Abstract
The clinical significance of disturbed mitochondrial function in the eye has emerged since mitochondrial DNA (mtDNA) mutation was described in Leber’s hereditary optic neuropathy. The spectrum of mitochondrial dysfunction has become apparent through increased understanding of the contribution of nuclear and somatic mtDNA mutations to mitochondrial dynamics and function. Common ophthalmic manifestations of mitochondrial dysfunction include optic atrophy, pigmentary retinopathy, and ophthalmoplegia. The majority of patients with ocular manifestations of mitochondrial disease also have variable central and peripheral nervous system involvement. Mitochondrial dysfunction has recently been associated with age-related retinal disease including macular degeneration and glaucoma. Therefore, therapeutic targets directed at promoting mitochondrial biogenesis and function offer a potential to both preserve retinal function and attenuate neurodegenerative processes.
Introduction
The importance of optimal mitochondrial function for ocular health has been clear since the first mitochondrial DNA (mtDNA) disease mutation was discovered in Leber’s hereditary optic neuropathy (LHON).Citation1 Other syndromic mtDNA diseases often have retinal involvement together with variable central nervous system pathology. A second major disease grouping classified as “mitochondrial” disease is due to mutations in nuclear genes that result in mitochondrial dysfunction, including autosomal dominant optic atrophy (ADOA), Friedreich’s ataxia, Mohr-Tranebjaerg syndrome, and Charcot-Marie-Tooth disease subtype CMT2A. These disorders commonly display optic neuropathy together with variable central nervous system involvement, and have been described in detail elsewhere.Citation2,Citation3 The neuro-ophthalmic manifestations of mitochondrial diseases have also been extensively reviewed by Newman et al.Citation4
With our increased awareness, the spectrum of “mitochondrial disease” has expanded from describing mtDNA disease, to diseases secondary to improper function of any protein located in the organelle resulting in abnormal mitochondrial function. Moreover, mitochondrial dysfunction is attracting growing attention as contributing to the pathogenesis of many common sporadic age-related neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and glaucoma.Citation5–Citation8 While controversy exists as to whether mitochondrial impairment in these diseases is primary or secondary to upstream disease pathways, the mitochondrion is emerging as pivotal in disease pathogenesis and as an important target of novel therapeutic approaches. The eye can therefore be viewed as a model for energetic impairment in the central nervous system, often being the first neuronal tissue affected by mitochondrial failure. The eye may become a model of therapeutic experimentation, with direct implications for degenerative brain diseases. Here we discuss the variable ocular involvement in inherited mitochondrial diseases, the possible role of mitochondrial dysfunction in the common age-related ophthalmic diseases, including glaucoma and age-related macular degeneration, and finally we review emerging therapeutic approaches to improving mitochondrial function.
Mitochondria and neurons
The accurate cliché of the mitochondrion being the “powerhouse of the cell” has been complicated by the growing recognition that this organelle is a central node of key cellular pathways governing not only intermediary metabolism, but also stress responses and cell death.Citation9 We first consider the traditional energetic role of the organelle and the effects on retinal neurons resulting from mtDNA mutations.
Neurons require large amounts of adenosine triphosphate (ATP) supplied by the mitochondria. Energetic needs are greatest at dendritic regions where ATP-dependent ion pumping reinstates the plasma membrane electrical potential consequent to impulse transmission.Citation10 Neuronal anatomy, with long processes extending from the cell body where mitochondria are synthesized, requires the purposeful transport of organelles along axons and dendrites to the sites of ATP usage. This transport is accomplished by energy-dependent bidirectional transport along microtubules. Kinesin moves mitochondria in the anterograde direction, whereas retrograde transport is via dynein motors.Citation11 Mitochondrial network dynamics is a growing area in mitochondrial research, with the discovery of genes involved in the constant fission and fusion of organelles. The optic atrophy 1 (autosomal dominant) gene (OPA1), the most common gene mutated in ADOA, encodes a dynamin-related GTPase of the mitochondrial inner membrane that directs fusion of this membrane. Why disruption of mitochondrial dynamics due to loss of OPA1 function results in specific loss of retinal ganglion cells remains unknown.
The simplistic idea that different degrees of energetic impairment lead to a hierarchy of neuronal populations being adversely affected does not adequately explain the pathological changes in the central nervous system in mitochondrial diseases. Retinal ganglion cells appear to be one of the most sensitive neurons to mitochondrial failure, although the reasons for this susceptibility remain unclear.
mtDNA and oxidative phosphorylation
Mitochondrial DNA is a circular, double-stranded DNA molecule residing in the mitochondrial matrix. It is the only non-nuclear DNA in mammalian cells, coding 13 of the approximately 90 protein subunits of the oxidative phosphorylation complexes. The majority of the protein machinery required for mitochondrial replication, transcription, translation, and assembly is encoded by nuclear genes,Citation12 whilst mtDNA contributes a 12SrRNA, a 16SrRNA, and 22 tRNAs.Citation13 The oxidative phosphorylation pathway produces the majority of ATP for use in all cells. It comprises five multisubunit enzyme complexes: complex I, the NADH:ubiquinone oxidoreductase (>45 subunits, seven from mtDNA); complex II, the succinate dehydrogenase (four nuclear subunits); complex III, the ubiquinone:cytochrome c oxidoreductase (11 subunits, one from mtDNA); complex IV, cytochrome c oxidase (13 subunits, three from mtDNA); and complex V, H+ATPsynthase (15 subunits, two from mtDNA). Electrons enter the respiratory chain at either complex I from the oxidation of NADH or complex II from oxidation of FADH2. As pairs of electrons travel via redox centers in complexes I, III, and IV, protons are extruded into the intermembrane space where the proton gradient is harnessed by complex V to phosphorylate ADP to ATP.Citation14
Dysfunction of oxidative phosphorylation consequent to mtDNA or nuclear gene mutations can result in a reduction in maximal ATP production rate and increased reactive oxygen species production by complexes I and III,Citation15,Citation16 heightening oxidative stress within the cell.Citation17 mtDNA is highly susceptible to damage by reactive oxygen species due in part to the lack of protective DNA binding histones,Citation18 limited DNA repair mechanisms,Citation19,Citation20 and the close proximity of mtDNA to the site of production of reactive oxygen species, the oxidative phosphorylation machinery. And unlike nuclear genes, mtDNA exists in hundreds to thousands of copies per cell, is replicated throughout life, and is maternally inherited. The extent to which mtDNA mutations produce pathologic changes in tissues depends on the balance between normal and mutant mtDNA populations in cells and tissues (heteroplasmy) and the resilience of tissues to impairment of oxidative phosphorylation (threshold effect), resulting in varied phenotypes and affected tissues in mitochondrial diseases.Citation21,Citation22 The differential expression of components of the electron transport chain in various tissues and the segregation of mitochondria during developmentCitation23 has also been implicated in tissue-specific diseases.Citation24
mtDNA haplogroups
The mtDNA genome accumulates mutations at a much higher rate than nuclear DNA, and during human evolution certain mutation groups (haplogroups) have evolved and become fixed in specific populations.Citation25 A haplogroup is a cluster of stable background mtDNA polymorphisms in individuals from a common female ancestor.Citation26 Specific mtDNA haplogroups have been associated with pseudoexfoliative glaucoma,Citation27,Citation28 primary angle-closure glaucoma,Citation29 age-related macular degeneration,Citation30–Citation32 and LHON.Citation32–Citation40
The implication of these associations is that the mtDNA sequence background may influence the clinical phenotype of a disease by subtle effects on oxidative phosphorylation function.Citation40 Interestingly, haplogroup associations in healthy populations have been demonstrated to influence maximum oxygen consumption, where healthy males carrying haplogroup J had lower efficiency of the electron transport chain and ATP production,Citation41 which may explain the increased prevalence of the J haplogroup in LHON patients.Citation40 At a molecular level, the haplogroup may influence the cellular response to stress, because LHON cells with point mutations m.11778G>A or m.14484T>C from mtDNA haplogroup J had markedly increased susceptibility to a neurotoxic metabolite compared with the same mutations on different mtDNA haplogroup backgrounds, such as the U and H haplogroups.Citation42 Certain haplogroups also delay the assembly kinetics of complexes I, II, and IV into oxidative phosphorylation supercomplexes,Citation43 where this cooperative stability is required to maintain efficient respiration.Citation44
Mitochondrial dynamics
If mitochondrial distribution in neurons is impaired, local energetic crisis may occur even if oxidative phosphorylation remains optimal. Within the eye, the optic nerve is structurally unique, consisting of retinal ganglion cell axons originating in the unmyelinated retinal nerve fiber layer which turn 90 degrees through a series of perforated collagen plates, known as the lamina cribrosa, at the optic nerve head, where mitochondrial density decreases as myelination begins.Citation45–Citation51
Mitochondrial shape and distribution are regulated by two opposing processes, ie, fission and fusion. Fission is mediated by dynamin-related protein 1 and fission 1 (mitochondrial outer membrane) homolog (Saccharomyces cerevisiae), whilst fusion is regulated by OPA1, mitofusin-1, and mitofusin-2. These processes are vital to maintain adequate mixtures of mtDNACitation52,Citation53 and to respond to localized metabolic demands.Citation54 Changes in the expression of fission/fusion proteins alter mitochondrial shape and size, and may promote or inhibit apoptotic signals leading to neuronal death. Neuronal cells have markedly high expression levels of dynamin-related protein 1 and OPA1 compared with non-neuronal cells.Citation55 Furthermore, knockdown of endogenous dynamin-related protein 1 significantly increases the mitochondrial length in all cell types, but only selectively causes apoptosis of cortical neurons,Citation55 indicating the sensitivity of specific neuronal populations to loss of control of mitochondrial dynamics.
Mitochondrial fission/fusion proteins and their regulators are associated with optic nerve loss. OPA1 mutation is the commonest cause of ADOA.Citation2–Citation4 Mutations in the outer membrane profusion protein, mitofusin-2, in Charcot-Marie-Tooth diseaseCitation56,Citation57 impair nerve conductive velocity in peripheral neurons, and lead to subacute optic atrophy in some pedigrees.Citation58 Evidence is emerging that involvement of single nucleotide polymorphisms of mitofusin-1 and mitofusin-2 genes is present in normal tension glaucoma.Citation59 Presenilin-associated rhomboid-like protease (PARL) regulates mitochondrial fusionCitation60 and processes OPA1 into a soluble form.Citation61 The T191C genetic variation of PARL in normal populations exhibited significant effects on mitochondrial content levels.Citation62 Single nucleotide polymorphisms of PARL have been associated with glaucoma, LHON, and mutations causing Parkinson’s disease.Citation59,Citation63,Citation64
Altering the fission-fusion balance has profound effects on mitochondrial function and structure. With pharmaceutical or genetic inhibition of fusion, there is a collapse of mitochondrial energy production as mitochondria consume ATPCitation65 which alters mitochondrial distribution toward fission,Citation54,Citation66 promoting apoptosis.Citation65,Citation67 Converse to mitochondrial fission, promoting mitochondrial fusion increases mitochondrial energy productionCitation68 which may be protective against injury.Citation69,Citation70 Increased mitochondrial fusion has been demonstrated to protect cells from autophagy by boosting mitochondrial cristae volume and enhancing mitochondrial ATP synthase activity.Citation65 Upregulation of OPA1 in mouse models was protective against retinal ganglion cell death in a glaucoma mouse line, DBA2 J,Citation71 and may be an important cellular defense mechanism against glaucomatous optic neuropathy.Citation72
Mitochondrial diseases and optic neuropathies
LHON is characterized by degeneration of retinal ganglion cells and their axons. LHON is the most common inherited mtDNA disease, and over 90% of cases are due to mutations in one of three mtDNA-encoded oxidative phosphorylation complex I genes, ie, G11778A/ND4, T14484C/ND6, and G3460A/ND1.Citation2–Citation4 These mtDNA mutations lead to decreased complex I enzyme ratesCitation73 and lowered ATP production,Citation74 which are hypothesized to sensitize retinal ganglion cells to apoptosis.Citation2,Citation3 Patients typically have normal vision until the age of 15–35 years, when a rapid loss of central vision occurs in one eye, then the second eye within one year. Visual loss progresses to 20/200 or worse, with visual field testing revealing central or centrocecal scotomas.Citation3 Axonal loss in the papillomacular bundle results in temporal atrophy of the optic nerve head. While maternal inheritance provides a strong diagnostic clue, penetrance of LHON is variable within kindreds, and males are overrepresented, with 80%–90% of affected individuals being male.Citation4
ADOA, also known as Kjer’s optic neuropathy,Citation75 also leads to vision impairment due to selective and specific degeneration of retinal ganglion cells and their axons in the optic nerve.Citation3,Citation76,Citation77 ADOA results from haploinsufficiency of the mitochondrial fusion protein, OPA1.Citation78 In ADOA, there is evidence of decreased mtDNA contentCitation79 and an increased prevalence of mtDNA mutations in severe cases of ADOA (ADOA+) where multiple deletions of mtDNA were identified.Citation80,Citation81 In addition to its role in maintaining mitochondrial cristae structure,Citation68,Citation82–Citation84 other functions of OPA1 may include ensuring adequate “mixing” of mtDNA.Citation54 Small hydrophobic peptide fragments from cleaved OPA1 may also contribute to mtDNA nucleoid attachment to the inner mitochondrial membrane, promoting mtDNA replication and distribution.Citation85
Disrupted mtDNA distribution, or cristae structure, is predicted to have secondary impacts on oxidative phosphorylation due to either inadequate mtDNA transcription or lack of inner membrane surface area for oxidative phosphorylation complex anchoring. It has been reported that decreased oxidative phosphorylation capacity in ADOA patients with OPA1 mutations correlates with relatively poor visual acuities, while related mutation carriers with normal vision appeared to have relatively preserved oxidative phosphorylation function.Citation86 This suggests that patients with preserved vision may harbor genetic variants that allow some compensation of oxidative phosphorylation function.
Syndromic mtDNA diseases with ocular involvement
Retinopathy
Several syndromic central nervous system diseases are also known to result from mtDNA mutations. While optic neuropathy is an occasional finding in these disorders, a pigmentary retinopathy is the commonest retinal pathology. This is best illustrated in the neurogenic atrophy and retinitis pigmentosa syndrome, which results from point mutations in the mtDNA ATPase-6 gene, commonly T8993G. Patients typically present with retinitis pigmentosa with or without optic neuropathy, and can develop dystonia.Citation87 Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) can result from many mtDNA point mutations, although the most common is the A3243G mutation in the tRNALeu gene. MELAS patients present with stroke-like episodes that lead to frequent retrochiasmal visual loss, but often also have pigmentary retinopathy without optic atrophy.Citation88 The spectrum of disease arising from the A3243G point mutation is also evidenced by its contribution to the pathogenesis of maternally inherited diabetes and deafness.Citation89,Citation90 This is a multisystemic disease characterized by sensorineural deafness, retinal abnormalities, and diabetes, commonly arising in the third to fourth decade of life.Citation89,Citation91 The retinal abnormalities, typically occurring in maternally inherited diabetes and deafness, are bilateral in the majority of cases, and involve two main phenotypes, as described by Rath et al.Citation92 The most common phenotype involves circumferential perifoveal atrophy with retinal pigment epithelium hyperpigmentation sparing of the fovea. The second phenotype is a pattern dystrophy characterized by relative sparing of the fovea, with diffuse granularity and pigment clumping, and retinal pigment epithelium within the retinal vascular arcades. However, despite the degree of atrophy, visual acuity is preserved, with a good prognosis.Citation93
Chronic progressive external ophthalmoplegia
Chronic progressive external ophthalmoplegia is a phenotype of mitochondrial myopathy with ophthalmic involvement. The presenting signs are of bilateral ptosis, which generally precedes development of ophthalmoplegia. Whilst visual acuity is preserved, progressive fibrosis of the extraocular muscles can manifest as diplopia.Citation4 Eyelid muscles are also affected and can contribute to exposure-related corneal disease. Chronic progressive external ophthalmoplegia is attributed to sporadic mutations, nuclear inherited mutations, and maternally inherited mutations of mtDNA. Most cases of chronic progressive external ophthalmoplegia arise sporadically from large-scale rearrangement of mtDNA resulting in defective oxidative phosphorylation.Citation4,Citation94 Nuclear inherited chronic progressive external ophthalmoplegia is attributed to multiple large-scale mutations in mtDNA affecting mtDNA replication and repair.Citation4 Maternally inherited chronic progressive external ophthalmoplegia commonly arises from point mutations in mtDNA tRNA genes.Citation95 The Kearns-Sayre syndrome is the severest form of chronic progressive external ophthalmoplegia, where the presenting feature is usually ptosis and ophthalmoplegia. Kearns-Sayre patients often develop a pigmentary retinopathy together with cardiac conduction defects and severe neurological signs, including ataxia.Citation96
Mendelian mitochondrial syndromes with ocular involvement
Several nuclear gene mitochondrial disorders have optic neuropathy as a secondary feature. These are considered briefly here because they result from mutations in mitochondrial proteins that indirectly interfere with oxidative phosphorylation, and so can add further insights into energetic failure and retinal pathogenesis.
Friedreich’s ataxia results from a GAA trinucleotide repeat expansion in the frataxin gene. Frataxin is a mitochondrial protein involved in Fe–S cluster assembly, disruption of which results in excessive mitochondrial iron levels and loss of retinal ganglion cells, giving rise to optic neuropathy and ataxia.Citation2,Citation4,Citation97 The oxidative phosphorylation pathway relies on the redox ability of Fe, via multiple Fe–S clusters, to perform electron transfer and thus energy transduction. Four of the five oxidative phosphorylation complexes contain either or both heme prosthetic groups or Fe–S clusters. The pattern of retinal ganglion cell loss in Friedreich’s ataxia is more diffuse than that seen in LHON and ADOA, not preferentially involving the papillomacular bundle but involving the optic radiations, and is slowly progressive.Citation2,Citation98,Citation99
Mohr-Tranebjaerg syndrome is an X-linked recessive disease characterized by deafness, dystonia, and optic atrophy.Citation100 It is caused by mutation of the deafness/dystonia peptide, also called TIMM8A.Citation101 TIMM8A is one of several proteins forming the translocase of the inner membrane complex, which together with the translocase of the outer membrane forms the machinery of mitochondrial protein import.Citation102,Citation103 Because the oxidative phosphorylation complexes are among the most abundant of mitochondrial proteins, perturbation of protein import is likely to impact on the oxidative phosphorylation pathway, although mitochondrial studies in patients have not yet been reported. Retinal ganglion cell loss appears to be similar to that reported for Friedreich’s ataxia, with diffuse involvement of the optic radiations.Citation104,Citation105
Hereditary spastic paraplegia is a disease grouping that can result from mutations in a number of nuclear genes. It is characterized by progressive spasticity of the lower limbs, frequently complicated by the presence of optic atrophy. One of these variants is caused by mutations in the SPG7 gene coding for paraplegin, an AAA-type metalloprotease of the mitochondrial inner membrane. Mutation of paraplegin has been reported to cause impairment of oxidative phosphorylation complex I, providing a pathogenetic link to LHON.Citation106 Pathological descriptions of the optic nerve have not been reported.
As mentioned above, when discussing mitochondrial dynamics, Charcot-Marie-Tooth disease subtype CMT2A has been associated with mutations in the mitochondrial fusion protein, mitofusin-2.Citation107 Charcot-Marie-Tooth disease is a common inherited peripheral neuropathy; the variant CMT2A also displays an optic neuropathy that typically develops a decade after onset of the neuropathy. Visual decline progresses rapidly, with bilateral central scotomas evident on fundus examination, reminiscent of LHON.Citation2,Citation4 Mitofusin-2 is a GTPase similar in structure to OPA1, but is located in the mitochondrial outer membrane. No consistent oxidative phosphorylation phenotype has been identified, although studies are limited.Citation2 Pathological reports indicate a pattern of retinal ganglion cell loss similar to LHON, with preferential involvement of the papillomacular bundle.Citation58
Oxidative phosphorylation defects and preferential retinal ganglion cell pathology
The very common but far from invariant finding of preferential retinal ganglion cell loss in mitochondrial diseases raises fascinating questions for pathogenesis. From the early “metabolic maps” provided by Kageyamia and Wong-Riley,Citation48 who pioneered histochemical reaction for the oxidative phosphorylation complex IV (cytochrome oxidase) to map mitochondrial density in the retina, it was clear that inner segments of the photoreceptor have the highest degree of enzymatic activity, followed by the inner and outer plexiform layers and the cell bodies of the retinal ganglion cells, and their axons forming the nerve fiber layer. Yet only in a minority of mitochondrial disease patients are the photoreceptors affected, such as found in the pigmentary retinopathies seen in MELAS, neurogenic atrophy, and retinitis pigmentosa, and Kearns-Sayre syndromes. The lack of outer retinal involvement in ADOA and CMT2A, disorders of mitochondrial fission/fusion, is circumstantial evidence that only neurons with long axons suffer from disordered mitochondrial fission and fusion.
Within the mtDNA diseases, how can different defects in the same pathway (oxidative phosphorylation) most commonly give rise to loss of retinal ganglion cells, yet sometimes cause photoreceptor loss in the absence of optic neuropathy? This may in part be explained by the high metabolic activity of the retinal pigment epithelium which forms the retina-brain barrier and is richly endowed with mitochondria.Citation10 Oxidative stress may be heightened by the high local oxygen concentrations due to proximity to the choroid and the daily outer segment phagocytosis by the retinal pigment epithelium that leads to accumulation of the phototoxin, N-retinyl-N-retinylidene ethanolamine. It is possible that the point mutations responsible for MELAS, neurogenic atrophy, and retinitis pigmentosa syndromes, and the mtDNA deletions associated with Kearns-Sayre syndrome/chronic progressive external ophthalmoplegia result in heightened oxidative stress compared with the LHON mutations. Experimentally, when retinal pigment epithelium oxidative phosphorylation is specifically ablated in postnatal mice using a cre-lox tFAM knockdown approach, the retinal pigment epithelium undergoes dedifferentiation and a secondary photoreceptor degeneration results.Citation108 Histopathological studies in limited cases of mtDNA-linked pigmentary retinopathy suggest a secondary disruption of photoreceptors consequent to retinal pigment epithelium failure.Citation4
Both LHON and ADOA can occur in more complex forms, with variable central nervous system involvement. Extraocular features of “LHON plus” syndromes include spastic dystonia, ataxia, or even more severe encephalopathies. It is interesting that mtDNA mutations identified in these cases are usually in complex I genes, are different from the primary LHON mutations, and have more severe defects in oxidative phosphorylation.Citation109–Citation111 It has become clear that ADOA can also be associated with extraocular features, with up to one in six OPA1-linked ADOA patients showing variable combinations of sensorineural deafness, ataxia, peripheral neuropathy, and ophthalmoplegia.Citation112
The clinical and biochemical evidence therefore supports the concept that subtle oxidative phosphorylation defects, especially in complex I genes, result in preferential retinal ganglion cell loss. More severe oxidative phosphorylation defects result in more severe disease, typically affecting the brainstem, basal ganglia or cerebellum, with or without optic neuropathy. The common involvement of sensorineural deafness and peripheral neuropathy in such patients reiterates the theme of neurons with long axons being more vulnerable to mitochondrial dysfunction. Refer to for a summary of mitochondrial disorders with ocular involvement which outlines the complexity of preferential retinal ganglion cell pathology.
Table 1 Mitochondrial diseases with ocular involvement
Age-related neurodegeneration, mitochondria, and the retina
mtDNA somatic mutation, oxidative phosphorylation, and aging
A lingering disadvantage of the semiautonomous nature of mitochondria in long-lived organisms is the necessity for continual replication of the mitochondrial genome. This leads to an age-related loss of mtDNA integrity, and a consequent decline in oxidative phosphorylation in postmitotic tissues.Citation113,Citation114 mtDNA is highly susceptible to damage from reactive oxygen species due to lack of DNA repair and protection mechanisms.Citation18,Citation20 Acquired mtDNA mutations through aging and oxidative stress are increasingly being attributed to diseases of the aging eye and brain.Citation115,Citation116 Evidence of increased point mutations, large-scale rearrangements, and mtDNA depletions has been demonstrated in age-related macular degeneration, subtypes of ADOA, and subtypes of glaucoma.
In subtypes of glaucoma, there is evidence of increased mtDNA mutations in peripheral blood and the trabecular meshwork, particularly in patients with primary open angle glaucoma,Citation117–Citation119 pseudoexfoliation glaucoma,Citation105 and congenital glaucoma.Citation120 Interestingly, mtDNA mutations did not occur, or were in very low abundance in other glaucoma subtypes (pigmented, juvenile, acute, neovascular, and chronic closed angle glaucoma),Citation119,Citation121 once again highlighting a combination of risk factors precipitating retinal ganglion cell loss.
Mouse models that accumulate high levels of mtDNA mutations owing to impairments in mitochondrial polymerase γ proofreading function have been shown to develop phenotypes consistent with accelerated aging. Our group recently demonstrated that mice with neuronal-specific mitochondrial polymerase γ mutations have increased mtDNA deletions and point mutations in their retinas, which increase the sensitivity of the retinal ganglion cell-dominant inner retinal function to acute intraocular pressure injury.Citation122 This insult is known to produce mechanical, metabolic, and oxidative stress in the retina.Citation7 These findings indicate that an accumulation of mtDNA mutations is associated with impairment in neural function and reduced capacity of neurons to resist external stress in vivo, suggesting a potential mechanism whereby the aging central nervous system can become more vulnerable to neurodegeneration.
Glaucoma
Glaucoma is a neurodegenerative disease of the optic nerve characterized by accelerated death of retinal ganglion cells and their axons, leading to progressive visual field loss. Two major risk factors are aging and increased intraocular pressure.Citation123,Citation124 Around 30%–40% of patients do not present with intraocular pressures above population means, indicating that glaucoma is a complex disease, and age-related mitochondrial failure has been hypothesized to play a role.Citation125–Citation127
Despite some clinical similarities, the pattern of retinal ganglion cell loss is considerably different in glaucoma to that seen in LHON and ADOA. In the latter diseases, the small fibers of the papillomacular bundle are preferentially affected, resulting in temporal atrophy and central vision loss. In primary open angle glaucoma, peripapillary atrophy classically leads to an arcuate, peripheral vision loss. However, this cannot be taken as evidence for a lack of oxidative phosphorylation involvement in some patients with glaucoma. As noted earlier for the nonsyndromic mitochondrial optic neuropathies, different patterns of optic nerve, retinal, and wider visual system pathology can result from mitochondrial dysfunction.
Age-related macular degeneration
Age-related macular degeneration is a late-onset neurodegenerative disease that shares several clinical and biological features associated with Alzheimer’s disease. In most cases, age-related macular degeneration involves the buildup of protein plaques, known as drusen, in the central macular region of the retina. Stress stimuli including oxidative stress, aging, genetic factors, and inflammation may drive both age-related macular degeneration and Alzheimer’s disease pathogenesis,Citation128 including the depositing of protein plaques in the retina or brain. Similarities in these two diseases are also seen with polymorphisms in the risk factor gene, APOE, being associated with age-related macular degeneration,Citation129,Citation130 as well as Alzheimer’s disease.Citation131,Citation132 The APOE gene regulates the homeostasis of triglycerides and cholesterol,Citation133 and loss of function of APOE has been linked to the deposit of senile plaques, mainly comprised of the amyloid beta peptide,Citation134 which build up in drusenCitation135,Citation136 and also colocalize with another risk factor for age-related macular degeneration, ie, complement protein.Citation137,Citation138 Evidence indicates the genotype of APOE may dictate the risk of Alzheimer’s disease and other chronic disorders, largely due to its impact on oxidative stress modulation.Citation139
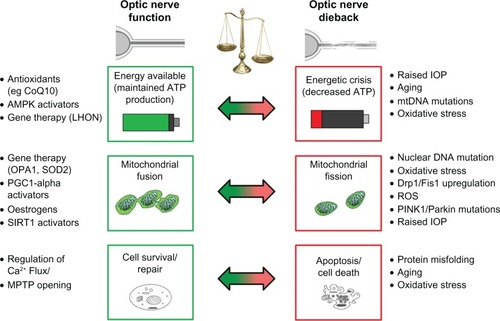
Age-related macular degeneration is divided into two major forms, ie, the “wet” form caused by leakage from choroidal neovascularization into the subretinal space, and the more common “dry” form associated with buildup of drusen in the macula.Citation140 There is an increased prevalence of large-scale mtDNA rearrangements and deletions in both the bloodCitation141 and retinasCitation142,Citation143 of patients with age-related macular degeneration. There are also increased rates of single nucleotide polymorphisms in the noncoding mtDNA control region (d-loop) in retinas with age-related macular degeneration,Citation30 which has been observed in Alzheimer’s disease and other oxidative stress conditions.Citation144 It is likely that an increased rate of mtDNA deletions and single nucleotide polymorphisms diminishes the number and density of mitochondria, which may partially explain the decreased mitochondrial density observed in age-related macular degeneration retinas.Citation145 The various components affecting mitochondrial function and the dynamics and mechanisms by which they contribute to mitochondrial disorders of the eye are summarized in .
Figure 1 Factors contributing to optic nerve degeneration.
Diagnostic approaches
In some cases, a clinical phenotype can be a strong predictor of the underlying molecular features. In both LHON and ADOA, evaluation of history, optic nerve head morphology, and optic nerve function can be predictive of disease.Citation146 However, both ADOA and LHON may be misdiagnosed as glaucoma on optic nerve head examination alone, and training programs may aid in accurate diagnosis of glaucomaCitation147 and other optic neuropathies.Citation148 When a mutation is suspected in retinal degenerative conditions, several considerations for patient counseling must be investigated. These include a positive family history of disease, typical clinical ophthalmic features for known mitochondrial diseases (including retinal and optic disc appearance, and optic nerve head appearance),Citation148 and laboratory investigationsCitation149,Citation150 which all aid identification of genetic involvement.
The complexity of mitochondrial disorders is enhanced by the dual genetic coding of mitochondrial oxidative phosphorylation from both nDNA and mtDNA. The mode of inheritance of mitochondrial disease can be maternal, autosomal dominant, autosomal recessive, or X-linked, which complicates genetic counseling for suspected patients and families.Citation151 Mitochondrial DNA mutations, either heritable or acquired through aging and as a consequence of oxidative stress, may underlie retinal ganglion cell loss. Identifying the type of mtDNA mutation is challenging, because mutations may be either heritable at known positions (eg, in LHON) or due to single nucleotide polymorphisms, point mutations, or large-scale deletions and rearrangements. Diagnosis of mtDNA mutation also faces the challenge that different tissues may harbor different levels of mutant and wild-type mtDNA (heteroplasmy).Citation21 In LHON patients, varying levels of heteroplasmy of mutant mtDNA were identified between hair follicles and blood cells,Citation152 and a LHON patient had higher levels of mutant mtDNA in their retina and optic nerves than in their blood cells at post mortem.Citation153 In age-related macular degeneration, post mortem analysis of retinas and peripheral blood demonstrated a greater accumulation of rearrangements and deletions of mtDNA in retinas.Citation141
Aside from genetic screening, functional laboratory investigations may give clues to the underlying pathogenesis of the mitochondrial disease. Typical laboratory investigations require sampling of biopsy tissue (eg, leucocytes, muscle, fibroblasts) for functional mitochondria studies.Citation149,Citation150 Detailed oxidative phosphorylation enzymatic studies may be undertaken from the tissue or cell line, but technique standardization is requiredCitation149,Citation154 due to interlaboratory variation in methods.Citation155 Mitochondrial function can also be determined noninvasively. Phosphorus magnetic resonance spectroscopy is well suited to gathering clinical data on skeletal muscle energetics, and is a very sensitive index of mitochondrial function which has been used to study bioenergetic defects in ADOA and LHON.Citation3,Citation81,Citation156–Citation158
Coupled with a positive clinical appearance, a better molecular understanding of the mitochondrial involvement in conditions of the eye will guide therapeutic development. We believe the future holds several avenues of therapeutic potential to ameliorate mitochondrial dysfunction, which are outlined below.
Avenues for neuroprotection
Gene therapy in the eye
Since the retina and optic nerve are part of the central nervous system, yet are relatively accessible, this provides an attractive opportunity to study the regenerative responses of adult neuronal cells to gene therapy. Current means of gene delivery may be via the use of replication-deficient viral vectors, recombinant plasmids, or electroporation. Gene replacement of defective genes (eg, LHON complex I), the therapeutic use of antioxidant genes (eg, superoxide dismutase, SOD) and increasing expression of haploinsufficient proteins (eg, OPA1 in ADOA) are currently under investigation. These may also hold promise in the treatment of other degenerative conditions, including glaucoma and optic neuritis. Haploinsufficiency of the OPA1 protein is the underlying pathogenic mechanism of retinal ganglion cell loss in human ADOA patients,Citation78 and OPA1 haploinsufficiency negatively affects retinal ganglion cell survival and function in animal models.Citation159–Citation161 Increasing OPA1 expression is being investigated as a means to ameliorate retinal ganglion cell loss. In cultured cells, the protective effect of increased OPA1 expression prevented cells undergoing apoptosisCitation82,Citation162 and protected against excitotoxicity.Citation163 Adenovirus-associated virus vector (AAV2) transfection of OPA1 into glaucomatous DBA/2 J mouse retinas protected against loss of retinal ganglion cells, with protection persisting up to two months after transfection.Citation71
Retinal ganglion cells are under constant oxidative stress, and boosting antioxidant defenses or providing neurotrophic factors may support retinal ganglion cell survival. Cells with the G1178A LHON mutation suffer increased oxidative stress levels.Citation77 AAV-mediated delivery of mitochondrial SOD (SOD2) improved cell survival when cells were cultured in galactose media where cells are forced to rely on oxidative phosphorylation for all ATP production.Citation164 When the SOD2 concept was transferred into an animal model with severe complex I defects in the retina induced by ribozyme-mediated knockdown of a nuclear complex I gene, the same group demonstrated protection of optic nerve fibers by preventing early apoptosis of retinal ganglion cells.Citation165 There have also been extensive animal trials on the AAV-mediated delivery of trophic factors (eg, ciliary neurotrophic factor) in promoting retinal ganglion cell growth and axonal regeneration in optic nerve transection or optic nerve crush models.Citation166
The ability to reverse, correct, or replace an inherited genetic mutation is a potential strength of gene therapy. Homologous recombination has not been convincingly demonstrated to occur in mtDNA, which has prevented the correction of heritable mtDNA mutations.Citation167–Citation170 This has also challenged modeling of mtDNA diseases in rodents and has required alternate approaches. LHON-like phenotypes were induced either by the intravitreal injection of rotenone, a toxic complex I inhibitor,Citation171–Citation173 or by delivery of mutant human ND subunits into the retina by either electroporationCitation174 or AAV-mediated delivery.Citation164
Various groups are now exploring the concept of replacing mutant complex I genes by allotropic rescue in animal models.Citation172,Citation174,Citation175 This concept involves the introduction of recoded human mtDNA genes, with an added mitochondrial import presequence into the nuclear genome of rodents by AAV delivery. However, these experiments face the uncertainty of whether introducing human mitochondrial genes into rodent cells would result in functional improvement of oxidative phosphorylation. Mismatching of nuclear and mitochondrial encoded oxidative phosphorylation subunits in cybrid models takes advantage of evolutionary divergence, which is sufficient to cause mitochondrial dysfunction.Citation176–Citation178 Thus, the successful delivery of human ND4 subunits into the mitochondrion does not necessarily provide a functional benefit to the murine mitochondria, nor does it guarantee correct assembly into functional oxidative phosphorylation complexes. These findings have also raised questions on whether AAV delivery of highly hydrophobic complex I subunits would be correctly assembled into mitochondrial membranes in vivo.Citation3,Citation179–Citation181
Although some of these results appear promising, much caution needs to be in place before commencing human trials because there are a number of concerns relating to the safety of gene rescue.Citation179,Citation182 To date, no human trials have commenced with AAV-mediated gene delivery into the retina, although patient selection and recruitment is actively taking place for LHON gene therapy trials, and a cohort of patients has already been identified and selected for suitability of inclusion in these trials.Citation183
Regulation of hormonal agents
Phenylbutyrate is a histone deacetylase inhibitor currently being used as a treatment in Parkinson’s disease due to its neuroprotective properties against oxidative cell death.Citation184–Citation187 The action of phenylbutyrate is mediated partly via activation of the protein DJ1, which was protective against mouse models of neurodegeneration induced by treatment with the oxidative phosphorylation complex I inhibitors rotenone or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.Citation187,Citation188 Phenylbutyrate treatment prior to ischemic intraocular pressure elevation (>100 mmHg) in the rat retina also protected against retinal ganglion cell loss and preserved retinal thickness.Citation189 The activation of DJ1 by phenylbutyrate stabilized nuclear factor erythroid 2-related factor, a master regulator of antioxidant transcription response,Citation190 as well as working in parallel with the pink/PARKIN pathway to protect mitochondrial function in the presence of oxidative stress.Citation191,Citation192
In response to histone deacetylase inhibition, estrogen receptor alpha (ERα) is activated and the DNA binding capacity of ERα is increased.Citation193 Deacetylation of ERα is also regulated by histone deacetylator 8 and sirtuin 1 homolog, which act together as independent enhancers of ERα activity.Citation193 Mitochondrial biogenesis involves signaling via numerous transcription factors and transcriptional coactivators, one being estrogen-related receptor (ERR)8α, that works in concert with the peroxisome proliferator-activated receptor gamma (PPARγ) coactivator (PGC1α) family.Citation194 Estrogen receptor signalling may be a means to activate mitochondrial transcriptionCitation195,Citation196 via estradiol treatment,Citation197 which upregulates mtDNA-encoded complex IV genes.Citation198,Citation199 Estrogens may have a vital role in maintaining mitochondrial function during stress, and have been shown to ameliorate the LHON complex I defect by enhancing mitochondrial biogenesis and improving mitochondrial energy production.Citation200 The putative antioxidant role of estrogens has also been supported by evidence from cybrid cell lines of LHON treated with 17β-estradiol, which demonstrated increased antioxidant SOD, stabilization of the mitochondrial membrane potential,Citation201 and more efficient mitochondrial biogenesis.Citation3 Estrogens also contribute to the regulation of glucose metabolism and insulin sensitivity via activation of ERα.Citation202,Citation203 The effect of estrogens on pathways regulating cellular energy levels has been demonstrated by the ability of 17β-estradiol to phosphorylate and activate AMP-activated protein kinase (AMPK), the master initiator of cellular catabolism, independently of AMP level.Citation204
As such, estrogens and novel nonhormonal analogs are of interest to protect neurons against acute brain injury and chronic neurodegeneration,Citation205 and the evidence suggests a protective mechanism against increased intraocular pressure in rodents.Citation206 There is also evidence that estrogen therapy (with estradiol) was able to increase retinal blood flow in rodent models,Citation207 as well as improving optic nerve head topography and optic nerve head blood flow in postmenopausal women on hormone replacement therapy,Citation207 which may protect the retina against injury. 17β-estradiol treatment also conferred neuroprotection in hippocampal neurons exposed to glutamate excitotoxicity by regulation of mitochondrial calcium load.Citation208
Regulation of mitochondrial biogenesis as a therapeutic target
The preservation of healthy mitochondrial function in neurodegeneration has guided emerging mitochondrial therapies towards examining mitochondrial biogenesis as a therapeutic target. Several reviews have highlighted the mechanisms of mitochondrial biogenesis and its role in neuroprotection.Citation209–Citation212
One master transcriptional regulator of mitochondrial biogenesis is PPARγ coactivator (PGC1α).Citation213 PGC1α regulates mitochondrial homeostasis through upregulation of mitochondrial biogenesis, respiration, and controlling the utilization of substrates for energy production.Citation214
The PPARs are ligand-controlled upstream regulators of PGC1α activity involved in transcription of genes responsible for fatty acid transport and mitochondrial oxidation. Synthetic PPAR agonists have been developed for the management of type 2 diabetes (PPARγ agonists, eg, thiazolidediones), and lipid disorders (PPARα agonists, eg, fibrates). The thiazolidedione class of medications has demonstrated a neuroprotective effect of two PPARγ ligands (troglitazone and 15d-PGJ2) in glutamate-induced cytotoxicity in a rat neuronal precursor cell culture model.Citation215 PPARγ agonists also demonstrated a role in neurorecovery following transient ischemic injury.Citation216–Citation218
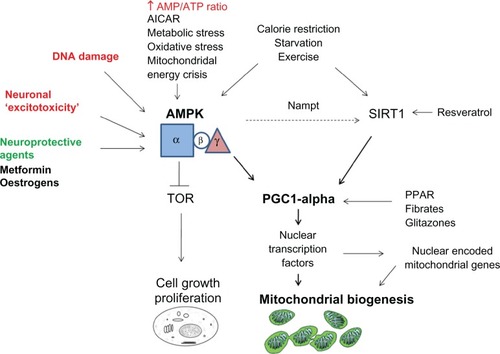
Given the high energy demand yet poor nutrient storage capability of central nervous system neurons,Citation219 it is not surprising that regulation of cellular energy is pivotal in the context of mitochondrial dysfunction in disorders of aging and the eye. AMPK is an evolutionary preserved cellular “calorimeter” common to all eukaryotes that acts as a cellular switch to activate catabolic pathways and turns off anabolic pathways.Citation220,Citation221 AMPK is intimately related to mitochondrial oxidative phosphorylation because the energy-sensing process is sensitive to changes in the ratio of cellular AMP to ATP.Citation222 Low ATP levels activate AMPK by phosphorylation of AMPK at Thr172 in the kinase domain,Citation223 and AMPK is implicated in promoting mitochondrial biogenesis through PGC1α. The glitazone class of medication has been shown to phosphorylate and thereby activate AMPK in muscle by increasing the AMP:ATP ratio.Citation224 Activated nuclear AMPK complexes are able to phosphorylate mitochondrial transcriptional coregulators directly, including PGC1α.Citation225 The potential for pharmacological activation of AMPK in promoting mitochondrial biogenesis and conferring neuroprotection has been suggested by in vitro studies using metformin, a biguanide commonly used in the management of type 2 diabetes.Citation226,Citation227 This highlights several mechanisms by which regulation of PGC1α can exert neuroprotection in the central nervous system.
The silent information regulator T1 (SIRT1) is also influential in mitochondrial function, biogenesis, and neuroprotection. SIRT1 is an enzyme that belongs to the sirtuin gene family. It acts as a metabolic sensor and mediates the regulation of oxidation and energy homeostasis genes through NAD+-dependent deacetylation of transcription factors.Citation214 Independent of deacetylase activity, sirtuins have been implicated in mitochondrial function through antioxidant propertiesCitation228 and regulation of oxidative stress genes.Citation229 Furthermore, SIRT1 activation deacetylates PGC1α, thereby activating mitochondrial biogenesis.Citation230 SIRT1 has also demonstrated immunomodulatory neuroprotection of retinal ganglion cells in models of autoimmune optic neuritis.Citation231 These mechanisms of SIRT1 activation have therefore set the foundation for potential neuroprotective interventions.
Calorie restriction is one such established method of improving mitochondrial biogenesis,Citation232 respiration,Citation233 and reduction of reactive oxygen species production through upregulation of SIRT1.Citation234 Calorie restriction has been shown to facilitate retinal ganglion cell recovery in acute intraocular pressure challenge and ischemia-reperfusion models.Citation235 Glucose restriction has been shown to activate AMPK, which then activated the gene for the NAD synthetic enzyme, Nampt.Citation236 This highlighted an important convergence between AMPK and SIRT1 in mitochondrial biogenesis ().Citation237 Similarly, resveratrol, a plant polyphenol, mimics the effects of calorie restriction, primarily through activation of SIRT1, but also activation of AMPK in several tissues.Citation238,Citation239 It has demonstrated neuroprotective effects in acute and chronic central nervous system injury.Citation53,Citation240,Citation241 Additionally, the antioxidant properties of resveratrol were shown to reduce the production of reactive oxygen species and inflammatory markers in the trabecular meshwork in primary open angle glaucoma,Citation242 and to protect against hydrogen peroxide-induced retinal pigment epithelial cell dysfunction that occurs with age-related macular degeneration.Citation243
Figure 2 Role of AMPK in neuroprotection.
Antioxidant and reactive oxygen species scavenging
Oxidative stress is a common feature of mitochondrial disease, and evidence points toward a pathogenic involvement in diseases of the eye, as discussed above. Ameliorating oxidative stress may be a potential therapeutic avenue to explore to protect the eye against the adverse effects of aging and mitochondrial dysfunction. There is evidence clinically that antioxidant and vitamin supplementing may benefit mitochondrial diseases, with most promise in the use of coenzyme Q10 and its derivatives, eg, idebenone. The rationale behind their use is to replace naturally depleted stores, to bypass defective electron transfer, and these agents also have naturally occurring antioxidant properties.Citation244 In the body, their use may lead to lowering of plasma lactate levels and reducing oxidative stress.Citation245 The use of coenzyme Q10 and derivatives is being investigated in children with various mitochondrial disordersCitation246 as well as more specifically in patients suffering MELASCitation247 and Parkinson’s disease.Citation248 Idebenone, a short-chain derivative of coenzyme Q10, has been used to slow the progression of Friedreich’s ataxia,Citation249,Citation250 and in conjunction with vitamin B2 and vitamin C to treat LHON. In a clinical trial of 28 LHON patients (14 placebo, 14 treatment), idebenone treatment has been demonstrated to speed the visual recovery period in LHON from 34 months to 17 monthsCitation251 and has proved effective in improving vision in an isolated patient caseCitation252 but not in two other patients.Citation253 Another large Phase II clinical trial involving up to 84 patients to test the effectiveness of idebenone in LHON is underway where an improvement in mean visual acuity is the primary outcome.Citation254 A trial of idebenone in reducing vision loss in ADOA reported potential benefits in some patients, where five out of seven patients experienced improvement in vision.Citation255 These data support the use of idebenone in the treatment of primary mitochondrial optic neuropathies, and may provide potential as a therapeutic in glaucoma and other mitochondrial optic neuropathies.
Conclusion
The most common ophthalmic manifestation of primary mitochondrial disease is optic atrophy, followed by pigmentary retinopathy and ophthalmoplegia. The majority of mitochondrial disease patients will have variable central nervous system involvement, with ataxias, sensorineural deafness, and peripheral neuropathies being among the most frequently encountered signs. A growing appreciation for the potential role of age-related mitochondrial dysfunction has focused increasing attention on the possible role of mitochondria in the common age-related retinal diseases, age-related macular degeneration and glaucoma. Therapeutic developments aimed at boosting mitochondrial function are gathering pace.
The retina is both a key target of mitochondrial pathology and an ideal model system to test neuroprotective and neuroregenerative therapies. It is likely that advances in retinal neuroprotection via modulation of mitochondrial function will have implications for age-related neurodegenerative brain diseases.
Disclosure
The authors report no conflicts of interest in this work.
References
- WallaceDCSinghGLottMTMitochondrial DNA mutation associated with Leber’s hereditary optic neuropathyScience19882424884142714303201231
- CarelliVLa MorgiaCValentinoMLBarboniPRoss-CisnerosFNSadunAARetinal ganglion cell neurodegeneration in mitochondrial inherited disordersBiochim Biophys Acta20091787551852819268652
- Yu-Wai-ManPGriffithsPGChinneryPFMitochondrial optic neuropathies – disease mechanisms and therapeutic strategiesProg Retin Eye Res20113028111421112411
- FraserJABiousseVNewmanNJThe neuro-ophthalmology of mitochondrial diseaseSurv Ophthalmol201055429933420471050
- CrouchPJCimdinsKDuceJABushAITrounceIAMitochondria in aging and Alzheimer’s diseaseRejuvenation Res200710334935717708691
- SchapiraAHGeggMMitochondrial contribution to Parkinson’s disease pathogenesisParkinsons Dis20112011159160
- KongGYVan BergenNJTrounceIACrowstonJGMitochondrial dysfunction and glaucomaJ Glaucoma20091829310019225343
- JarrettSGLewinASBoultonMEThe importance of mitochondria in age-related and inherited eye disordersOphthalmic Res201044317919020829642
- WallaceDCMitochondrial DNA mutations in disease and agingEnviron Mol Mutagen201051544045020544884
- Wong-RileyMEnergy metabolism of the visual systemEye and Brain201029911623226947
- ChengAHouYMattsonMPMitochondria and neuroplasticityASN Neuro201025e0004520957078
- SarasteMOxidative phosphorylation at the fin de siecleScience199928354071488149310066163
- ClaytonDATranscription and replication of mitochondrial DNAHum Reprod200015Suppl 21117
- WallaceDCFanWProcaccioVMitochondrial energetics and therapeuticsAnnu Rev Pathol2010529734820078222
- CadenasEBoverisARaganCIStoppaniAOProduction of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondriaArch Biochem Biophys19771802248257195520
- TurrensJFBoverisAGeneration of superoxide anion by the NADH dehydrogenase of bovine heart mitochondriaBiochem J198019124214276263247
- WeiYHLuCYLeeHCPangCYMaYSOxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory functionAnn N Y Acad Sci19988541551709928427
- RichterCParkJWAmesBNNormal oxidative damage to mitochondrial and nuclear DNA is extensiveProc Natl Acad Sci U S A19888517646564673413108
- ClaytonDADodaJNFriedbergECThe absence of a pyrimidine dimer repair mechanism in mammalian mitochondriaProc Natl Acad Sci U S A1974717277727814212385
- CroteauDLStierumRHBohrVAMitochondrial DNA repair pathwaysMutat Res1999434313714810486588
- DiMauroSSchonEAMitochondrial respiratory-chain diseasesN Engl J Med2003348262656266812826641
- McKenzieMLiolitsaDHannaMGMitochondrial disease: mutations and mechanismsNeurochem Res200429358960015038606
- St JohnJCFacucho-OliveiraJJiangYKellyRSalahRMitochondrial DNA transmission, replication and inheritance: a journey from the gamete through the embryo and into offspring and embryonic stem cellsHum Reprod Update201016548850920231166
- HoeggerMJLievenCJLevinLADifferential production of superoxide by neuronal mitochondriaBMC Neurosci20089418182110
- CannRLGenetic clues to dispersal in human populations: retracing the past from the presentScience200129155091742174811249820
- Yu-Wai-ManPGriffithsPGHudsonGChinneryPFInherited mitochondrial optic neuropathiesJ Med Genet200946314515819001017
- Abu-AmeroKKCabreraVMLarrugaJMOsmanEAGonzalezAMAl-ObeidanSAEurasian and Sub-Saharan African mitochondrial DNA haplogroup influences pseudoexfoliation glaucoma development in Saudi patientsMol Vis20111754354721364909
- WolfCGramerEMuller-MyhsokBPasuttoFWissingerBWeisschuhNMitochondrial haplogroup U is associated with a reduced risk to develop exfoliation glaucoma in the German populationBMC Genet201011820109175
- Abu-AmeroKKMoralesJBosleyTMMohamedGHCabreraVMThe role of mitochondrial haplogroups in glaucoma: a study in an Arab populationMol Vis20081451852218385785
- UdarNAtilanoSRMemarzadehMMitochondrial DNA haplogroups associated with age-related macular degenerationInvest Ophthalmol Vis Sci20095062966297419151382
- SanGiovanniJPArkingDEIyengarSKMitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degenerationPLoS One200945e550819434233
- TorroniAPetrozziMD’UrbanoLHaplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484Am J Hum Genet1997605110711219150158
- YuDJiaXZhangAMMitochondrial DNA sequence variation and haplogroup distribution in Chinese patients with LHON and m.14484T.CPLoS One2010510e1342620976138
- ZhangMZhouXLiCMitochondrial haplogroup M9a specific variant ND1 T3394C may have a modifying role in the phenotypic expression of the LHON-associated ND4 G11778A mutationMol Genet Metab20101011–319219920728388
- ZhouXZhangHZhaoFVery high penetrance and occurrence of Leber’s hereditary optic neuropathy in a large Han Chinese pedigree carrying the ND4 G11778A mutationMol Genet Metab2010100437938420627642
- JiYZhangAMJiaXMitochondrial DNA haplogroups M7b1’2 and M8a affect clinical expression of Leber hereditary optic neuropathy in Chinese families with the m.11778G.a mutationAm J Hum Genet200883676076819026397
- CarelliVAchilliAValentinoMLHaplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of Leber hereditary optic neuropathy pedigreesAm J Hum Genet200678456457416532388
- SudoyoHSuryadiHLertritPPramoonjagoPLyrawatiDMarzukiSAsian-specific mtDNA backgrounds associated with the primary G11778 A mutation of Leber’s hereditary optic neuropathyJ Hum Genet2002471159460412436196
- LamminenTHuoponenKSistonenPmtDNA haplotype analysis in Finnish families with Leber hereditary optic neuroretinopathyEur J Hum Genet1997552712799412783
- HudsonGCarelliVSpruijtLClinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup backgroundAm J Hum Genet200781222823317668373
- MarcuelloAMartinez-RedondoDDahmaniYHuman mitochondrial variants influence on oxygen consumptionMitochondrion200991273018952007
- GhelliAPorcelliAMZannaCThe background of mitochondrial DNA haplogroup J increases the sensitivity of Leber’s hereditary optic neuropathy cells to 2,5-hexanedione toxicityPLoS One2009411e792219936068
- PelloRMartinMACarelliVMitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial diseaseHum Mol Genet200817244001401118806273
- KacserHBurnsJAMolecular democracy: who shares the controls?Biochem Soc Trans19797511491160389705
- AndrewsRMGriffithsPGJohnsonMATurnbullDMHistochemical localisation of mitochondrial enzyme activity in human optic nerve and retinaBr J Ophthalmol199983223123510396204
- BristowEAGriffithsPGAndrewsRMJohnsonMATurnbullDMThe distribution of mitochondrial activity in relation to optic nerve structureArch Ophthalmol2002120679179612049585
- HollanderHMakarovFStefaniFHStoneJEvidence of constriction of optic nerve axons at the lamina cribrosa in the normotensive eye in humans and other mammalsOphthalmic Res19952752963098552370
- KageyamaGHWong-RileyMTThe histochemical localization of cytochrome oxidase in the retina and lateral geniculate nucleus of the ferret, cat, and monkey, with particular reference to retinal mosaics and ON/OFF-center visual channelsJ Neurosci1984410244524596092560
- MincklerDSBuntAHJohansonGWOrthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkeyInvest Ophthalmol Vis Sci197716542644167096
- MutsaersSECarrollWMFocal accumulation of intra-axonal mitochondria in demyelination of the cat optic nerveActa Neuropathol19989621391439705128
- BalaratnasingamCMorganWHJohnstoneVCringleSJYuDYHeterogeneous distribution of axonal cytoskeleton proteins in the human optic nerveInvest Ophthalmol Vis Sci20095062824283819168905
- AttardiGRole of mitochondrial DNA in human agingMitochondrion200221–2273716120306
- InoueHJiangXFKatayamaTOsadaSUmesonoKNamuraSBrain protection by resveratrol and fenofibrate against stroke requires peroxisome proliferator-activated receptor alpha in miceNeurosci Lett2003352320320614625020
- ChenHChanDCCritical dependence of neurons on mitochondrial dynamicsCurr Opin Cell Biol200618445345916781135
- UoTDworzakJKinoshitaCDrp1 levels constitutively regulate mitochondrial dynamics and cell survival in cortical neuronsExp Neurol2009218227428519445933
- KijimaKNumakuraCIzuminoHMitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2AHum Genet20051161–2232715549395
- LawsonVHGrahamBVFlaniganKMClinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 geneNeurology200565219720416043786
- ZuchnerSDe JonghePJordanovaAAxonal neuropathy with optic atrophy is caused by mutations in mitofusin 2Ann Neurol200659227628116437557
- WolfCGramerEMuller-MyhsokBEvaluation of nine candidate genes in patients with normal tension glaucoma: a case control studyBMC Med Genet2009109119754948
- McQuibbanGASauryaSFreemanMMitochondrial membrane remodelling regulated by a conserved rhomboid proteaseNature2003423693953754112774122
- PellegriniLScorranoLA cut short to death: PARL and OPA1 in the regulation of mitochondrial morphology and apoptosisCell Death Differ20071471275128417464328
- CurranJEJowettJBAbrahamLJGenetic variation in PARL influences mitochondrial contentHum Genet2010127218319019862556
- PhasukkijwatanaNKunhapanBStankovichJGenome-wide linkage scan and association study of PARL to the expression of LHON families in ThailandHum Genet20101281394920407791
- McQuibbanGABulmanDEThe PARLance of Parkinson diseaseAutophagy20117779079221471738
- GomesLCDi BenedettoGScorranoLDuring autophagy mitochondria elongate, are spared from degradation and sustain cell viabilityNat Cell Biol201113558959821478857
- YaffeMPThe machinery of mitochondrial inheritance and behaviorScience199928354071493149710066164
- KarbowskiMArnoultDChenHChanDCSmithCLYouleRJQuantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosisJ Cell Biol2004164449349914769861
- ChenHChanDCEmerging functions of mammalian mitochondrial fusion and fissionHum Mol Genet200514Spec No 2R283R28916244327
- ChenYMcMillan-WardEKongJIsraelsSJGibsonSBMitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen speciesJ Cell Sci2007120Pt 234155416618032788
- Jahani-AslACheungECNeuspielMMitofusin 2 protects cerebellar granule neurons against injury-induced cell deathJ Biol Chem200728233237882379817537722
- JuWKKimKYDuong-PolkKXLindseyJDEllismanMHWeinrebRNIncreased optic atrophy type 1 expression protects retinal ganglion cells in a mouse model of glaucomaMol Vis2010161331134220664796
- DaiYWeinrebRNKimKYInducible nitric oxide synthase-mediated alteration of mitochondrial OPA1 expression in ocular hypertensive ratsInvest Ophthalmol Vis Sci20115252468247621220562
- BrownMDTrounceIAJunASAllenJCWallaceDCFunctional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 Leber’s hereditary optic neuropathy mitochondrial DNA mutationJ Biol Chem200027551398313983610976107
- KorstenAde CooIFSpruijtLde WitLESmeetsHJSluiterWPatients with Leber hereditary optic neuropathy fail to compensate impaired oxidative phosphorylationBiochim Biophys Acta20101797219720319836344
- KjerPInfantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish familiesActa Ophthalmol Suppl1959164Suppl 54114713660776
- CarelliVLa MorgiaCIommariniLMitochondrial optic neuropathies: how two genomes may kill the same cell type?Biosci Rep2007271–317318417479363
- CarelliVRoss-CisnerosFNSadunAAMitochondrial dysfunction as a cause of optic neuropathiesProg Retin Eye Res2004231538914766317
- MarchbankNJCraigJELeekJPDeletion of the OPA1 gene in a dominant optic atrophy family: evidence that haploinsufficiency is the cause of diseaseJ Med Genet2002398e4712161614
- KimJYHwangJMKoHSSeongMWParkBJParkSSMitochondrial DNA content is decreased in autosomal dominant optic atrophyNeurology200564696697215781809
- Amati-BonneauPValentinoMLReynierPOPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypesBrain2008131Pt 233835118158317
- HudsonGAmati-BonneauPBlakelyELMutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenanceBrain2008131Pt 232933718065439
- CipolatSMartins de BritoODal ZilioBScorranoLOPA1 requires mitofusin 1 to promote mitochondrial fusionProc Natl Acad Sci U S A200410145159271593215509649
- FrezzaCCipolatSMartins de BritoOOPA1 controls apoptotic cristae remodeling independently from mitochondrial fusionCell2006126117718916839885
- OlichonABaricaultLGasNLoss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosisJ Biol Chem2003278107743774612509422
- ElachouriGVidoniSZannaCOPA1 links human mitochondrial genome maintenance to mtDNA replication and distributionGenome Res2011211122020974897
- Van BergenNJCrowstonJGKearnsLSMitochondrial oxidative phosphorylation compensation may preserve vision in patients with OPA1-linked autosomal dominant optic atrophyPLoS One201166e2134721731710
- Mäkelä-BengsPSuomalainenAMajanderACorrelation between the clinical symptoms and the proportion of mitochondrial DNA carrying the 8993 point mutation in the NARP syndromePediatr Res19953756346397603783
- SueCMMitchellPCrimminsDSMoshegovCByrneEMorrisJGPigmentary retinopathy associated with the mitochondrial DNA 3243 point mutationNeurology1997494101310179339682
- Van den OuwelandJMLemkesHHRuitenbeekWMutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafnessNat Genet1992153683711284550
- ReardonWRossRJSweeneyMGDiabetes mellitus associated with a pathogenic point mutation in mitochondrial DNALancet19923408832137613791360090
- MichaelidesMJenkinsSABamiouDEMacular dystrophy associated with the A3243G mitochondrial DNA mutation. Distinct retinal and associated features, disease variability, and characterization of asymptomatic family membersArch Ophthalmol2008126332032818332310
- RathPPJenkinsSMichaelidesMCharacterisation of the macular dystrophy in patients with the A3243G mitochondrial DNA point mutation with fundus autofluorescenceBr J Ophthalmol200892562362918441172
- MassinPVirally-MonodMVialettesBPrevalence of macular pattern dystrophy in maternally inherited diabetes and deafness. GEDIAM GroupOphthalmology199910691821182710485557
- ZevianiMMoraesCTDiMauroSDeletions of mitochondrial DNA in Kearns-Sayre syndromeNeurology199851615259855494
- NishigakiYTadesseSBonillaEA novel mitochondrial tRNA(Leu(UUR)) mutation in a patient with features of MERRF and Kearns-Sayre syndromeNeuromuscul Disord200313433434012868503
- IsashikiYNakagawaMOhbaNRetinal manifestations in mitochondrial diseases associated with mitochondrial DNA mutationActa Ophthalmol Scand19987616139541428
- CarrollWMKrissABaraitserMBarrettGHallidayAMThe incidence and nature of visual pathway involvement in Friedreich’s ataxia. A clinical and visual evoked potential study of 22 patientsBrain198010324134347397485
- FortunaFBarboniPLiguoriRVisual system involvement in patients with Friedreich’s ataxiaBrain2009132Pt 111612318931386
- GivreSJWallMKardonRHVisual loss and recovery in a patient with Friedreich ataxiaJ Neuroophthalmol200020422923311130744
- BinderJHofmannSKreiselSClinical and molecular findings in a patient with a novel mutation in the deafness-dystonia peptide (DDP1) geneBrain2003126Pt 81814182012805099
- JinHMayMTranebjaergLA novel X-linked gene, DDP, shows mutations in families with deafness (DFN-1), dystonia, mental deficiency and blindnessNat Genet19961421771808841189
- NeupertWHerrmannJMTranslocation of proteins into mitochondriaAnnu Rev Biochem20077672374917263664
- KoehlerCMLeuenbergerDMerchantSRenoldAJunneTSchatzGHuman deafness dystonia syndrome is a mitochondrial diseaseProc Natl Acad Sci U S A19999652141214610051608
- PonjavicVAndreassonSTranebjaergLLubsHAFull-field electroretinograms in a family with Mohr-Tranebjaerg syndromeActa Ophthalmol Scand19967466326359017058
- TranebjaergLJensenPKVan GhelueMNeuronal cell death in the visual cortex is a prominent feature of the X-linked recessive mitochondrial deafness-dystonia syndrome caused by mutations in the TIMM8a geneOphthalmic Genet200122420722311803487
- CasariGDe FuscoMCiarmatoriSSpastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloproteaseCell19989369739839635427
- ZuchnerSMersiyanovaIVMugliaMMutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2ANat Genet200436544945115064763
- ZhaoCVollrathDmTOR pathway activation in age-related retinal diseaseAging (Albany NY)20113434634721483039
- HowellNKubackaIXuMMcCulloughDALeber hereditary optic neuropathy: involvement of the mitochondrial ND1 gene and evidence for an intragenic suppressor mutationAm J Hum Genet19914859359422018041
- JunASTrounceIABrownMDShoffnerJMWallaceDCUse of transmitochondrial cybrids to assign a complex I defect to the mitochondrial DNA-encoded NADH dehydrogenase subunit 6 gene mutation at nucleotide pair 14459 that causes Leber hereditary optic neuropathy and dystoniaMol Cell Biol19961637717778622678
- TarnopolskyMABakerSKMyintTMaxnerCERobitailleJRobinsonBHClinical variability in maternally inherited Leber hereditary optic neuropathy with the G14459 A mutationAm J Med Genet A2004124A437237614735584
- Yu-Wai-ManPGriffithsPGGormanGSMulti-system neurological disease is common in patients with OPA1 mutationsBrain2010133Pt 377178620157015
- BealMFAging, energy, and oxidative stress in neurodegenerative diseasesAnn Neurol19953833573667668820
- WallaceDCA mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicineAnnu Rev Genet20053935940716285865
- WangALLukasTJYuanMNeufeldAHAge-related increase in mitochondrial DNA damage and loss of DNA repair capacity in the neural retinaNeurobiol Aging201031112002201019084291
- KannOKovacsRMitochondria and neuronal activityAm J Physiol Cell Physiol20072922C641C65717092996
- Abu-AmeroKKMoralesJBosleyTMMitochondrial abnormalities in patients with primary open-angle glaucomaInvest Ophthalmol Vis Sci20064762533254116723467
- IzzottiASaccaSCLongobardiMCartigliaCMitochondrial damage in the trabecular meshwork of patients with glaucomaArch Ophthalmol2010128672473020547950
- IzzottiALongobardiMCartigliaCSaccaSCMitochondrial damage in the trabecular meshwork occurs only in primary open-angle glaucoma and in pseudoexfoliative glaucomaPLoS One201161e1456721283745
- TanwarMDadaTSihotaRDadaRMitochondrial DNA analysis in primary congenital glaucomaMol Vis20101651853320361014
- Abu-AmeroKKMoralesJOsmanMNBosleyTMNuclear and mitochondrial analysis of patients with primary angle-closure glaucomaInvest Ophthalmol Vis Sci200748125591559618055808
- KongYXVan BergenNTrounceIAIncrease in mitochondrial DNA mutations impairs retinal function and renders the retina vulnerable to injuryAging Cell201110457258321332926
- QuigleyHABromanATThe number of people with glaucoma worldwide in 2010 and 2020Br J Ophthalmol200690326226716488940
- DimitrovPNMukeshBNMcCartyCATaylorHRFive-year incidence of bilateral cause-specific visual impairment in the Melbourne Visual Impairment ProjectInvest Ophthalmol Vis Sci200344125075508114638700
- KongYXCrowstonJGVingrysAJTrounceIABuiVBFunctional changes in the retina during and after acute intraocular pressure elevation in miceInvest Ophthalmol Vis Sci200950125732574019643960
- OsborneNNMitochondria: Their role in ganglion cell death and survival in primary open angle glaucomaExp Eye Res201090675075720359479
- ChrysostomouVTrounceIACrowstonJGMechanisms of retinal ganglion cell injury in aging and glaucomaOphthalmic Res201044317317820829641
- KaarnirantaKSalminenAHaapasaloASoininenHHiltunenMAge-related macular degeneration (AMD): Alzheimer’s disease in the eye?J Alzheimers Dis201124461563121297256
- KlaverCCKliffenMvan DuijnCMGenetic association of apolipoprotein E with age-related macular degenerationAm J Hum Genet19986312002069634502
- BairdPNGuidaEChuDTVuHTGuymerRHThe epsilon2 and epsilon4 alleles of the apolipoprotein gene are associated with age-related macular degenerationInvest Ophthalmol Vis Sci20044551311131515111582
- CorderEHSaundersAMStrittmatterWJGene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset familiesScience199326151239219238346443
- DingJDLinJMaceBETargeting age-related macular degeneration with Alzheimer’s disease based immunotherapies: anti-amyloid-beta antibody attenuates pathologies in an age-related macular degeneration mouse modelVis Res200848333934517888483
- MahleyRWApolipoprotein E: cholesterol transport protein with expanding role in cell biologyScience198824048526226303283935
- BlennowKde LeonMJZetterbergHAlzheimer’s diseaseLancet2006368953338740316876668
- DentchevTMilamAHLeeVMTrojanowskiJQDunaiefJLAmyloid-beta is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinasMol Vis2003918419012764254
- LuiblVIsasJMKayedRGlabeCGLangenRChenJDrusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomersJ Clin Invest2006116237838516453022
- AndersonDHTalagaKCRivestAJBarronEHagemanGSJohnsonLVCharacterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degenerationExp Eye Res200478224325614729357
- JohnsonLVLeitnerWPRivestAJStaplesMKRadekeMJAndersonDHThe Alzheimer’s A beta-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degenerationProc Natl Acad Sci U S A20029918118301183512189211
- Jofre-MonsenyLMinihaneAMRimbachGImpact of apoE genotype on oxidative stress, inflammation and disease riskMol Nutr Food Res200852113114518203129
- GassJDAgarwalALavinaAMTawansyKAFocal inner retinal hemorrhages in patients with drusen: an early sign of occult choroidal neovascularization and chorioretinal anastomosisRetina200323674175114707822
- KenneyMCAtilanoSRBoyerDCharacterization of retinal and blood mitochondrial DNA from age-related macular degeneration patientsInvest Ophthalmol Vis Sci20105184289429720357205
- KarunadharmaPPNordgaardCLOlsenTWFerringtonDAMitochondrial DNA damage as a potential mechanism for age-related macular degenerationInvest Ophthalmol Vis Sci201051115470547920505194
- LinHXuHLiangFQMitochondrial DNA damage and repair in retinal pigment epithelium associated with aging and age-related macular degenerationInvest Ophthalmol Vis Sci20115263521352921273542
- CoskunPEBealMFWallaceDCAlzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replicationProc Natl Acad Sci U S A200410129107261073115247418
- FeherJKovacsIArticoMCavallottiCPapaleABalacco GabrieliCMitochondrial alterations of retinal pigment epithelium in age-related macular degenerationNeurobiol Aging200627798399315979212
- O’NeillECMackeyDAConnellPPHewittAWDanesh-MeyerHVCrowstonJGThe optic nerve head in hereditary optic neuropathiesNat Rev Neurol20095527728719488085
- O’NeillECKongYXConnellPPGaze behavior among experts and trainees during optic disc examination: Does how we look affect what we see?Invest Ophthalmol Vis Sci20115273976398321467174
- O’NeillECDanesh-MeyerHVKongGXOptic disc evaluation in optic neuropathies: the optic disc assessment projectOphthalmology2011118596497021126771
- ThorburnDRChowCWKirbyDMRespiratory chain enzyme analysis in muscle and liverMitochondrion200445–636337516120398
- TrounceIAKimYLJunASWallaceDCAssessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell linesMethods Enzymol19962644845098965721
- KirbyDMThorburnDRApproaches to finding the molecular basis of mitochondrial oxidative phosphorylation disordersTwin Res Hum Genet200811439541118637740
- YenMYYenTCPangCYLiuJHWeiYHMitochondrial DNA mutation in Leber’s hereditary optic neuropathyInvest Ophthalmol Vis Sci1992338256125661634353
- HowellNXuMHalvorsonSBodis-WollnerIShermanJA heteroplasmic LHON family: tissue distribution and transmission of the 11778 mutationAm J Hum Genet19945512032068023847
- MedjaFAlloucheSFrachonPDevelopment and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosisMitochondrion20099533133919439198
- GellerichFNMayrJAReuterSSperlWZierzSThe problem of interlab variation in methods for mitochondrial disease diagnosis: enzymatic measurement of respiratory chain complexesMitochondrion200445–642743916120404
- BarbiroliBMontagnaPCortelliPDefective brain and muscle energy metabolism shown by in vivo 31P magnetic resonance spectroscopy in nonaffected carriers of 11778 mtDNA mutationNeurology1995457136413697617199
- LodiRTaylorDJTabriziSJIn vivo skeletal muscle mitochondrial function in Leber’s hereditary optic neuropathy assessed by 31P magnetic resonance spectroscopyAnn Neurol19974245735799382468
- ChinneryPFJohnsonMAWardellTMThe epidemiology of pathogenic mitochondrial DNA mutationsAnn Neurol200048218819310939569
- HeiduschkaPSchnichelsSFuhrmannNElectrophysiological and histologic assessment of retinal ganglion cell fate in a mouse model for OPA1-associated autosomal dominant optic atrophyInvest Ophthalmol Vis Sci20105131424143119834041
- ShahrestaniPLeungHTLePKHeterozygous mutation of Drosophila Opa1 causes the development of multiple organ abnormalities in an age-dependent and organ-specific mannerPLoS One200948e686719718456
- WhiteKEDaviesVJHoganVEOPA1 deficiency associated with increased autophagy in retinal ganglion cells in a murine model of dominant optic atrophyInvest Ophthalmol Vis Sci20095062567257119234344
- IshiharaNFujitaYOkaTMiharaKRegulation of mitochondrial morphology through proteolytic cleavage of OPA1EMBO J200625132966297716778770
- Jahani-AslAPilon-LaroseKXuWThe mitochondrial inner membrane GTPase, optic atrophy 1 (Opa1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicityJ Biol Chem201128664772478221041314
- QiXSunLHauswirthWWLewinASGuyJUse of mitochondrial antioxidant defenses for rescue of cells with a Leber hereditary optic neuropathy-causing mutationArch Ophthalmol2007125226827217296905
- QiXLewinASSunLHauswirthWWGuyJSOD2 gene transfer protects against optic neuropathy induced by deficiency of complex IAnn Neurol200456218219115293270
- HellstromMHarveyARRetinal ganglion cell gene therapy and visual system repairCurr Gene Ther201111211613121291357
- PinkertCATrounceIAGeneration of transmitochondrial mice: development of xenomitochondrial mice to model neurodegenerative diseasesMethods Cell Biol20078054956917445713
- TraceyIDunnJFRaddaGKA 31P-magnetic resonance spectroscopy and biochemical study of the mo(vbr) mouse: potential model for the mitochondrial encephalomyopathiesMuscle Nerve19972011135213599342151
- IrwinMHJohnsonLWPinkertCAIsolation and microinjection of somatic cell-derived mitochondria and germline heteroplasmy in transmitochondrial miceTransgenic Res19998211912310481311
- CollombetJMWheelerVCVogelFCoutelleCIntroduction of plasmid DNA into isolated mitochondria by electroporation. A novel approach toward gene correction for mitochondrial disordersJ Biol Chem19972728534253479030609
- ZhangXJonesDGonzalez-LimaFMouse model of optic neuropathy caused by mitochondrial complex I dysfunctionNeurosci Lett200232629710012057837
- MarellaMSeoBBThomasBBMatsuno-YagiAYagiTSuccessful amelioration of mitochondrial optic neuropathy using the yeast NDI1 gene in a rat animal modelPLoS One201057e1147220628600
- RojasJCGonzales-LimaFMitochondrial optic neuropathy: in vivo model of neurodegeneration and neuroprotective strategiesEye and Brain201022137
- EllouzeSAugustinSBouaitaAOptimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunctionAm J Hum Genet200883337338718771762
- GuyJQiXKoilkondaRDEfficiency and safety of AAV-mediated gene delivery of the human ND4 complex I subunit in the mouse visual systemInvest Ophthalmol Vis Sci20095094205421419387075
- PogozelskiWKFletcherLDCassarCADunnDATrounceIAPinkertCAThe mitochondrial genome sequence of Mus terricolor: comparison with Mus musculus domesticus and implications for xenomitochondrial mouse modelingGene20084181–2273318501533
- McKenzieMTrounceIExpression of Rattus norvegicus mtDNA in Mus musculus cells results in multiple respiratory chain defectsJ Biol Chem200027540315143151910908563
- McKenzieMChiotisMPinkertCATrounceIAFunctional respiratory chain analyses in murid xenomitochondrial cybrids expose coevolutionary constraints of cytochrome b and nuclear subunits of complex IIIMol Biol Evol20032071117112412777531
- Perales-ClementeEFernandez-SilvaPAcin-PerezRPerez-MartosAEnriquezJAAllotopic expression of mitochondrial-encoded genes in mammals: achieved goal, undemonstrated mechanism or impossible task?Nucleic Acids Res201139122523420823090
- KoilkondaRDGuyJLeber’s hereditary optic neuropathy – gene therapy: From benchtop to bedsideJ Ophthalmol2011 Article ID 179412
- SadunAAMorgiaCLCarelliVLeber’s hereditary optic neuropathyCurr Treat Options Neurol201113110911721063922
- Figueroa-MartinezFVazquez-AcevedoMCortes-HernandezPWhat limits the allotopic expression of nucleus-encoded mitochondrial genes? The case of the chimeric Cox3 and Atp6 genesMitochondrion201111114715420854934
- LamBLFeuerWJAbukhalilFPorciattiVHauswirthWWGuyJLeber hereditary optic neuropathy gene therapy clinical trial recruitment: year 1Arch Ophthalmol201012891129113520837795
- YingMXuRWuXSodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLAJ Biol Chem200628118125801258616407196
- GardianGBrowneSEChoiDKNeuroprotective effects of phenylbutyrate in the N171-182Q transgenic mouse model of Huntington’s diseaseJ Biol Chem2005280155656315494404
- MinamiyamaMKatsunoMAdachiHSodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophyHum Mol Genet200413111183119215102712
- ZhouWBercuryKCummiskeyJLuongNLebinJFreedCRPhenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson diseaseJ Biol Chem201128617149411495121372141
- GardianGYangLClerenCCalingasanNYKlivenyiPBealMFNeuroprotective effects of phenylbutyrate against MPTP neurotoxicityNeuromolecular Med20045323524115626823
- JengYYLinNTChangPHRetinal ischemic injury rescued by sodium 4-phenylbutyrate in a rat modelExp Eye Res200784348649217178414
- ClementsCMMcNallyRSContiBJMakTWTingJPDJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2Proc Natl Acad Sci U S A200610341150911509617015834
- ThomasKJMcCoyMKBlackintonJDJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagyHum Mol Genet2011201405020940149
- IrrcherIAleyasinHSeifertELLoss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamicsHum Mol Genet201019193734374620639397
- WilsonBJTremblayAMDebloisGSylvain-DroletGGiguereVAn acetylation switch modulates the transcriptional activity of estrogen-related receptor alphaMol Endocrinol20102471349135820484414
- ScarpullaRCTranscriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cellsGene20022861818911943463
- CammarataPRChuSMoorAWangZYangSHSimpkinsJWSubcellular distribution of native estrogen receptor alpha and beta subtypes in cultured human lens epithelial cellsExp Eye Res200478486187115037120
- YangSHLiuRPerezEJMitochondrial localization of estrogen receptor betaProc Natl Acad Sci U S A2004101124130413515024130
- MattinglyKAIvanovaMMRiggsKAWickramasingheNSBarchMJKlingeCMEstradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesisMol Endocrinol200822360962218048642
- ChenJQEsheteMAlworthWLYagerJDBinding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elementsJ Cell Biochem200493235837315368362
- HsiehYCYuHPSuzukiTUpregulation of mitochondrial respiratory complex IV by estrogen receptor-beta is critical for inhibiting mitochondrial apoptotic signaling and restoring cardiac functions following trauma-hemorrhageJ Mol Cell Cardiol200641351152116859701
- GiordanoCMontopoliMPerliEOestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathyBrain2011134Pt 122023420943885
- MoorANGottipatiSMalletRTA putative mitochondrial mechanism for antioxidative cytoprotection by 17beta-estradiolExp Eye Res200478593394415051475
- GorresBKBomhoffGLMorrisJKGeigerPCIn vivo stimulation of oestrogen receptor increases insulin-stimulated skeletal muscle glucose uptakeJ Physiol2011589Pt 82041205421486807
- RogersNHWitczakCAHirshmanMFGoodyearLJGreenbergASEstradiol stimulates Akt, AMP-activated protein kinase (AMPK) and TBC1D1/4, but not glucose uptake in rat soleusBiochem Biophys Res Commun2009382464665019265681
- SchulzEAnterEZouMHKeaneyJFJrEstradiol-mediated endothelial nitric oxide synthase association with heat shock protein 90 requires adenosine monophosphate-dependent protein kinaseCirculation2005111253473348015967841
- SimpkinsJWYiKDYangSHDykensJAMitochondrial mechanisms of estrogen neuroprotectionBiochim Biophys Acta20101800101113112019931595
- RussoRCavaliereFWatanabeC17-Beta-estradiol prevents retinal ganglion cell loss induced by acute rise of intraocular pressure in ratProg Brain Res200817358359018929136
- DeschenesMCDescovichDMoreauMPostmenopausal hormone therapy increases retinal blood flow and protects the retinal nerve fiber layerInvest Ophthalmol Vis Sci20105152587260020019375
- NilsenJDiaz BrintonRMechanism of estrogen-mediated neuroprotection: regulation of mitochondrial calcium and Bcl-2 expressionProc Natl Acad Sci U S A200310052842284712604781
- LeeSVan BergenNJKongGYMitochondrial dysfunction in glaucoma and emerging bioenergetic therapiesExp Eye Res2010293100
- KimIRodriguez-EnriquezSLemastersJJSelective degradation of mitochondria by mitophagyArch Biochem Biophys2007462224525317475204
- GoldmanSJTaylorRZhangYJinSAutophagy and the degradation of mitochondriaMitochondrion201010430931520083234
- WenzTWilliamsSLBacmanSRMoraesCTEmerging therapeutic approaches to mitochondrial diseasesDev Disabil Res Rev201016221922920818736
- PuigserverPSpiegelmanBMPeroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulatorEndocr Rev2003241789012588810
- CantoCAuwerxJPGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditureCurr Opin Lipidol20092029810519276888
- AounPSimpkinsJWAgarwalNRole of PPAR-gamma ligands in neuroprotection against glutamate-induced cytotoxicity in retinal ganglion cellsInvest Ophthalmol Vis Sci20034472999300412824244
- ZhaoYPatzerAGohlkePHerdegenTCulmanJThe intracerebral application of the PPARgamma-ligand pioglitazone confers neuroprotection against focal ischaemia in the rat brainEur J Neurosci200522127828216029218
- CulmanJZhaoYGohlkePHerdegenTPPAR-gamma: therapeutic target for ischemic strokeTrends Pharmacol Sci200728524424917416424
- FongWHTsaiHDChenYCWuJSLinTNAnti-apoptotic actions of PPAR-gamma against ischemic strokeMol Neurobiol2010412–318018620127524
- FehmHLKernWPetersAThe selfish brain: competition for energy resourcesProg Brain Res200615312914016876572
- KahnBBAlquierTCarlingDHardieDGAMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolismCell Metab200511152516054041
- HardieDGAMP-activated/SNF1 protein kinases: conserved guardians of cellular energyNat Rev Mol Cell Biol200781077478517712357
- PoelsJSpasicMRCallaertsPNorgaKKExpanding roles for AMP-activated protein kinase in neuronal survival and autophagyBioessays200931994495219644919
- SugdenCCrawfordRMHalfordNGHardieDGRegulation of spinach SNF1-related (SnRK1) kinases by protein kinases and phosphatases is associated with phosphorylation of the T loop and is regulated by 5′-AMPPlant J199919443343910504565
- FryerLGParbu-PatelACarlingDThe anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathwaysJ Biol Chem200227728252262523211994296
- FulcoMSartorelliVComparing and contrasting the roles of AMPK and SIRT1 in metabolic tissuesCell Cycle20087233669367919029811
- CorreiaSCarvalhoCSantosMSMetformin protects the brain against the oxidative imbalance promoted by type 2 diabetesMed Chem20084435836418673148
- El-MirMYDetailleDR-VillanuevaGNeuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neuronsJ Mol Neurosci2008341778718040888
- PfisterJAMaCMorrisonBED’MelloSROpposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activityPLoS One2008312e409019116652
- BrunetASweeneyLBSturgillJFStress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylaseScience200430356662011201514976264
- CivitareseAECarlingSHeilbronnLKCalorie restriction increases muscle mitochondrial biogenesis in healthy humansPLoS Med200743e7617341128
- ShindlerKSVenturaERexTSElliottPRostamiASIRT1 activation confers neuroprotection in experimental optic neuritisInvest Ophthal-mol Vis Sci200748836023609
- Lopez-LluchGHuntNJonesBCalorie restriction induces mitochondrial biogenesis and bioenergetic efficiencyProc Natl Acad Sci U S A200610361768177316446459
- NisoliETonelloCCardileACalorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOSScience2005310574631431716224023
- DescampsORiondelJDucrosVRousselAMMitochondrial production of reactive oxygen species and incidence of age-associated lymphoma in OF1 mice: effect of alternate-day fastingMech Ageing Dev2005126111185119116126250
- KawaiSIVoraSDasSGachieEBeckerBNeufeldAHModeling of risk factors for the degeneration of retinal ganglion cells after ischemia/reperfusion in rats: effects of age, caloric restriction, diabetes, pigmentation, and glaucomaFASEB J20011571285128711344115
- FulcoMCenYZhaoPGlucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of NamptDev Cell200814566167318477450
- GuarenteLConnecting the dots: Linking sirtuins and AMPK in metabolism and agingDev Cell2011204e1
- ZangMXuSMaitland-ToolanKAPolyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient miceDiabetes20065582180219116873680
- DasguptaBMilbrandtJResveratrol stimulates AMP kinase activity in neuronsProc Natl Acad Sci U S A2007104177217722217438283
- LagougeMArgmannCGerhart-HinesZResveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alphaCell200612761109112217112576
- ParkerJAArangoMAbderrahmaneSResveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neuronsNat Genet200537434935015793589
- LunaCLiGLitonPBResveratrol prevents the expression of glaucoma markers induced by chronic oxidative stress in trabecular meshwork cellsFood Chem Toxicol200947119820419027816
- KingREKentKDBomserJAResveratrol reduces oxidation and proliferation of human retinal pigment epithelial cells via extracellular signal-regulated kinase inhibitionChem Biol Interact2005151214314915698585
- QuinziiCMLopezLCNainiADiMauroSHiranoMHuman CoQ10 deficienciesBiofactors2008321–411311819096106
- RodriguezMCMacDonaldJRMahoneyDJPariseGBealMFTarnopolskyMABeneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disordersMuscle Nerve200735223524217080429
- StacpoolePW dTFeigenbaumASKerrDSPhase III trial of coenzyme Q10 in mitochondrial disease www.clinicaltrials.gov, NCT00432744
- HiranoMEngelstadKJeromeRStudy of Idebenone in the treatment of Mitochondrial Encephalopathy Lactic Acidosis and Stroke-Like Episodes (MELAS) www.clinicaltrials.gov, NCT00887562
- KerrDSTreatment of mitochondrial electron transport chain disorders: a review of clinical trials over the past decadeMol Genet Metab201099324625520060349
- MeierTBuyseGIdebenone: an emerging therapy for Friedreich ataxiaJ Neurol2009256Suppl 1253019283347
- SchulzJBDi ProsperoNAFischbeckKClinical experience with high-dose idebenone in Friedreich ataxiaJ Neurol2009256Suppl 1424519283350
- MashimaYKigasawaKWakakuraMOguchiYDo idebenone and vitamin therapy shorten the time to achieve visual recovery in Leber hereditary optic neuropathy?J Neuroophthalmol200020316617011001192
- CarelliVBarboniPZacchiniALeber’s hereditary optic neuropathy (LHON) with 14484/ND6 mutation in a North African patientJ Neurol Sci199816021831889849804
- BarnilsNMesaEMunozSFerrer-ArtolaAArrugaJResponse to idebenone and multivitamin therapy in Leber’s hereditary optic neuropathyArch Soc Esp Oftalmol2007826377380 Spanish17573650
- RouleauJKTChinneryPStudy to assess efficacy, safety and tolerability of idebenone for Leber’s hereditary optic neuropathy www.clinicaltrials.gov, NCT00747487
- BarboniPLa MorgiaCCarbonelliMRecovery of visual acuity in dominant optic atrophy after idebenone treatmentPresented at the Association for Research in Vision and Ophthalmology Visionary Genomics meetingFort Lauderdale, FLMay 1–5, 2011