
Abstract
Background
This international, phase 2, open-label, multicenter study (ClinicalTrials.gov Identifier: NCT01609933) was conducted to evaluate the safety and efficacy of an enhanced regimen consisting of the direct-acting antivirals (DAAs) ombitasvir, paritaprevir, and ritonavir administered for 24 weeks, combined with pegylated interferon-α2a plus ribavirin (pegIFN-α2a/RBV) for 48 weeks, in patients with chronic hepatitis C virus (HCV) genotype 1 infection who had experienced virologic failure with a prior DAA regimen. This study was undertaken at a time when options were limited for the retreatment of patients who had failed prior DAA therapy.
Methods and results
Thirty-two patients were enrolled; the majority were male (78%) and White (94%), and the median age was 54.5 years. Twelve weeks after the last dose of study drug, sustained virologic response was achieved in 81.3% of patients. Five patients prematurely discontinued the study drugs and one patient relapsed. Safety and tolerability were similar to prior studies of pegIFN-α2a/RBV alone.
Conclusion
Given the availability of highly efficacious DAA regimens that are both IFN- and RBV-free, this regimen is no longer relevant in today’s HCV treatment landscape.
The direct-acting antiviral (DAA) combination ombitasvir/paritaprevir/ritonavir ± dasabuvir (OBV/PTV/r±DSV), with or without ribavirin (RBV), is approved for patients infected with hepatitis C virus (HCV) genotype (GT) 1 with compensated liver disease, including cirrhosis.Citation1–Citation3 When dosed for 12 or 24 weeks according to the labeled indications, the regimen achieves a sustained virologic response rate at 12 weeks post-treatment (SVR12) >90% with a favorable safety profile in all approved populations.Citation1–Citation3
During the development of OBV/PTV/r±DSV, there was concern that treatment failure could result in multi-class drug resistance and reduce the chance for successful retreatment. In the absence of data supporting an effective retreatment regimen, an international, phase 2, open-label, multicenter study (NCT01609933) was undertaken between December 2012 and May 2017 to evaluate the safety and efficacy of an enhanced regimen consisting of DAAs coadministered with pegylated interferon-α2a plus RBV (pegIFN-α2a/RBV) in patients with chronic HCV GT1 infection (HCV RNA concentration ≥2,000 IU/mL) who had failed DAA therapy in a previous AbbVie or Abbott clinical trial. The DAAs included a higher dose of PTV (200 mg daily) with ritonavir 100 mg and OBV 25 mg. The inclusion criteria are:
Subject must have voluntarily signed and dated an informed consent, approved by an Institutional Review Board/Ethics Committee, prior to the initiation of any study-specific procedure.
Subject must have been able to understand and adhere to the study visit schedule and all other protocol requirements.
Subject must have experienced virologic failure as defined in a previous AbbVie/Abbott DAA combination study.
Female subjects must not be of childbearing potential, defined as:
Postmenopausal for at least 2 years prior to screening (defined as amenorrheic for longer than 2 years, age appropriate, and confirmed by a follicle stimulating hormone (FSH) level indicating a postmenopausal state), or
Surgically sterile (defined as bilateral tubal ligation, bilateral oophorectomy or hysterectomy) or had a vasectomized partner(s), or
Practicing total abstinence from sexual intercourse (minimum 1 complete menstrual cycle), or
Sexually active with female partners only–of childbearing potential and sexually active with male partner(s):
Currently using at least 1 effective method of birth control at the time of screening and agree to practice 2 effective methods of birth control while receiving study drugs (as outlined in the subject information and consent form or other subject information documents), starting with Study Day 1 and for 7 months after stopping study drug or as directed by the local RBV label (use of estrogen-containing hormonal contraceptives, including oral, injectable, implantable, patch, and ring varieties was not permitted during DAA treatment).
Females must have had negative results for pregnancy tests performed:
At screening by urine specimen within 42 days prior to initial study drug administration, and
At baseline (prior to dosing) by urine specimen.
Female subjects with a borderline human chorionic gonadotropin result at Day 1 may have enrolled into the study if they either:
Had a documented history of bilateral tubal ligation, hysterectomy, bilateral oophorectomy; or
Were confirmed to be postmenopausal defined as amenorrheic for longer than 2 years, age appropriate, and confirmed by a FSH level indicating a postmenopausal state at screening.
Sexually active males had to be surgically sterile or had male partners only or if sexually active with female partner(s) of childbearing potential had to agree to practice 2 effective forms of birth control (as outlined in the subject information and consent form or other subject information documents) throughout the course of the study, starting with Study Day 1 and for 7 months after stopping study drug or as directed by the local RBV label. Contraceptives containing ethinyl estradiol were considered effective if used by the female partners of male subjects.
Subject had to be considered an appropriate candidate for pegIFN, RBV, OBV/PTV/r therapy in the opinion of the investigator.
Subject was infected with HCV GT1 at the screening visit.
Subject had a plasma HCV RNA level ≥2000 IU/mL at the pre-screening visit.
In addition to Inclusion Criteria 1 through 10, subjects diagnosed with cirrhosis also had to meet the following criteria:
| 11) | Compensated cirrhosis defined as Child-Pugh score of ≤6 at screening. | ||||
| 12) | Absence of hepatocellular carcinoma (HCC) based on a negative ultrasound, computed tomography (CT) scan, or magnetic resonance imaging (MRI) performed within 3 months prior to screening or during the screening period. | ||||
The exclusion criteria are:
In subjects with a prior null or partial response to pegIFN/RBV treatment at any time prior to pre-screening for this study or any prior failure with pegIFN/RBV plus telaprevir, the presence of variants relative to the appropriate prototypic reference sequence (H77 for 1a or Con1 for 1b) at any of the following positions: NS3 155, 156, or 168; or NS5A 28, 29, 30, 31, 32, 58, or 93.
Prior use of an investigational HCV therapy or approved DAA not included in this protocol or the previous AbbVie/Abbott DAA combination study.
Females who were pregnant or planning to become pregnant, or breastfeeding, or males whose partners were pregnant or planning to become pregnant within 7 months (or as per local RBV label) after their last dose of RBV.
Positive result of a urine drug screen at the screening visit for opiates, barbiturates, amphetamines, cocaine, benzodiazepines, phencyclidine, and propoxyphene with the exception of a positive result (including methadone), associated with documented short-term use or chronic stable use of a prescribed medication in that class.
Positive test for hepatitis B surface antigens or anti-human immunodeficiency virus antibodies.
Use of known strong inducers (eg, phenobarbital, rifampin, carbamazepine, St. John’s wort) of cytochrome P450 3A within 2 weeks prior to study drug administration.
Use of any medications listed in within 2 weeks or 10 half-lives, of the medication, whichever was longer, prior to study drug administration.
Table 1 Medications contraindicated for use with the study drug regimen
Discontinuation of antiviral therapy due to intolerance or a DAA- or RBV-associated adverse event (AE) in the previous AbbVie/Abbott DAA combination study (excluding intolerance or AEs associated with telaprevir).
Beck Depression Inventory II (BDI-II) score of >21 at screening. Subjects with BDI-II score >21 were not permitted to rescreen.
Clinically significant abnormal electrocardiogram (ECG), ECG with QT interval corrected for heart rate using Fridericia’s correction formula (QTcF) >470 msec at screening or Day 1 visits.
Screening laboratory analyses showed any of the following abnormal laboratory results:
Alanine aminotransferase (ALT) >7 × upper limit of normal (ULN);
Aspartate aminotransferase (AST) >7 × ULN;
Calculated creatinine clearance (using Cockcroft-Gault method) <60 mL/minute;
Albumin < lower limit of normal (LLN);
International normalized ratio (INR) >1.7;
Subjects with a known inherited blood disorder and INR >1.7 may be enrolled with permission of the AbbVie Study Designated Physician;
Hemoglobin <LLN;
Platelets <90,000 cells per mm3;
Absolute neutrophil count <1500 cells/μL (<1200 cells/μL for subjects of black/African descent);
Indirect bilirubin >1.5 × ULN and direct bilirubin >ULN;
Thyroid stimulating hormone values outside the normal range.
The use of colony stimulating factors, such as granulocyte colony stimulating factor or erythropoietin within 2 months of the screening period.
Recent history of drug (within 6 months prior to study drug administration) or alcohol abuse that, in the opinion of the investigator, could preclude adherence to the protocol.
Clinically significant abnormalities, other than HCV infection, based on the results of a medical history, physical examination, vital signs, laboratory profile, and a 12-lead ECG that made the subject an unsuitable candidate for this study in the opinion of the investigator.
Evidence of infection with other HCV genotypes other than GT1 at the screening visit.
Current enrollment in another clinical study or previous enrollment in this study. Concurrent participation in non-interventional, epidemiologic, or registry trials may have been permitted with approval by the AbbVie Therapeutic Area Medical Director.
History of solid organ transplantation.
In addition to Exclusion Criteria 1–17, subjects with compensated cirrhosis must not have met the following criteria:
| 18) | Any current or past clinical evidence of Child-Pugh B or C classification or clinical history of liver decompensation such as ascites (noted on physical examination), variceal bleeding, or hepatic encephalopathy. | ||||
| 19) | Serum alpha-fetoprotein >100 ng/mL at screening. | ||||
| 20) | A screening ultrasound suspicious for HCC and confirmed with a subsequent CT scan or MRI during the screening period. | ||||
The study protocol was approved by the independent ethics committee/institutional review board at each site. A full list can be found in the Supplementary material. The study was conducted in accordance with the Good Clinical Practice guidelines and the ethical principles of the Declaration of Helsinki; all patients provided written consent. Patients (N=32), including those with compensated cirrhosis, initially received 24 weeks’ treatment with OBV, PTV, and ritonavir with pegIFN-α2a 180 μg weekly and weight-based RBV. After 24 weeks of combination dosing, patients were to receive pegIFN-α2a and RBV for another 24 weeks. All 32 patients received ≥1 dose of study drugs and 26 patients completed the DAA-dosing period (see for the patient flowchart). Plasma HCV RNA levels were determined for each sample collected by the central laboratory using the COBAS® TaqMan® (Roche, Indianapolis, IN, USA) real-time reverse transcriptase polymerase chain reaction assay v2.0. The lower limit of detection was 15 IU/mL and the lower limit of quantitation (LLOQ) was 25 IU/mL.
Figure 1 Patient flow chart.
Abbreviations: AE, adverse event; OBV, ombitasvir; pegIFN-α2a, pegylated interferon-α2a; PTV, paritaprevir; RBV, ribavirin.
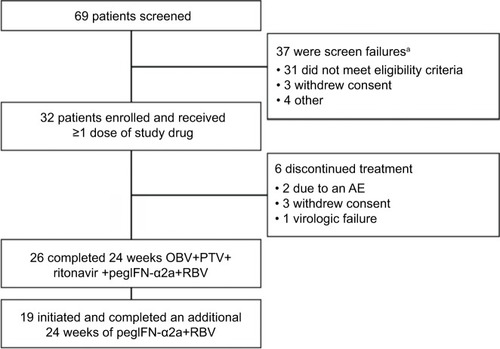
Most patients were male (78%), White (94%), had minimal fibrosis (F0–F1, 73%), and HCV GT1a infection (66%) (). Half (50%) of patients had on-treatment virologic failure with initial DAA therapy, and half had post-treatment relapse. Of the 26 patients who completed DAA dosing, 19 went on to receive 24 weeks of additional treatment with pegIFN-α2a and RBV.
Table 2 Baseline demographics and clinical characteristics
SVR12, the primary endpoint (HCV RNA < LLOQ 12 weeks after the last dose of study drug), was achieved in 81.3% of patients (n/N=26/32; 95% CI, 64.7–91.1). Of the six patients who did not achieve SVR12, one GT1a-infected patient had relapsed by post-treatment Week (PTW) 4 (HCV RNA ≥ LLOQ [defined as two consecutive HCV RNA measurements ≥ LLOQ] at any point after HCV RNA < LLOQ), and five patients prematurely discontinued study drugs (two due to an AE; three withdrew consent). In a sensitivity analysis excluding patients with non-virologic failure, SVR12 was achieved in 96.3% of patients (n/N=26/27; 95% CI, 81.7–99.3). SVR was maintained at PTW24 (SVR24) in 78.1% of patients (n/N=25/32; 95% CI, 61.2–89.0); an additional GT1a-infected patient relapsed between PTW12 and PTW24.
During the 24-week period of DAA dosing, 90.6% of patients had ≥1 treatment-emergent AE (AEs occurring after treatment initiation until 30 days post treatment; ). The three most commonly reported AEs (≥10.0%) were headache (43.8%), fatigue (34.4%), and nausea (28.1%), and most were considered by the investigator to be related to pegIFN-α2a (81.3%) and/or RBV (84.4%). Most patients had AEs of mild or moderate severity; five patients had ≥1 severe AE. One patient had a serious AE: a 53-year-old White male who had a fatal myocardial infarction on Day 51 that the investigator considered unrelated to study drugs. An additional patient discontinued treatment on Day 24 due to anxiety and migraine considered possibly related to study drugs. No patient had worsening laboratory values of grade 3 or 4 severity, and no cases of hepatic decompensation, hepatic failure, or hepatocellular carcinoma were reported.
Table 3 AEs and post-baseline laboratory abnormalities after initiating treatment with OBV, PTV, and ritonavir plus pegIFN-α2a/RBV for 12 weeks (recorded until 30 days post treatment)
Among the 19 patients who received the additional pegIFN-α2a and RBV dosing, AEs were reported in 68.4% patients (n/N=13/19; onset >30 days post-DAA treatment until 30 days post treatment with pegIFN-α2a/RBV). The most common AEs (≥10.0%) were decreased neutrophil count (15.8%), neutropenia (10.5%), and decreased white blood cell count (10.5%).
Resistance analyses were conducted for the two patients who relapsed. Amino acid substitutions relative to prototypic reference sequence at the following signature amino acid positions in GT1a were analyzed: 36, 43, 55, 56, 80, 155, 156, 168 in NS3; 24, 28, 29, 30, 31, 32, 58, 62, 92, 93 in NS5A. Variants in NS5A and NS3 were identified by population and/or clonal nucleotide sequencing. The patient who relapsed at PTW4 did not have baseline substitutions in NS3 but had treatment-emergent Y56H and D168A at PTW4, which persisted through PTW48; M28T in NS5A was also detected at baseline and persisted through PTW48. The patient who relapsed at PTW24 had Q80K in NS3 and Q30R in NS5A at baseline, which persisted through PTW48, with no treatment-emergent substitutions.
High SVR rates were achieved with OBV, PTV, and ritonavir coadministered with pegIFN-α2a/RBV in patients with chronic HCV GT1 infection who had virologic failure with prior DAA therapy in this study, with a safety and tolerability profile consistent with those of pegIFN plus RBV. This study was intended to offer an intensified retreatment option to patients who participated in a prior AbbVie or Abbott DAA trial but did not achieve SVR. It was initiated when the strategy for retreatment after DAA failure was not well characterized, and next-generation DAAs with activity against resistant strains were not yet available; accordingly, the most effective options for these patients included pegIFN and RBV.Citation4 There are now approved regimens with acceptable efficacy for the retreatment of patients with prior DAA failure that do not contain IFN or RBV.Citation5 The regimen used in this trial offers no clinical benefit over next-generation DAA regimens, and it was not developed further.
Data sharing statement
AbbVie is committed to responsible data sharing regarding the clinical trials it sponsored. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (eg, protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinicaltrials-data-and-information-sharing/data-and-informationsharing-with-qualified-researchers.html.
Acknowledgments
Paritaprevir was identified by AbbVie and Enanta. The authors would like to express their gratitude to the patients who participated in this study and their families, as well as all of the trial investigators and their research staff. Editorial support was provided by Fiona Powell, PhD, and Paul Mac-Callum, PhD, of Fishawack Communications Ltd.; funded by AbbVie. AbbVie sponsored the study; contributed to its design; and participated in the collection, analysis, and interpretation of the data and in the writing, reviewing, and approval of the publication.
Disclosure
David Bernstein has acted as a consultant for and received research grants from AbbVie and Gilead. Daniel E Cohen and Rakesh Tripathi are employees of AbbVie and may hold stock or options. The authors report no other conflicts of interest in this work.
References
- PAK VIEKIRA (ombitasvir, paritaprevir, and ritonavir tablets; dasabuvir tablets) [prescribing information]North Chicago, ILAbbVie, Inc. approved December 2014. Available from. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/206619s017lbl.pdfAccessed August 28, 2018
- VIEKIRAX (ombitasvir, paritaprevir, and ritonavir tablets) [summary of product characteristics]North Chicago, ILAbbVie, Inc. approved January 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003839/WC500183997.pdfAccessed August 28, 2018
- EXVIERA (dasabuvir tablets) [summary of product characteristics]North Chicago, ILAbbVie, Inc. approved January 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003837/WC500182233.pdfAccessed August 28, 2018
- FerenciPTreatment options for anti-HCV treatment-experienced patientsClin Liv Dis2012124950
- American Association for the Study of Liver Diseases and the Infectious Diseases Society of AmericaHCV guidance: recommendations for testing, managing, and treating hepatitis C2018 Available from: https://www.hcvguidelines.org/sites/default/files/full-guidance-pdf/HCVGuidance_May_24_2018b.pdfAccessed August 28, 2018