obtain therapeutic levels of utrophin. Targeted upregulation of either peroxisome proliferator–activated receptor cofactor 1-alpha (PGC-1α),Citation164,Citation165 its downstream effector peroxisome proliferator–activated receptor beta/delta (PPARβ/δ), GA-binding protein (GABP) α/β, active calcineurin (CnA*), or associated nuclear factors of activated T cells (NFAT) in mdx mice illustrate a twofold increase in utrophin mRNA levels (reviewed in Fairclough et al).Citation162 Encouragingly, many of these targets illustrate promoter-based synergism,Citation166,Citation167 indicating a multitargeting utrophin approach is feasible. However, it is important to note that, at present, pharmacological optimization of individual targets holds varying promise. For example, the biological benefit of activating PPARβ/δ using the histone deacetylase inhibitor valproic acid (VPA) and its derivatives may be outweighed by concerns over their developmental toxicity.Citation168 Similarly, synthetic PPAR ligands are under evaluation for non-DMD therapies,Citation169 but complications including off-target kinase activationCitation170 and severe side effects have led in some cases to recall and reformulation.Citation171 A more promising approach is administration of the adenosine monophosphate analog 5-aminoimidazole-4-carboxamide ribotide (AICAR). AICAR activates PGC-1α and PPARβ/δ via AMP-activated protein kinase (AMPK),Citation172 potentially affording greater therapeutic effect by enhancing synergism between PGC-1α with the α subunit of GABP.Citation166 Encouragingly, AICAR administration elevates sarcolemmal utrophin and β-DG protein levels and fast-to-slow muscle-fiber transitionCitation173 in mdx similar to using PPARβ/δ agonist GW501516Citation167 and AAV-mediated PGC-1α delivery.Citation164,Citation165 As AICAR is in frequent clinical use,Citation174 its future clinical assessment in DMD patients thus seems likely.
An alternate promoter-based utrophin strategy involves artificial zinc-finger proteins fused with effector domains (ZF-ATF), which is currently being evaluated in mdx (see Passananti et al).Citation175 However, the current favored strategy is identification of orally deliverable compounds with utrophin upregulation capabilities (reviewed in Moorwood et al).Citation161 Proof of principle was established by functional screening of chemical scaffold candidates, resulting in optimization of an orally bioavailable 2-arylbenzoxazole derivative – SMT-C1100 (BMN195).Citation176 Daily SMT-C1100 administration in mdx improved membrane integrity and demonstrated synergism with prednisolone.Citation176 However, a move into Phase I safety trials in healthy individuals was discontinued due to insufficient levels of SMT C1100 in plasma,Citation177 a difficulty being addressed by reformulation. However, the lack of safety issues with SMT-C1100 is encouraging, evidenced by complementary studies using compound libraries of FDA-approved and natural substances,Citation161 pharmacological interest (Zalicus and PTC Therapeutics),Citation154 and development of improved screening assays.Citation178
Protein-based therapy: TAT-utrophin and biglycan
Direct protein replacement of utrophin in dystrophin-deficient muscle uses deliverable chimeras constructed by fusing the transactivator of transcription (TAT) protein transduction domain (PTD) of human immunodeficiency virus (HIV-1)Citation179 with micro-utrophin (µUtr) protein (TAT-µUtr).Citation180 Intraperitoneal injection of TAT-µUtr in mdx established functional sarcolemmal µUtr–glycoprotein complexes, leading to improved membrane integrity and contractile function.Citation180 As similar levels of functional improvement in dko mice establish this approach as an attractive therapeutic possibility,Citation181 rigorous optimization is being performed prior to preclinical safety and toxicology studies.Citation182 Pending results, clinical TAT-µUtr trials (Retrophin, compound RE-001)Citation182 are anticipated in late 2012.
A related protein-based pharmacological candidate is biglycan, a small leucine-rich proteoglycan found at elevated levels within the ECM of DMD patient skeletal muscle.Citation183 Biglycan is a critical regulator of sarcolemmal proteins such as nNOS, components of the DAPC, and utrophin in particular, during the muscular response to cell damage and apoptosis.Citation184 Single systemic administration of recombinant human biglycan (rhBGN) was well tolerated and sufficient to counteract mdx pathology by enhancing UAPC stabilisation, as an identical dose regime was ineffective in dko mice.Citation185 To mitigate off-target effects, active rhBGN is currently being manufactured without biglycan-associated complex carbohydrate side chains for preclinical evaluation (Tivorsan, compound TVN-102).Citation183
α7-integrin upregulation/laminin-111
Integrin/laminin complexes act as mechanosignaling anchors, linking ECM laminin and fibronectin with intercellular cytoskeletal components.Citation186 Similar to the structural and signaling role of the DAPC, α7β1-integrin/laminin-211 complexes act as crucial enablers of muscle development, repair, regeneration, and integrity in skeletal muscle.Citation187,Citation188 Indeed the degree of functional redundancy between integrin/laminin complexes and the DAPC coupled with endogenous elevation of sarcolemmal α7β1 protein in DMD patients and mdx mice indicated that α7-integrin upregulation may stem DMD muscle pathology.Citation187 Efficacy of functional compensation was established by transgenic overexpression of α7-integrin in dko mice, which was effective in extending longevity (threefold), reducing kyphosis and increasing mobility as a result of increased sarcolemmal α7β1 protein.Citation189 While not preventing initial degeneration, α7-integrin upregulation appears to mediate sarcolemmal stability after subsequent regeneration by promoting SC proliferation, adherence, and activation.Citation189,Citation190 Favorably, inducing α7-integrin over-expression does not demonstrate visible toxicity or affect in vivo global gene-expression profiles,Citation190 and, as a result, small-compound screening for α7-integrin upregulators has been initiated.
Similar to utrophin, compound screening for α7-integrin provides a relatively uncomplicated means to develop orally bioavailable molecules to complement other DMD therapies or benefit patients ineligible for strategies such as exon skipping. VPA was identified as an α7-integrin upregulation compound using a cell-based assay, and intraperitoneal injection of VPA in dko mice results in decreased fibrosis, hypertrophy, and increases sarcolemmal integrity.Citation191 However, contrary to observations from in vitro studies, α7-integrin levels remain unchanged.Citation191 This discrepancy may be explained by the observation that VPA and α7-integrin both act, albeit independently, via the acutely transforming retrovirus AKT8 in rodent T-cell lymphoma (Akt)/mammalian target of rapamycin (mTOR) signaling pathwayCitation191 to positively regulate skeletal muscle hypertrophy.Citation192 Thus, in vivo VPA administration alone appears sufficient to trigger Akt- mediated signaling independently of α7-integrin.Citation191 However, as previously outlined in the utrophin section, the potential toxicity of VPA required to achieve clinical benefit remains a concern.Citation168
An alternative candidate identified via small-compound screening is laminin-111 (LAM-111).Citation193 Intramuscular or systemic LAM-III injection in mdx has enabled the sufficient induction of α7-integrin to achieve both sarcolem-mal stabilisationCitation193 and increased regenerative capacity.Citation194 Although this study has been countered by the failure of enhancing heterodimer LAM-111 formation in improving dystrophic skeletal muscle morphology in mdx mice,Citation195 the use of validated α7-integrin effectors has clinical promise. To preclude compounds with toxicity concerns, current pharmacological strategies are based on FDA-approved drug libraries (Prothelia)Citation196 or synergistic approved drug approaches (Zalicus).Citation197 Further, a number of lead compounds are currently at the preclinical (LAM-111/PRT-01 and PRT-20) and discovery (PRT-300) stages.Citation196
Targeting secondary DMD pathology resulting from dystrophin deficiency
Although the pathological presentation of dystrophin deficiency has been traditionally classified according to phenotypical and biochemical parameters such as fibrosis, necrosis, oxidative stress, and inflammation, it is increasingly apparent that the molecular processes that underlie these processes are intimately linked.Citation198 As a result, pharmacological intervention devised to alleviate one of these parameters may result in either assisting or even hindering one another. This is apparent with long-term corticosteroid treatment, which is thought to act by positively modifying both inflammation and Ca2+ homeostasis.Citation21 With this in mind, we categorize the progression of pharmacological intervention approaches according to their original aim of targeting a specific pathological or cellular defect, while outlining their links to others.
Reactive oxygen species and intracellular Ca2+ influx
Fragility of the DAPC leads to stretch-induced membrane permeability and Ca2+ influx, which activates proteases and enhances mitochondrial production of ROS,Citation17 which in turn regulates the nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) complex.Citation199 Although the synthetic membrane sealant polyaxamer 188 (PA188) shows promise in reducing membrane permeability in mdxCitation200 and GRMD models,Citation201 the primary strategy to aid sarcolemmal integrity involves supplementing the antioxidant response in DMD patients to normalize redox balance and protect against oxida-tive stress (reviewed in Tidball and Wehling-Henricks).Citation202
Dietary supplementation using glutamine, which is normally depleted in DMD patients,Citation203 prevents glucocorticoid-mediated upregulation of the transforming growth factor beta (TGF-β) family member myostatin (see section on fibrosis)Citation204 and protects against oxidative stress in mdx.Citation205 However, similar to combinatorial approaches using creatine and alanine, reproducibility in clinical trials is modest at best (reviewed in Fairclough et al).Citation162 In contrast, the commercial antioxidant melatonin improves muscle redox status and reduces inflammation in mdx5cvCitation206 and DMD patients,Citation207 while prenatal administration of epigallocatechin gallate effectively reduces ROS-mediated NF-κB activation in mdxCitation208,Citation209 to the extent that Phase I/II DMD clinical trials are planned.Citation210
Another promising supplement is N-acetylcysteine (NAC), a direct ROS scavenger and indirect L-cysteine precursor.Citation211 NAC treatment in postnecrotic mdx-decreased nuclear NF-κB protine enhances sarcolemmal UAPC formation, reduces stretch-induced damage,Citation212 and protects against damage and necrosis after only a week of treatment.Citation213 Further, linking antioxidants to lipophilic cations has human efficacy and improves effectiveness of antioxidants in disease models.Citation214 A recent study directly addressed calcium influx in mdx and dko muscle via oral gavage of BGP-15 (O-[3-piperidino-2-hydroxy-1-propyl]nicotinic amidoxime), increasing expression of heat shock protein 72, which binds and preserves sarcoplasmic reticulum Ca2+-ATPase SERCA under cellular stress.Citation215 As BGP-15 administration has been effective in slowing progression, preserving strength, and improving muscle function in dko,Citation215 this methodology holds immense therapeutic potential for DMD.
Mitochondrial permeability transition pore
Sustained increase of [Ca2+]i and the resultant redox imbalance in dystrophic muscle induces formation of the mitochondrial permeability transition pore (MPTP), leading to a self-perpetuating cycle of ROS, TNF-α, and NF-κB and eventual cell death (reviewed in Lemasters et al).Citation216 Attempts to desensitize mitochondria using cyclosporine A (CsA)-mediated blockage of cyclophilin DCitation217,Citation218 in mdx decreased necrosis in some studiesCitation219 but not others.Citation220 This result was reflected in DMD clinical trials where a small study reported improvements in muscle-force generation,Citation221 but a following, larger randomized trial illustrated little benefit.Citation222 To circumvent the effect of long-term CsA treatment on both the immune system and calcineurin signaling, use of a nonimmunosuppressing CsA analog (Debio-025) was recently shown to enhance MPTP blockade over CsA or prednisolone in mdx, and is under assessment for other muscular dystrophies (reviewed in Fairclough et al).Citation162
Necrosis
Dysregulation of the NO/cyclic guanosine monophosphate (cGMP) pathway has been implicated in the loss of contractile performance and sarcolemmal integrity in dystrophic muscle.Citation223 Increased [Ca2+]i levels in dystrophic muscle leads to increased catalysis of NO (a free radical scavenger and regulator of the NO/cGMP signaling pathway) from L-arginine by NOS, whereas limiting [Ca2+]i initiates NO-facilitated relaxation.Citation224 Further, loss of the DAPC in mdx and DMD muscle prevents anchoring of nNOS at the sarcolemma, which also decreases NO levels and results in myofiber damage.Citation10 Conversely, transgenic NOS restoration in mdx reduces ROS-mediated activation, decreases muscle damage/fibrosis and enhances formation of UAPC complexes,Citation225 and NO-releasing agents such as the nonsteroidal anti-inflammatory drug HCT-1026 can prevent mdx muscle inflammation and damage to a greater extent than prednisolone.Citation226 In addition, several NO donors including MyoNovin, isorbide dinitrate, or the analog guaifenesin dinitrateCitation227 act to alleviate multiple aspects of mdx pathology by activation of SCsCitation224 and/or via alleviating glucocorticoid side effects.Citation228 Similar benefits are observed in mdx when catalysis of GMP to cGMP is either increased by transgenic meansCitation223 or by using phosphodiesterase 5 (PDE5) inhibitors to prevent degradation of cGMP.Citation229 Further, PDE5 inhibitors deactivate hypertrophy signaling pathways triggered by pressure load, including those deregulated in DMD, such as calcineurin/NFAT, phosphoinositide-3 kinase/Akt, and extracellular signal–regulated kinase 1/2 cascades.Citation229 Given that commercially available cGMP-specific PDE5 inhibitors can prevent and even reverse contraction-induced myofiber damage (tadalafil)Citation230 or cardiac hypertrophy (sildenafil)Citation231 in mdx mice, both are at the recruitment stage for DMD clinical trials.Citation232
Protein-degradation inhibitors
Cysteine calpains are also activated in response to [Ca2+]i in DMD,Citation233 including the muscle-specific isoform absent in limb-girdle muscular dystrophy-2A.Citation234 Interestingly, calpain modulation in dystrophic muscle primarily disrupts regulatory mechanisms influenced by calpains rather than increase proteolytic activity.Citation234 Alleviating mdx muscle degeneration and necrosis by targetting calpain overactivity has been successful, using natural (calpastatin) or pharmacological means, including leupeptin, prodrug BN82270, dystrypsin (camostat mesylate) and enhanced-uptake cell-penetrating alpha-keto-amide calpain inhibitors (reviewed in Fairclough et al).Citation162 However, despite links between calpain inhibition and TGF-β downregulation,Citation235 calpastatin overexpressionCitation236 or administration of leupeptin-carnitine conjugate C101 was ineffective in mdxCitation236 and GRMDCitation237 models. Further, calpain inhibitors are also endogenously countered via elevated proteasome activity,Citation164,Citation236 which may preclude their pharmacological use. Alternatively, proteasomal targeting by systemic administration of nonspecific (MG-132) and specific (bortezomib) ubiquitin ligase inhibitors effectively reduces NF-κB–mediated inflammation and restores DAPC components to the sarcolemma in mdxCitation238–Citation240 and DMD explants.Citation241 Therefore, their synergistic use with compounds that restore redox balance/provide functional compensation is possible if a balance between benefit and side effects from long-term use can be achieved.
Inflammation
Prior to the onset of visible muscle damage, increased TNF-α leads to induction of IκB kinase (IKK) mediated NF-κB signaling,Citation242 a major contributor to the inflammatory and necrotic response of DMD myofibers.Citation243 Indeed, NF-κB activation leads to aberrant signaling inexorably linked to increased ROS, and this synergism significantly contributes to the preliminary wave of inflammation in secondary DMD pathology.Citation244 Further, glucocorticoids exert positive effects through NF-κB inhibition,Citation245 indicating that specific pharmacological targeting of NF-κB may have therapeutic benefit.
Direct TNF-α inhibition using the anti–TNF-α antibody infliximab, or depletion of circulating TNF-α levels using receptor decoy protein (etanercept) decrease fibrosis and necrosis and improve muscle function in mdx (reviewed in Fairclough et al).Citation162 Blocking downstream targets of NF-κB signaling such as cyclooxygenase-2 (COX-2) by curcumin improves sarcolemmal integrity and muscle strength in mdx, in addition to decreasing CK and levels of factors involved in the inflammatory process, including TNF-α and NF-κB.Citation246,Citation247 Unfortunately, the progressive increase of NF-κB levels in dystrophic muscle become increasingly resistant to curcumin,Citation247 indicating that targeted COX-2 inhibition may not provide benefit in a clinical setting. Nevertheless, a DMD Phase I safety trial is planned for the COX-2 inhibitor Flavocoxid.Citation69 In contrast to curcumin, direct targeting of IKK using rAAV-mediated intramuscular administration of dominant negative IKK protein improves regeneration in older but not younger mdx mice.Citation248 Impressively, systemic delivery of IKK inhibitory peptide (NF-κB essential modulator binding domain [NBD]) increases regeneration, reduces necrosis, and improves contractile function in mdx and dko diaphragm.Citation249 Optimizing intracellular delivery of NBD by fusion with a cationic cell–penetrating octalysine peptideCitation250 (8K-NBD) leads to further improvement in mdx histology.Citation251 Further, the ability of 8K-NBD to enhance benefits provided by AAV9-mDYS deliveryCitation252 indicates that IKK-mediated NF-κB inhibition may assist in treating residual fibrosis and necrosis observed with gene-transfer approaches.
Fibrosis
Pathological fibrosis in DMD muscle correlates with increased TGF-β signaling,Citation254 which hallmarks increased type I collagen productionCitation253 and the upregulation of several key intracellular markers of inflammation and oxidative stress.Citation253 TGF-β antagonists suramin and decorin have shown efficacy in promoting muscle recovery by attenuating sarcolemmal damage, decreasing fibrosis, and enhancing muscle regeneration (reviewed in Burks and Cohn).Citation255 Oral administration of nontoxic antifibrotics such as Bowman–Birk inhibitor and imatinib inhibit upstream and downstream TGF-β effectors, respectively, to affect a phenotype similar to direct antagonists in mdx.Citation256–Citation258 The plant alkaloid halofuginone (granted orphan drug status for DMD as HT-100)Citation259 acts as a potent inhibitor of TGF-β profibrotic signaling to recapitulate these parameters in mdx by enhancing myotube fusionCitation259 and function in initialCitation260 and established fibrosis,Citation261 negating the necessity for accurate therapeutic timing. It is also interesting to note that the TNF-α receptor decoy etanercept (see previous section) also reduces type I collagen and TGF-β mRNA,Citation262 indicating TNF-α blockade approaches may be effective in modulating TGF-β–mediated fibrosis.
TGF-β signaling can also be indirectly mediated by blocking bone morphogenic protein (BMP) ligands or the renin–angiotensin system (RAS), as both are continuously elevated in mdx and DMD skeletal muscle (reviewed in Burks and Cohn).Citation255 BMP antagonists noggin, dorsomorphin, and LDN-193189 enhance differentiation in human myoblastsCitation263 and intramuscular AAV delivery of the most potent and selective antagonist, noggin (“ad-noggin”), in dko mice enhances regeneration and alleviated dystrophic pathology.Citation263 However, repressing BMP signaling may influence toxicity and severity of side effects, parameters that preclude long-term in vivo administration of dorsomorphin.Citation263 RAS inhibition is considered a recent promising approach, where administration of angiotensin converting enzyme inhibitors (ACEi), antagonists of the angiotensin II (ATII type I) receptor, or TGF-β–neutralising antibodies (such as ID-11) demonstrate improvements in mdx pathology such as reduced fibrosis, increased muscle strength, and enhanced respiratory function.Citation264–Citation266 Further, ATII receptor agonists may counter effects mediated by NF-κB, such as inflammation- and oxida-tive stress–related muscle damage.Citation267 As early intervention using combined ACEi/ATII type I antagonists preserves muscle function in dko to an extent currently unparalleled by other pharmacological strategies,Citation267 preclinical evaluation is highly anticipated.
Blocking secreted myostatin,Citation268 a TGF-β–related negative regulator of muscle growth,Citation269 also increases muscle-fiber size. Myostatin-null mice illustrate robust muscular hypertrophy and hyperplasia by deregulation of myoblast proliferation and differentiation.Citation268 Similar improvements in mdx have been achieved using neutralizing antibodies, myostatin propeptide (MRPO) follistatin-derived peptides and the soluble extracellular form of the myostatin activin type-II receptor (reviewed in Burks and Cohn).Citation255 Unfortunately, Phase II trials of recombinant ActRIIB decoy (ACE-031) were suspended due to safety issues,Citation270 and PhaseI/II clinical trials of antimyostatin MYO-029 antibody (stamulumab), although well tolerated, did not improve muscle strength.Citation271 These findings impact alternate approaches, as clinical trials are not planned for AAV8-mediated MRPO delivery validated in the GRMD model.Citation272 However, myostatin-blockade approaches have benefited from exon-skipping methodologies developed for dystrophin, where induction of mdx muscle hypertrophy using destructive 2OMP/PMO myostatin pre-mRNA targetingCitation273 has advanced to 2′OMePS-based strategies simultaneously targeting myostatin and dystrophin.Citation274
Muscle-growth strategies also involve exogenous delivery of insulin-like growth factor I (IGF-I), which stimulates SC proliferation and differentiation during muscle regeneration.Citation275 Subcutaneous injection or viral expression of human IGF-I (rhIGF-I) or polyethylene glycol-modified IGF-I analogs (PEG-IGF-I) in mdx increase muscle strength and resistance to fatigue,Citation276,Citation277 but are ineffective against mechanical injury and myofiber degeneration.Citation278 Further, PEG-IGF-I administration in dko and older mdx mice highlight somewhat limited potential to ameliorate severe or established pathophysiology, and the authors suggest delivery should be initiated only for mild muscle pathologies.Citation277 Nevertheless, an IGF-I Phase I clinical trial is currently at the recruitment stage.Citation69
Conclusion
In recent years, significant progress has been made in the discovery of novel DMD therapeutic strategies and the continued development of established gene- and cell-based protocols. This is due, in part, to the continued understanding of molecular mechanisms that underlie DMD pathogenesis and the ability to establish efficacy using an increasing array of animal models. The development of a definitive DMD therapy is increasingly likely to involve synergism between adjunctive pharmaceuticals with gene-based approaches (such as exon skipping) to target multiple aspects of dystrophic pathology. Further, advances in cell-based technology show distinct promise in aiding efforts to correct endogenous dystrophin by their ability to act as autologous delivery vehicles. The successful move to clinical trials in each field has not only highlighted important aspects in the treatment and management of DMD but has also provided useful information for future design to accurately determine the age and state of the disease where treatment has clinically meaningful benefit. It is also increasingly apparent that accurate genetic diagnosis is key, given the increasing development of mutation-specific molecular therapies. As outlined in this review, many challenges lie ahead in the development and delivery of DMD therapeutics, and the specific approach(es) that will eventually result in success is unclear. However, it is clear that despite various hurdles, the incredible progress in therapeutic design in recent years has led to improved methodologies with immense translational potential.
Disclosure
KED is a consultant for Summit Plc. and is on the Scientific Advisory Board of Prosensa Plc. The authors report no other conflicts of interest in this work.
References
- DubowitzVMuscle disorders in childhoodMajor Probl Clin Pediatr197816iiixiii1282661378
- EmeryAMuscular dystrophy – the factsNeuromuscul Disord1995565218580734
- BushbyKFinkelRBirnkrantDJDiagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial managementLancet Neurol201091779319945913
- MonacoAPNeveRLColletti-FeenerCBertelsonCJKurnitDMKunkelLMIsolation of candidate cDNAs for portions of the Duchenne muscular dystrophy geneNature198632360896466503773991
- HoffmanEPBrownRHJrKunkelLMDystrophin: the protein product of the Duchenne muscular dystrophy locusCell19875169199283319190
- KoenigMHoffmanEPBertelsonCJMonacoAPFeenerCKunkelLMComplete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individualsCell19875035095173607877
- LevineBAMoirAJPatchellVBPerrySVThe interaction of actin with dystrophinFEBS Lett199026311591622185033
- Ibraghimov-BeskrovnayaOErvastiJMLeveilleCJSlaughterCASernettSWCampbellKPPrimary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrixNature199235563626967021741056
- DaviesKENowakKJMolecular mechanisms of muscular dystrophies: old and new playersNat Rev Mol Cell Biol200671076277316971897
- BrenmanJEChaoDSXiaHAldapeKBredtDSNitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarco-lemma in Duchenne muscular dystrophyCell19958257437527545544
- Sadoulet-PuccioHMRajalaMKunkelLMDystrobrevin and dystrophin: an interaction through coiled-coil motifsProc Natl Acad Sci U S A1997942312413124189356463
- YangBJungDRafaelJAChamberlainJSCampbellKPIdentification of alpha-syntrophin binding to syntrophin triplet, dystrophin, and utrophinJ Biol Chem199527010497549787890602
- LynchGSRafaelJAChamberlainJSFaulknerJAContraction-induced injury to single permeabilized muscle fibers from mdx, transgenic mdx, and control miceAm J Physiol Cell Physiol20002794C1290C129411003610
- Abdel-SalamEAbdel-MeguidIKorraaSSMarkers of degeneration and regeneration in Duchenne muscular dystrophyActa Myol20092839410020476668
- FoidartMFoidartJMEngelWKCollagen localization in normal and fibrotic human skeletal muscleArch Neurol19813831521577469848
- MorandiLMoraMGussoniETedeschiSCornelioFDystrophin analysis in Duchenne and Becker muscular dystrophy carriers: correlation with intracellular calcium and albuminAnn Neurol19902856746791979724
- NetheryDCallahanLAStofanDMatteraRDiMarcoASupinskiGPLA(2) dependence of diaphragm mitochondrial formation of reactive oxygen speciesJ Appl Physiol2000891728010904037
- McDouallRMDunnMJDubowitzVExpression of class I and class II MHC antigens in neuromuscular diseasesJ Neurol Sci1989892–32132262926449
- De PaepeBCreusKKMartinJJDe BleeckerJLUpregulation of chemokines and their receptors in Duchenne muscular dystrophy: potential for attenuation of myofiber necrosisMuscle Nerve Epub642012
- IchimTEAlexandrescuDTSolanoFMesenchymal stem cells as anti-inflammatories: implications for treatment of Duchenne muscular dystrophyCell Immunol20102602758219917503
- DeSilvaSDrachmanDBMellitsDKunclRWPrednisone treatment in Duchenne muscular dystrophy. Long-term benefitArch Neurol19874488188223632394
- MoxleyRT3rdAshwalSPandyaSPractice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology SocietyNeurology2005641132015642897
- WillmannRPossekelSDubach-PowellJMeierTRueggMAMammalian animal models for Duchenne muscular dystrophyNeuromuscul Disord200919424124919217290
- BulfieldGSillerWGWightPAMooreKJX chromosome-linked muscular dystrophy (mdx) in the mouseProc Natl Acad Sci U S A1984814118911926583703
- SicinskiPGengYRyder-CookASBarnardEADarlisonMGBarnardPJThe molecular basis of muscular dystrophy in the mdx mouse: a point mutationScience19892444912157815802662404
- StedmanHHSweeneyHLShragerJBThe mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophyNature199135263355365391865908
- ChapmanVMMillerDRArmstrongDCaskeyCTRecovery of induced mutations for X chromosome-linked muscular dystrophy in miceProc Natl Acad Sci U S A1989864129212962919177
- Helderman-van den EndenATStraathofCSAartsma-RusABecker muscular dystrophy patients with deletions around exon 51; a promising outlook for exon skipping therapy in Duchenne patientsNeuromuscul Disord201020425125420153965
- ArakiENakamuraKNakaoKTargeted disruption of exon 52 in the mouse dystrophin gene induced muscle degeneration similar to that observed in Duchenne muscular dystrophyBiochem Biophys Res Commun199723824924979299538
- Bremmer-BoutMAartsma-RusAde MeijerEJTargeted exon skipping in transgenic hDMD mice: a model for direct preclinical screening of human-specific antisense oligonucleotidesMol Ther200410223224015294170
- MegeneyLAKablarBGarrettKAndersonJERudnickiMAMyoD is required for myogenic stem cell function in adult skeletal muscleGenes Dev19961010117311838675005
- GradyRMGrangeRWLauKSRole for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophiesNat Cell Biol19991421522010559919
- RaymackersJMDebaixHColson-Van SchoorMConsequence of parvalbumin deficiency in the mdx mouse: histological, biochemical and mechanical phenotype of a new double mutantNeuromuscul Disord200313537638712798793
- GuoCWillemMWernerAAbsence of alpha 7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophyHum Mol Genet200615698999816476707
- ChandrasekharanKYoonJHXuYA human-specific deletion in mouse Cmah increases disease severity in the mdx model of Duchenne muscular dystrophySci Transl Med201024242ra54
- DeconinckAERafaelJASkinnerJAUtrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophyCell19979047177279288751
- ZhouLRafael-FortneyJAHuangPHaploinsufficiency of utrophin gene worsens skeletal muscle inflammation and fibrosis in mdx miceJ Neurol Sci20082641–210611117889902
- SheltonGDEngvallECanine and feline models of human inherited muscle diseasesNeuromuscul Disord200515212713815694134
- SharpNJKornegayJNVan CampSDAn error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophyGenomics19921311151211577476
- ShimatsuYKatagiriKFurutaTCanine X-linked muscular dystrophy in Japan (CXMDJ)Exp Anim2003522939712806883
- WinandNCooperBMolecular characterization of severe Duchenne-type muscular dystrophy in a family of rottweiler dogsProceedings of Molecular Mechanisms of Neuromuscular DiseaseTucsonUniversity of Arizona1994
- SchatzbergSJOlbyNJBreenMMolecular analysis of a spontaneous dystrophin ‘knockout’ dogNeuromuscul Disord19999528929510407848
- JonesBRBrennanSMooneyCTMuscular dystrophy with truncated dystrophin in a family of Japanese Spitz dogsJ Neurol Sci2004217214314914706216
- WalmsleyGLArechavala-GomezaVFernandez-FuenteMA duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skippingPLoS One201051e864720072625
- CooperBJWinandNJStedmanHThe homologue of the Duchenne locus is defective in X-linked muscular dystrophy of dogsNature198833461781541563290691
- ShimatsuYYoshimuraMYuasaKMajor clinical and histopathological characteristics of canine X-linked muscular dystrophy in Japan, CXMDJActa Myol200524214515416550932
- YokotaTLuQLPartridgeTEfficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogsAnn Neurol200965666767619288467
- Aartsma-RusAJansonAAKamanWEAntisense-induced multiexon skipping for Duchenne muscular dystrophy makes more senseAm J Hum Genet2004741839214681829
- VosJHvan der Linde-SipmanJSGoedegebuureSADystrophy-like myopathy in the catJ Comp Pathol19869633353413722476
- KlymiukNThirionCBurkhardtK238 tailored pig model of duchenne muscular dystrophyReprod Fertil Dev2011241231
- WinandNJEdwardsMPradhanDBerianCACooperBJDeletion of the dystrophin muscle promoter in feline muscular dystrophyNeuromuscul Disord199445–64334457881288
- PartridgeTModels of dystrophinopathy, pathological mechanisms and assessment of therapiesBrownSCLucyJADystrophin: Gene, Protein and Cell BiologyCambridgeCambridge University Press1997310321
- MeregalliMFariniAColleoniFCassinelliLTorrenteYThe role of stem cells in muscular dystrophiesCurr Gene Ther201212319220522463740
- PartridgeTAGroundsMSloperJCEvidence of fusion between host and donor myoblasts in skeletal muscle graftsNature19782735660306308652035
- MoulyVAamiriAPerieSMyoblast transfer therapy: is there any light at the end of the tunnel?Acta Myol200524212813316550930
- HuardJRoyRBouchardJPMalouinFRichardsCLTremblayJPHuman myoblast transplantation between immunohistocompatible donors and recipients produces immune reactionsTransplant Proc1992246304930511466052
- SkukDParadisMGouletMTremblayJPIschemic central necrosis in pockets of transplanted myoblasts in nonhuman primates: implications for cell-transplantation strategiesTransplantation200784101307131518049116
- PriceFDKurodaKRudnickiMAStem cell based therapies to treat muscular dystrophyBiochim Biophys Acta20071772227228317034994
- JankowskiRJDeasyBMHuardJMuscle-derived stem cellsGene Ther200291064264712032710
- CollinsCAOlsenIZammitPSStem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell nicheCell2005122228930116051152
- WebsterCBlauHMAccelerated age-related decline in replicative life-span of Duchenne muscular dystrophy myoblasts: implications for cell and gene therapySomat Cell Mol Genet19901665575652267630
- TorrenteYBelicchiMMarchesiCAutologous transplantation of muscle-derived CD133+ stem cells in Duchenne muscle patientsCell Transplant200716656357717912948
- GussoniESoneokaYStricklandCDDystrophin expression in the mdx mouse restored by stem cell transplantationNature1999401675139039410517639
- MuXXiangGRathboneCRSlow-adhering stem cells derived from injured skeletal muscle have improved regenerative capacityAm J Pathol2011179293194121684246
- DarabiRGehlbachKBachooRMFunctional skeletal muscle regeneration from differentiating embryonic stem cellsNat Med200814213414318204461
- JacksonWMNestiLJTuanRSPotential therapeutic applications of muscle-derived mesenchymal stem and progenitor cellsExpert Opin Biol Ther201010450551720218920
- MaXZhangSZhouJClone-derived human AF-amniotic fluid stem cells are capable of skeletal myogenic differentiation in vitro and in vivoJ Tissue Eng Regen Med20126859861322396316
- KongKYRenJKrausMFinklesteinSPBrownRHJrHuman umbilical cord blood cells differentiate into muscle in sjl muscular dystrophy miceStem Cells200422698199315536189
- ClinicalTrials.gov [homepage on the Internet]Clinical Trials for Duchenne muscular dystrophyBethesda, MDUS National Library of Medicine2012 [cited August 8, 2012]. Available from: http://clinicaltrials.gov/ct2/results?term=dmdAccessed August 18, 2012
- SampaolesiMTorrenteYInnocenziACell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblastsScience2003301563248749212855815
- DezawaMIshikawaHItokazuYBone marrow stromal cells generate muscle cells and repair muscle degenerationScience2005309573231431716002622
- Nitahara-KasaharaYHayashita-KinohHOhshima-HosoyamaSLong-term engraftment of multipotent mesenchymal stromal cells that differentiate to form myogenic cells in dogs with Duchenne muscular dystrophyMol Ther201220116817721934652
- BerrySELiuJChaneyEJKaufmanSJMultipotential mesoangioblast stem cell therapy in the mdx/utrn−/− mouse model for Duchenne muscular dystrophyRegen Med20072327528817511564
- OptiStem.org [homepage on the Internet]Progress reportsOtpiStem2012 [cited August 8, 2012]. Available from: http://www.optistem.org/progress-reportsAccessed August 18, 2012
- HarperSQHauserMADelloRussoCModular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophyNat Med20028325326111875496
- SakamotoMYuasaKYoshimuraMMicro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgeneBiochem Biophys Res Commun200229341265127212054513
- BowlesDEMcPheeSWLiCPhase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vectorMol Ther201220244345522068425
- SetoJTRamosJNMuirLChamberlainJSOdomGLGene replacement therapies for duchenne muscular dystrophy using adeno-associated viral vectorsCurr Gene Ther201212313915122533379
- YuasaKYoshimuraMUrasawaNInjection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene productsGene Ther200714171249126017581597
- KooTMalerbaAAthanasopoulosTDelivery of AAV2/9-microdystrophin genes incorporating helix 1 of the coiled-coil motif in the C-terminal domain of dystrophin improves muscle pathology and restores the level of alpha1-syntrophin and alpha-dystrobrevin in skeletal muscles of mdx miceHum Gene Ther201122111379138821453126
- KornegayJNLiJBoganJRWidespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogsMol Ther20101881501150820517298
- OhshimaSShinJHYuasaKTransduction efficiency and immune response associated with the administration of AAV8 vector into dog skeletal muscleMol Ther2009171738018941441
- MendellJRCampbellKRodino-KlapacLDystrophin immunity in Duchenne’s muscular dystrophyN Engl J Med2010363151429143720925545
- AcsadiGDicksonGLoveDRHuman dystrophin expression in mdx mice after intramuscular injection of DNA constructsNature199135263388158181881437
- DankoIFritzJDLatendresseJSHerweijerHSchultzEWolffJADystrophin expression improves myofiber survival in mdx muscle following intramuscular plasmid DNA injectionHum Mol Genet1993212205520618111373
- DecrouyARenaudJMDavisHLLundeJADicksonGJasminBJMini-dystrophin gene transfer in mdx4cv diaphragm muscle fibers increases sarcolemmal stabilityGene Ther1997454014089274716
- PichavantCChapdelainePCerriDGBizarioJCTremblayJPElectrotransfer of the full-length dog dystrophin into mouse and dystrophic dog musclesHum Gene Ther201021111591160120553115
- RomeroNBBraunSBenvenisteOPhase I study of dystrophin plasmid-based gene therapy in Duchenne/Becker muscular dystrophyHum Gene Ther200415111065107615610607
- DuanDMyodys, a full-length dystrophin plasmid vector for Duchenne and Becker muscular dystrophy gene therapyCurr Opin Mol Ther2008101869418228186
- BachrachELiSPerezALSystemic delivery of human microdystrophin to regenerating mouse dystrophic muscle by muscle progenitor cellsProc Natl Acad Sci U S A2004101103581358614993597
- PichavantCChapdelainePCerriDGExpression of dog microdystrophin in mouse and dog muscles by gene therapyMol Ther20101851002100920179674
- KimuraELiSGregorevicPFallBMChamberlainJSDystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expressionMol Ther201018120621319888194
- SampaolesiMBlotSD’AntonaGMesoangioblast stem cells ameliorate muscle function in dystrophic dogsNature2006444711957457917108972
- FengSWChenFCaoJRestoration of muscle fibers and satellite cells after isogenic MSC transplantation with microdystrophin gene deliveryBiochem Biophys Res Commun201241911622321394
- IkezawaMCaoBQuZDystrophin delivery in dystrophin-deficient DMDmdx skeletal muscle by isogenic muscle-derived stem cell transplantationHum Gene Ther200314161535154614577915
- MoissetPASkukDAsselinISuccessful transplantation of genetically corrected DMD myoblasts following ex vivo transduction with the dystrophin minigeneBiochem Biophys Res Commun1998247194999636661
- MoissetPAGagnonYKarpatiGTremblayJPExpression of human dystrophin following the transplantation of genetically modified mdx myoblastsGene Ther1998510134013469930339
- HoshiyaHKazukiYAbeSA highly stable and nonintegrated human artificial chromosome (HAC) containing the 2.4 Mb entire human dystrophin geneMol Ther200917230931719034264
- NakahataTAwayaTChangHDerivation of engraftable myogenic precursors from murine ES/iPS cells and generation of disease-specific iPS cells from patients with Duchenne muscular dystrophy (DMD) and other diseasesRinsho Shinkeigaku2010501188921921492
- KazukiYHiratsukaMTakiguchiMComplete genetic correction of ips cells from Duchenne muscular dystrophyMol Ther201018238639319997091
- TedescoFSHoshiyaHD’AntonaGStem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophySci Transl Med201139696ra78
- RandoTADisatnikMHZhouLZRescue of dystrophin expression in mdx mouse muscle by RNA/DNA oligonucleotidesProc Natl Acad Sci U S A200097105363536810805797
- BertoniCRandoTADystrophin gene repair in mdx muscle precursor cells in vitro and in vivo mediated by RNA-DNA chimeric oligonucleotidesHum Gene Ther200213670771811936970
- BartlettRJStockingerSDenisMMIn vivo targeted repair of a point mutation in the canine dystrophin gene by a chimeric RNA/DNA oligonucleotideNat Biotechnol200018661562210835598
- AlexeevVIgouchevaOYoonKSimultaneous targeted alteration of the tyrosinase and c-kit genes by single-stranded oligonucleotidesGene Ther20029241667167512457280
- BertoniCLauCRandoTARestoration of dystrophin expression in mdx muscle cells by chimeraplast-mediated exon skippingHum Mol Genet200312101087109912719373
- BertoniCMorrisGERandoTAStrand bias in oligonucleotide- mediated dystrophin gene editingHum Mol Genet200514222123315563511
- MaguireKSuzukiTDiMatteoDParekh-OlmedoHKmiecEGenetic correction of splice site mutation in purified and enriched myoblasts isolated from mdx5cv miceBMC Mol Biol2009101519236710
- KoleRSazaniPAntisense effects in the cell nucleus: modification of splicingCurr Opin Mol Ther20013322923411497345
- BenchaouirRGoyenvalleASplicing modulation mediated by small nuclear RNAs as therapeutic approaches for muscular dystrophiesCurr Gene Ther201212317919122515846
- DunckleyMGManoharanMVillietPEperonICDicksonGModification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotidesHum Mol Genet199877108310909618164
- MannCJHoneymanKChengAJAntisense-induced exon skipping and synthesis of dystrophin in the mdx mouseProc Natl Acad Sci U S A2001981424711120883
- Arechavala-GomezaVAnthonyKMorganJMuntoniFAntisense oligonucleotide-mediated exon skipping for duchenne muscular dystrophy: progress and challengesCurr Gene Ther201212315216022533380
- LuQLRabinowitzAChenYCSystemic delivery of anti-sense oligoribonucleotide restores dystrophin expression in body-wide skeletal musclesProc Natl Acad Sci U S A2005102119820315608067
- AokiYNakamuraAYokotaTIn-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouseMol Ther201018111995200520823833
- YokotaTHoffmanETakedaSAntisense oligo-mediated multiple exon skipping in a dog model of duchenne muscular dystrophyMethods Mol Biol201170929931221194037
- MitrpantCFletcherSIversenPLWiltonSDBy-passing the nonsense mutation in the 4 CV mouse model of muscular dystrophy by induced exon skippingJ Gene Med2009111465619006096
- YinHLuQWoodMEffective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx miceMol Ther2008161384517968354
- Prosensa.eu [homepage on the Internet]Pre-clinical portfolioLeiden, The NetherlandsProsensa; 2012 [cited August 8, 2012]. Available from: http://prosensa.eu/technology-and-products/pre-clinical-portfolioAccessed August 18, 2012
- van DeutekomJCJansonAAGinjaarIBLocal dystrophin restoration with antisense oligonucleotide PRO051N Engl J Med2007357262677268618160687
- NeriMTorelliSBrownSDystrophin levels as low as 30% are sufficient to avoid muscular dystrophy in the humanNeuromuscul Disord20071711–1291391817826093
- GoemansNMTuliniusMvan den AkkerJTSystemic administration of PRO051 in Duchenne’s muscular dystrophyN Engl J Med2011364161513152221428760
- Sareptatherapeutics.com [homepage on the Internet]Duchenne muscular dystrophy: drug candidates/clinical trialsCambridge, MASerepta Therapeutics2012 [cited August 8, 2012]. Available from: http://www.sareptatherapeutics.com/our-programs/rare-diseases/duchenne-muscular-dystrophyAccessed August 18, 2012
- Arechavala-GomezaVGrahamIRPopplewellLJComparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscleHum Gene Ther200718979881017767400
- KinaliMArechavala-GomezaVFengLLocal restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept studyLancet Neurol200981091892819713152
- CirakSArechavala-GomezaVGuglieriMExon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation studyLancet2011378979159560521784508
- De AngelisFGSthandierOBerarducciBChimeric snRNA molecules carrying antisense sequences against the splice junctions of exon 51 of the dystrophin pre-mRNA induce exon skipping and restoration of a dystrophin synthesis in Delta 48–50 DMD cellsProc Natl Acad Sci U S A200299149456946112077324
- BrunCSuterDPauliCU7snRNAs induce correction of mutated dystrophin pre-mRNA by exon skippingCell Mol Life Sci200360355756612737315
- GoyenvalleAVulinAFougerousseFRescue of dystrophic muscle through U7 snRNA-mediated exon skippingScience200430657021796179915528407
- DentiMARosaAD’AntonaGChimeric adeno-associated virus/antisense U1 small nuclear RNA effectively rescues dystrophin synthesis and muscle function by local treatment of mdx miceHum Gene Ther200617556557416716113
- GoyenvalleABabbsAWrightJRescue of severely affected dystrophin/utrophin-deficient mice through scAAV-U7snRNA-mediated exon skipping. Hum Mol GenetMar 132012211125592571
- GoyenvalleAWrightJBabbsAWilkinsVGarciaLDaviesKEEngineering multiple U7snRNA constructs to induce single and multiexon-skipping for Duchenne muscular dystrophyMol Ther20122061212122122354379
- BishLTSleeperMMForbesSCLong-term restoration of cardiac dystrophin expression in golden retriever muscular dystrophy following rAAV6-mediated exon skippingMol Ther201220358058922146342
- BarbashIMCecchiniSFaraneshAZMRI roadmap-guided transendocardial delivery of exon-skipping recombinant adeno-associated virus restores dystrophin expression in a canine model of Duchenne muscular dystrophyGene Ther Epub532012
- ChapdelainePPichavantCRousseauJPaquesFTremblayJPMeganucleases can restore the reading frame of a mutated dystrophinGene Ther201017784685820393509
- Aartsma-RusAFokkemaIVerschuurenJTheoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutationsHum Mutat200930329329919156838
- HeemskerkHde WinterCvan KuikPPreclinical PK and PD studies on 2′-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse modelMol Ther20101861210121720407428
- Aartsma-RusAvan OmmenGJLess is more: therapeutic exon skipping for Duchenne muscular dystrophyLancet Neurol200981087387519713153
- ‘t HoenPAde MeijerEJBoerJMGeneration and characterization of transgenic mice with the full-length human DMD geneJ Biol Chem200828395899590718083704
- MitrpantCAdamsAMMeloniPLMuntoniFFletcherSWiltonSDRational design of antisense oligomers to induce dystrophin exon skippingMol Ther20091781418142619293776
- O’LearyDASharifOAndersonPIdentification of small molecule and genetic modulators of AON-induced dystrophin exon skipping by high-throughput screeningPLoS One2009412e834820020055
- MoultonHMCell-penetrating peptides enhance systemic delivery of antisense morpholino oligomersMethods Mol Biol201286740741422454076
- MalikVRodino-KlapacLRViolletLGentamicin-induced readthrough of stop codons in Duchenne muscular dystrophyAnn Neurol201067677178020517938
- BushbyKGenetics and the muscular dystrophiesDev Med Child Neurol2000421178078411104352
- ManuvakhovaMKeelingKBedwellDMAminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation systemRNA2000671044105510917599
- PalmerEWilhelmJMShermanFPhenotypic suppression of nonsense mutants in yeast by aminoglycoside antibioticsNature19792775692148150366439
- Barton-DavisERCordierLShoturmaDILelandSESweeneyHLAminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx miceJ Clin Invest1999104437538110449429
- LoufraniLDubrocaCYouDAbsence of dystrophin in mice reduces NO-dependent vascular function and vascular density: total recovery after a treatment with the aminoglycoside gentamicinArterioscler Thromb Vasc Biol200424467167614751810
- PolitanoLNigroGNigroVGentamicin administration in Duchenne patients with premature stop codon. Preliminary resultsActa Myol2003221152112966700
- WagnerKRHamedSHadleyDWGentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutationsAnn Neurol200149670671111409421
- YoshizawaSFourmyDPuglisiJDStructural origins of gentamicin antibiotic actionEMBO J19981722643764489822590
- DunantPWalterMCKarpatiGLochmullerHGentamicin fails to increase dystrophin expression in dystrophin-deficient muscleMuscle Nerve200327562462712707984
- YukiharaMItoKTanoueOEffective drug delivery system for duchenne muscular dystrophy using hybrid liposomes including gentamicin along with reduced toxicityBiol Pharm Bull201134571271621532162
- A New Approach to Drug Discovery [webpage on the Internet]. Therapeutic areasSouth Plainfield, NJPTC Therapeutics2012 [cited August 8, 2012]. Available from: http://www.ptcbio.com/therapeutic_areasAccessed August 18, 2012
- WelchEMBartonERZhuoJPTC124 targets genetic disorders caused by nonsense mutationsNature20074477140879117450125
- ArakawaMShiozukaMNakayamaYNegamycin restores dystrophin expression in skeletal and cardiac muscles of mdx miceJ Biochem2003134575175814688241
- LoveDRHillDFDicksonGAn autosomal transcript in skeletal muscle with homology to dystrophinNature1989339621955582541343
- TinsleyJMBlakeDJRocheAPrimary structure of dystrophin-related proteinNature199236064045915931461283
- KhuranaTSWatkinsSCChafeyPImmunolocalization and developmental expression of dystrophin related protein in skeletal muscleNeuromuscul Disord1991131851941822793
- FisherRTinsleyJMPhelpsSRNon-toxic ubiquitous over-expression of utrophin in the mdx mouseNeuromuscul Disord200111871372111595513
- MoorwoodCLozynskaOSuriNNapperADDiamondSLKhuranaTSDrug discovery for Duchenne muscular dystrophy via utrophin promoter activation screeningPLoS One2011610e2616922028826
- FaircloughRJPerkinsKJDaviesKEPharmacologically targeting the primary defect and downstream pathology in duchenne muscular dystrophyCurr Gene Ther201212320624422571500
- GramoliniAOBelangerGThompsonJMChakkalakalJVJasminBJIncreased expression of utrophin in a slow vs a fast muscle involves posttranscriptional eventsAm J Physiol Cell Physiol20012814C1300C130911546668
- SelsbyJTMorineKJPendrakKBartonERSweeneyHLRescue of dystrophic skeletal muscle by PGC-1alpha involves a fast to slow fiber type shift in the mdx mousePLoS One201271e3006322253880
- HandschinCKobayashiYMChinSSealePCampbellKPSpiegelmanBMPGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophyGenes Dev200721777078317403779
- AngusLMChakkalakalJVMejatACalcineurin-NFAT signaling, together with GABP and peroxisome PGC-1{alpha}, drives utrophin gene expression at the neuromuscular junctionAm J Physiol Cell Physiol20052894C908C91715930144
- MiuraPChakkalakalJVBoudreaultLPharmacological activation of PPARbeta/delta stimulates utrophin A expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx miceHum Mol Genet200918234640464919744959
- LampenASiehlerSEllerbeckUGottlicherMNauHNew molecular bioassays for the estimation of the teratogenic potency of valproic acid derivatives in vitro: activation of the peroxisomal proliferator-activated receptor (PPARdelta)Toxicol Appl Pharmacol1999160323824910544058
- Lopez-SorianoJChielliniCMaffeiMGrimaldiPAArgilesJMRoles of skeletal muscle and peroxisome proliferator-activated receptors in the development and treatment of obesityEndocr Rev200627331832916556851
- GardnerOSDewarBJGravesLMActivation of mitogen-activated protein kinases by peroxisome proliferator-activated receptor ligands: an example of nongenomic signalingMol Pharmacol200568493394116020742
- BalakumarPKathuriaSSubmaximal PPARgamma activation and endothelial dysfunction: new perspectives for the management of cardiovascular disordersBr J Pharmacol201216671981199222404217
- LeickLFentzJBiensoRSPGC-1{alpha} is required for AICAR-induced expression of GLUT4 and mitochondrial proteins in mouse skeletal muscleAm J Physiol Endocrinol Metab20102993E456E46520628026
- LjubicicVKhogaliSRenaudJMJasminBJChronic AMPK stimulation attenuates adaptive signaling in dystrophic skeletal muscleAm J Physiol Cell Physiol20113021C110C12121940670
- PoldRJensenLSJessenNLong-term AICAR administration and exercise prevents diabetes in ZDF ratsDiabetes200554492893415793229
- PassanantiCCorbiNOnoriADi CertoMGMatteiETransgenic mice expressing an artificial zinc finger regulator targeting an endogenous geneMethods Mol Biol201064918320620680835
- TinsleyJMFaircloughRJStorerRDaily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mousePLoS One201165e1918921573153
- Biomarin Pharmaceutical IncBioMarin and Summit plc sign worldwide licensing agreement for Duchenne muscular dystrophy program [press release]Novato, CABiomarin Pharmaceutical Inc2008722 Available from: http://phx.corporate-ir.net/phoenix.zhtml?c=106657&p=irol-newsArticle&ID=1177621&highlight=Accessed July 22, 2008
- FaircloughRFSquireSEPotterACIdentification of new chemical compounds which upregulate utrophin for the therapy of Duchenne muscular dystrophyProceedings of the Muscular Dystrophy Campaign UK Neuromuscular Translational Research Conference2012 Mar 22–23Newcastle, UK
- SchwarzeSRHoAVocero-AkbaniADowdySFIn vivo protein transduction: delivery of a biologically active protein into the mouseScience199928554331569157210477521
- SonnemannKJHeun-JohnsonHTurnerAJBaltgalvisKALoweDAErvastiJMFunctional substitution by TAT-utrophin in dystrophin-deficient micePLoS Med200965e100008319478831
- CallJAErvastiJMLoweDATAT-muUtrophin mitigates the pathophysiology of dystrophin and utrophin double-knockout miceJ Appl Physiol2011111120020521565990
- Retrophin. RE-001: Retrophin’s investigational agent for Duchenne muscular dystrophyNew York, NYRetrophin LLC2011 [cited August 8, 2012]. Available from: http://www.retrophin.com/pipeline.phpAccessed September 25, 2012
- Tivorsan PharmaceuticalsMDA awards $1 million to Tivorsan Pharmaceuticals for accelerating pivotal pre-clinical work on Tvn-102 as a potential muscular dystrophy treatment [press release]Providence, RITivorsan Pharmaceuticals, Inc201215 Available from: http://www.tivorsan.com/wp-content/uploads/2012/01/PR_MDATivorsan_1M_Final_1_5_12.pdfAccessed January 5, 2012
- ZanottiSNegriTCappellettiCDecorin and biglycan expression is differentially altered in several muscular dystrophiesBrain2005128Pt 112546255516183658
- AmentaARYilmazABogdanovichSBiglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx miceProc Natl Acad Sci U S A2010108276276721187385
- HynesROIntegrins: versatility, modulation, and signaling in cell adhesionCell199269111251555235
- HodgesBLHayashiYKNonakaIWangWArahataKKaufmanSJAltered expression of the alpha7beta1 integrin in human and murine muscular dystrophiesJ Cell Sci1997110Pt 22287328819427295
- MayerUSaherGFasslerRAbsence of integrin alpha 7 causes a novel form of muscular dystrophyNat Genet19971733183239354797
- BurkinDJWallaceGQNicolKJKaufmanDJKaufmanSJEnhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic miceJ Cell Biol200115261207121811257121
- LiuJBurkinDJKaufmanSJIncreasing alpha 7 beta 1-integrin promotes muscle cell proliferation, adhesion, and resistance to apoptosis without changing gene expressionAm J Physiol Cell Physiol20082942C627C64018045857
- GurpurPBLiuJBurkinDJKaufmanSJValproic acid activates the PI3K/Akt/mTOR pathway in muscle and ameliorates pathology in a mouse model of Duchenne muscular dystrophyAm J Pathol20091743999100819179609
- BodineSCStittTNGonzalezMAkt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivoNat Cell Biol20013111014101911715023
- RooneyJEGurpurPBBurkinDJLaminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophyProc Natl Acad Sci U S A2009106197991799619416897
- RooneyJEGurpurPBYablonka-ReuveniZBurkinDJLaminin-111 restores regenerative capacity in a mouse model for alpha7 integrin congenital myopathyAm J Pathol2009174125626419074617
- Coral-VazquezRCohnRDMooreSADisruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophyCell199998446547410481911
- Prothelia [webpage on the Internet]Therapeutics for muscular dystrophyMilford, MAProthelia Inc2012 [cited August 8, 2012]. Available from: http://www.prothelia.com/livesite/pages/pipelineAccessed September 25, 2012
- CombinatoRxCombinatoRx and Charley’s Fund/Nash Avery Foundation collaborate to develop novel agents for Duchenne muscular dystrophy [press release]CombinatoRx, Incorporated2007 [cited August 8, 2012]. Available from: http://phx.corporate-ir.net/phoenix.zhtml?c=148036&p=irol-newsArticle&ID=1074719&highlight=Accessed September 25, 2012
- RandoTAOxidative stress and the pathogenesis of muscular dystrophiesAm J Phys Med Rehabil200281Suppl 11S175S18612409822
- MorganMJLiuZGCrosstalk of reactive oxygen species and NF-kappaB signalingCell Res201121110311521187859
- SpurneyCFGuerronADYuQMembrane sealant Poloxamer P188 protects against isoproterenol induced cardiomyopathy in dystrophin deficient miceBMC Cardiovasc Disord2011112021575230
- TownsendDTurnerIYasudaSChronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogsJ Clin Invest201012041140115020234088
- TidballJGWehling-HenricksMThe role of free radicals in the pathophysiology of muscular dystrophyJ Appl Physiol200710241677168617095633
- HankardRMaurasNHammondDHaymondMDarmaunDIs glutamine a ‘conditionally essential’ amino acid in Duchenne muscular dystrophy?Clin Nutr199918636536910634922
- SalehianBMahabadiVBilasJTaylorWEMaKThe effect of glutamine on prevention of glucocorticoid-induced skeletal muscle atrophy is associated with myostatin suppressionMetabolism20065591239124716919545
- MokEConstantinBFavreauFl-Glutamine administration reduces oxidized glutathione and MAP kinase signaling in dystrophic muscle of mdx micePediatr Res200863326827318287965
- HibaouiYReutenauer-PatteJPatthey-VuadensORueggUTDorchiesOMMelatonin improves muscle function of the dystrophic mdx5Cv mouse, a model for Duchenne muscular dystrophyJ Pineal Res201151216317121486366
- ChahbouniMEscamesGVenegasCMelatonin treatment normalizes plasma pro-inflammatory cytokines and nitrosative/oxidative stress in patients suffering from Duchenne muscular dystrophyJ Pineal Res201048328228920210854
- SahJFBalasubramanianSEckertRLRorkeEAEpigallocatechin-3-gallate inhibits epidermal growth factor receptor signaling pathway. Evidence for direct inhibition of ERK1/2 and AKT kinasesJ Biol Chem200427913127551276214701854
- ChenPCWheelerDSMalhotraVOdomsKDenenbergAGWongHRA green tea-derived polyphenol, epigallocatechin-3-gallate, inhibits IkappaB kinase activation and IL-8 gene expression in respiratory epitheliumInflammation200226523324112238566
- BuyseGMGoemansNvan den HauweMIdebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: results from a 12 month, double-blind, randomized placebo-controlled trialNeuromuscul Disord201121639640521435876
- AruomaOIHalliwellBHoeyBMButlerJThe antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acidFree Radic Biol Med1989665935972546864
- WhiteheadNPPhamCGervasioOLAllenDGN-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx miceJ Physiol200858672003201418258657
- TerrillJRRadley-CrabbHGGroundsMDArthurPGN-Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosisNeuromuscul Disord201122542743422206641
- SmithRAMurphyMPMitochondria-targeted antioxidants as therapiesDiscov Med2011115710611421356165
- GehrigSMvan der PoelCSayerTAHsp72 preserves muscle function and slows progression of severe muscular dystrophyNature2012484739439439822495301
- LemastersJJTheruvathTPZhongZNieminenALMitochondrial calcium and the permeability transition in cell deathBiochim Biophys Acta20091787111395140119576166
- McGuinnessOYafeiNCostiACromptonMThe presence of two classes of high-affinity cyclosporin A binding sites in mitochondria. Evidence that the minor component is involved in the opening of an inner-membrane Ca(2+)-dependent poreEur J Biochem199019426716792176603
- MatasJYoungNTBourcier-LucasCIncreased expression and intramitochondrial translocation of cyclophilin-D associates with increased vulnerability of the permeability transition pore to stress-induced opening during compensated ventricular hypertrophyJ Mol Cell Cardiol200946342043019094991
- De LucaANicoBLiantonioAA multidisciplinary evaluation of the effectiveness of cyclosporine a in dystrophic mdx miceAm J Pathol2005166247748915681831
- WellerBMassaRKarpatiGCarpenterSGlucocorticoids and immunosuppressants do not change the prevalence of necrosis and regeneration in mdx skeletal musclesMuscle Nerve19911487717741891001
- SharmaKRMynhierMAMillerRGCyclosporine increases muscular force generation in Duchenne muscular dystrophyNeurology1993433 Pt 15275328450995
- KirschnerJSchesslJScharaUTreatment of Duchenne muscular dystrophy with ciclosporin A: a randomised, double-blind, placebo-controlled multicentre trialLancet Neurol20109111053105920801085
- KhairallahMKhairallahRJYoungMESildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiencyProc Natl Acad Sci U S A2008105197028703318474859
- StamlerJSMeissnerGPhysiology of nitric oxide in skeletal musclePhysiol Rev200181120923711152758
- WehlingMSpencerMJTidballJGA nitric oxide synthase transgene ameliorates muscular dystrophy in mdx miceJ Cell Biol2001155112313111581289
- BrunelliSScioratiCD’AntonaGNitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapyProc Natl Acad Sci U S A2007104126426917182743
- WangGBurczynskiFJHasinoffBBZhangKLuQAndersonJEDevelopment of a nitric oxide-releasing analogue of the muscle relaxant guaifenesin for skeletal muscle satellite cell myogenesisMol Pharm20096389590419317416
- ScioratiCGalvezBGBrunelliSEx vivo treatment with nitric oxide increases mesoangioblast therapeutic efficacy in muscular dystrophyJ Cell Sci2006119Pt 245114512317158915
- TakimotoEChampionHCLiMChronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophyNat Med200511221422215665834
- AsaiASahaniNKanekiMOuchiYMartynJAYasuharaSEPrimary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophyPLoS One200728e80617726536
- AdamoCMDaiDFPercivalJMSildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophyProc Natl Acad Sci U S A201010744190791908320956307
- MokELetellierGCuissetJMLack of functional benefit with glutamine versus placebo in Duchenne muscular dystrophy: a randomized crossover trialPLoS One200945e544819421321
- KumamotoTFujimotoSItoTHorinouchiHUeyamaHTsudaTProteasome expression in the skeletal muscles of patients with muscular dystrophyActa Neuropathol2000100659560211078210
- TidballJGSpencerMJCalpains and muscular dystrophiesInt J Biochem Cell Biol20003211510661889
- SawadaHNagahiroKKikukawaYTherapeutic effect of camostat mesilate on Duchenne muscular dystrophy in mdx miceBiol Pharm Bull20032671025102712843632
- BriguetAErbMCourdier-FruhIEffect of calpain and proteasome inhibition on Ca2+-dependent proteolysis and muscle histopathology in the mdx mouse. FASEB JDec2008221241904200
- ChildersMKBoganJRBoganDJChronic administration of a leupeptin-derived calpain inhibitor fails to ameliorate severe muscle pathology in a canine model of duchenne muscular dystrophyFront Pharmacol201128922291646
- BonuccelliGSotgiaFSchubertWProteasome inhibitor (MG-132) treatment of mdx mice rescues the expression and membrane localization of dystrophin and dystrophin-associated proteinsAm J Pathol200316341663167514507673
- BonuccelliGSotgiaFCapozzaFGazzerroEMinettiCLisantiMPLocalized treatment with a novel FDA-approved proteasome inhibitor blocks the degradation of dystrophin and dystrophin-associated proteins in mdx miceCell Cycle20076101242124817495527
- GazzerroEAsseretoSBonettoATherapeutic potential of proteasome inhibition in Duchenne and Becker muscular dystrophiesAm J Pathol201017641863187720304949
- AsseretoSStringaraSSotgiaFPharmacological rescue of the dystrophin-glycoprotein complex in Duchenne and Becker skeletal muscle explants by proteasome inhibitor treatmentAm J Physiol Cell Physiol20062902C577C58216192300
- AcharyyaSVillaltaSABakkarNInterplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophyJ Clin Invest2007117488990117380205
- PorrecaEGuglielmiMDUnciniAHaemostatic abnormalities, cardiac involvement and serum tumor necrosis factor levels in X-linked dystrophic patientsThromb Haemost199981454354610235436
- WhiteheadNPYeungEWAllenDGMuscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen speciesClin Exp Pharmacol Physiol200633765766216789936
- De BosscherKSchmitzMLVanden BergheWPlaisanceSFiersWHaegemanGGlucocorticoid-mediated repression of nuclear factor-kappaB-dependent transcription involves direct interference with transactivationProc Natl Acad Sci U S A1997942513504135099391055
- PanYChenCShenYCurcumin alleviates dystrophic muscle pathology in mdx miceMol Cells200825453153718460899
- DurhamWJArbogastSGerkenELiYPReidMBProgressive nuclear factor-kappaB activation resistant to inhibition by contraction and curcumin in mdx miceMuscle Nerve200634329830316718687
- TangYReayDPSalayMNInhibition of the IKK/NF-kappaB pathway by AAV gene transfer improves muscle regeneration in older mdx miceGene Ther201017121476148320720575
- PetersonJMKlineWCananBDPeptide-based inhibition of NF-kappaB rescues diaphragm muscle contractile dysfunction in a murine model of Duchenne muscular dystrophyMol Med2011175–650851521267511
- TilstraJRehmanKKHennonTPlevySEClemensPRobbinsPDProtein transduction: identification, characterization and optimizationBiochem Soc Trans200735Pt 481181517635154
- ReayDPYangMWatchkoJFSystemic delivery of NEMO binding domain/IKKgamma inhibitory peptide to young mdx mice improves dystrophic skeletal muscle histopathologyNeurobiol Dis201143359860821624467
- ReayDPNiizawaGAWatchkoJFEffect of NF-kappaB inhibition on AAV9 minidystrophin gene transfer to the mdx mouseMol Med201218146647622231732
- LeaskAAbrahamDJTGF-beta signaling and the fibrotic responseFASEB J200418781682715117886
- BernasconiPTorchianaEConfalonieriPExpression of transforming growth factor-beta 1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokineJ Clin Invest1995962113711447635950
- BurksTNCohnRDRole of TGF-beta signaling in inherited and acquired myopathiesSkelet Muscle2011111921798096
- BizarioJCCerriDGRodriguesLCImatinib mesylate ameliorates the dystrophic phenotype in exercised mdx miceJ Neuroimmunol20092121–29310119508953
- HuangPZhaoXSFieldsMRansohoffRMZhouLImatinib attenuates skeletal muscle dystrophy in mdx miceFASEB J20092382539254819289603
- MorrisCASelsbyJTMorrisLDPendrakKSweeneyHLBowman-Birk inhibitor attenuates dystrophic pathology in mdx miceJ Appl Physiol201010951492149920847128
- RoffeSHagaiYPinesMHalevyOHalofuginone inhibits Smad3 phosphorylation via the PI3K/Akt and MAPK/ERK pathways in muscle cells: effect on myotube fusionExp Cell Res201031661061106920060825
- TurgemanTHagaiYHuebnerKPrevention of muscle fibrosis and improvement in muscle performance in the mdx mouse by halofuginoneNeuromuscul Disord2008181185786818672370
- HuebnerKDJassalDSHalevyOPinesMAndersonJEFunctional resolution of fibrosis in mdx mouse dystrophic heart and skeletal muscle by halofuginoneAm J Physiol Heart Circ Physiol20082944H1550H156118263710
- GosselinLEWilliamsJEDeeringMBrazeauDKourySMartinezDALocalization and early time course of TGF-beta 1 mRNA expression in dystrophic muscleMuscle Nerve200430564565315389721
- ShiSHoogaarsWMde GorterDJBMP antagonists enhance myogenic differentiation and ameliorate the dystrophic phenotype in a DMD mouse modelNeurobiol Dis201141235336020940052
- CohnRDvan ErpCHabashiJPAngiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic statesNat Med200713220421017237794
- SpurneyCFSaliAGuerronADLosartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx miceJ Cardiovasc Pharmacol Ther2011161879521304057
- NelsonCAHunterRBQuigleyLAInhibiting TGF-beta activity improves respiratory function in mdx miceAm J Pathol201117862611262121641384
- Rafael-FortneyJAChimanjiNSSchillKEEarly treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in Duchenne muscular dystrophy miceCirculation2011124558258821768542
- McPherronACLawlerAMLeeSJRegulation of skeletal muscle mass in mice by a new TGF-beta superfamily memberNature1997387662883909139826
- LangleyBThomasMBishopASharmaMGilmourSKambadurRMyostatin inhibits myoblast differentiation by down-regulating MyoD expressionJ Biol Chem200227751498314984012244043
- WahlMACE-031 clinical trials in Duchenne MD stopped for nowTucson, AZQuest2011 [cited August 8, 2012]. Available from: http://quest.mda.org/news/ace-031-clinical-trials-duchenne-md-stopped-nowAccessed August 18, 2012
- WagnerKRFleckensteinJLAmatoAAA phase I/IItrial of MYO-029 in adult subjects with muscular dystrophyAnn Neurol200863556157118335515
- QiaoCLiJZhengHHydrodynamic limb vein injection of adeno-associated virus serotype 8 vector carrying canine myostatin propeptide gene into normal dogs enhances muscle growthHum Gene Ther200920111018828709
- KangJKMalerbaAPopplewellLFosterKDicksonGAntisense-induced myostatin exon skipping leads to muscle hypertrophy in mice following octa-guanidine morpholino oligomer treatmentMol Ther201119115916420924365
- KemaladewiDUHoogaarsWMvan HeiningenSHDual exon skipping in myostatin and dystrophin for Duchenne muscular dystrophyBMC Med Genomics201143621507246
- Barton-DavisERShoturmaDIMusaroARosenthalNSweeneyHLViral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle functionProc Natl Acad Sci U S A1998952615603156079861016
- GregorevicPPlantDRLeedingKSBachLALynchGSImproved contractile function of the mdx dystrophic mouse diaphragm muscle after insulin-like growth factor-I administrationAm J Pathol200216162263227212466140
- GehrigSMvan der PoelCHoeflichANaimTLynchGSMetzgerFTherapeutic potential of PEGylated insulin-like growth factor I for skeletal muscle disease evaluated in two murine models of muscular dystrophyGrowth Horm IGF Res2012222697522424862
- AbmayrSGregorevicPAllenJMChamberlainJSPhenotypic improvement of dystrophic muscles by rAAV/microdystrophin vectors is augmented by Igf1 codeliveryMol Ther200512344145016099410