
Abstract
Autophagy is an intracellular lysosomal degradation process performed by the cells to maintain energy balance. The autophagy response plays an important role in the progression of liver disease due to hepatitis virus infection, alcoholic liver disease, nonalcoholic fatty liver disease, liver cirrhosis, and hepatocellular carcinoma (HCC). An increased autophagy response also contributes to the pathogenesis of liver disease through modulation of innate and adaptive immune responses; a defective cellular autophagy response leads to the development of HCC. Recent progress in the field indicates that autophagy modulation provides a novel targeted therapy for human liver cancer. The purpose of this review is to update our understanding of how the cellular autophagy response impacts the pathophysiology of liver disease and HCC treatment.
Introduction
Autophagy is a lysosomal degradation mechanism important for cell survival under conditions of starvation, stress, or infection. The mechanism evolved as a means of regenerating energy from intracellular materials (cytoplasm, organelles, protein aggregates, etc) to meet energy requirements in low-nutrient conditions.Citation1 Autophagy, is one such mechanism and is induced by a variety of stimuli, including cytokine stimulation, stress, diverse pathogens, accumulation of misfolded proteins, and damaged organelles.Citation2 The importance of autophagy in liver homeostasis and energy conservation has been verified in animal models. For example, inhibition of autophagy in mouse models has been observed to impair lysosomal degradation in hepatocytes, resulting in a fourfold increase in liver weight.Citation3 Likewise, nutrient starvation experiments in mice have shown that autophagy is responsible for degradation of 35% of total proteins in the liver within 24 hours.Citation4 These data illustrate the importance of autophagy in the maintenance of liver functions and liver weight. Autophagy also plays a major role in the modulation of innate and adaptive immune responses in the pathogenesis of chronic liver diseases, including diseases due to cancer, diabetes, neurodegeneration, and aging.
Initiation and termination of autophagy are linked to cellular nutrient-sensing mechanisms.Citation5,Citation6 For example, the molecule AMP-kinase (AMPK) senses cellular energy requirements through AMP to ATP ratios in the cell cytoplasm. High AMP levels reflect low energy states in the cell, and under these conditions, AMPK can initiate autophagy through inactivation of mTOR1 (a mechanistic target of rapamycin complex 1) or by phosphorylation of ULK1/2 protein. Another autophagy-inducing signal is related to inhibition of mTOR1 by depletion of amino acid levels in the cytoplasm. It is now believed that inhibition of mTOR1 due to low energy states in the cell activates autophagy, whereas activation of mTOR1 due to high energy states inhibits cellular autophagy.
Three different types of autophagy response have been described in the mammalian cells: macroautophagy, chaperon-mediated autophagy (CMA), and microautophagy.Citation7 The differences among these three types of autophagy are illustrated in . In macroautophagy, a portion of cytosol is engulfed by a double-membrane structure called an autophagosome, which fuses with a lysosome to become an autophagolysosome; the contents of the autophagolysosome are degraded by lysosomal enzymes (proteases, lipases, nucleases, and glycases) in a process coordinated by 37 ATG proteins. Several cellular compartments, including the endoplasmic reticulum (ER), Golgi/trans-Golgi apparatus, and plasma membrane, participate in autophagosome formation. CMA is responsible for the degradation of cytosolic proteins under conditions of stress. All CMA substrates contain a consensus pentapeptide motif (KFERQ) that is recognized by a cytosolic chaperone, for example, HSC70;Citation8 HSC70 binds to Lamp2a, which results in the direct translocation of unfolded protein substrate across lysosomal membranes and subsequent degradation of the cytosolic proteins. In microautophagy, cytosolic material is directly engulfed by the lysosome via membrane rearrangement. Recently, microautophagy has been renamed on the basis of the cargo it degrades, as mitophagy, pexophagy, reticulophagy, and ribophagy.
Figure 1 Three different types of autophagy response allow degradation of cytosolic content and organelles in the lysosome.
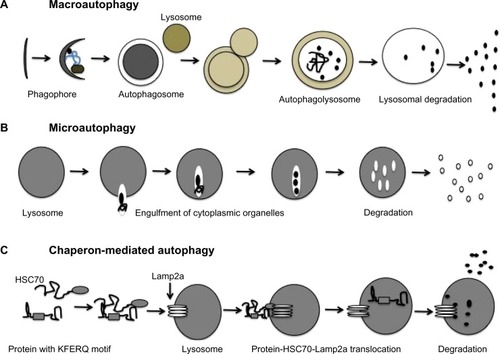
This review focuses mainly on macroautophagy (autophagy) and its role in the pathogenesis of liver diseases, cirrhosis, and hepatocellular carcinoma (HCC).
Molecular interactions in autophagy
In general, autophagy is coordinated by five different steps, known as initiation, nucleation, elongation, maturation, and degradation (). Decreasing mTOR1 levels caused by low nutrient levels, such as low levels of amino acid, lipids, and sugars, activate autophagy signaling. During basal-level autophagy, Unc-51-like kinase (ULK1/2), ATG13, FIP200, and ATG101, exist in an inactive complex with mTOR1. During initiation, a decrease in mTOR activity leads to phosphorylation and translocation of the ULK complex (ULK1–ATG13–FIP200–ATG101) from the cytoplasm to the ER.Citation6,Citation9,Citation10 The interaction of the ULK complex with ER-resident proteins leads to initiation of autophagy.Citation6 The second step, nucleation, occurs as the ULK complex enlarges, due to interactions with class III phosphatidylinositol 3-kinase (PI3K) consisting of either Beclin 1–ATG14L–PI3KCIII–p150–Ambra1 or Beclin 1–UVRAG–PI3KCIII–p150–Bif1.Citation11 Additional ER-resident proteins (DFCPI and WIPI) facilitate the nucleation and creation of a curved double-membrane structure. It has been shown that the autophagy process can be inhibited if Beclin 1 forms a complex with Rubicon or antiapoptotic protein (called Bcl-2).Citation12 The third step, called elongation, is primarily mediated by ubiquitin-like protein conjugation systems. The ATG12–ATG5 complex associates with ATG16 to form ATG12–ATG5–ATG16 (ATG16L), which localizes at the autophagosomal membrane. The ATG16L complex then promotes LC3 lipidation by PE and membrane insertion. The LC3–PE is localized at the inner and outer membranes of the autophagosome. During elongation, the LC3 protein on the autophagosome can interact with misfolded and polyubiquitinated proteins through autophagy receptor proteins (p62, NBR1, or NIX).Citation13,Citation14 Proteins degraded through autophagy are recognized by autophagy receptors such as p62 and NBR1 that interact with ubiquitin-like protein LC3, which modifies the target proteins for delivery to the lysosome. The fourth step, maturation, is related to autophagosome completion. The autophagosome undergoes two maturation steps. First, the autophagosome fuses with multivesicular endosomes to form an amphisome, where proton pumps are acquired for acidification. Second, the amphisomes fuse with a lysosome to become an autolysosome. Finally, the cellular materials present inside the autolysosome are degraded by the action of different lysosomal enzymes into amino acids, lipids, and sugars. The degradation products, such as amino acids, lipids, and sugars, are released from the autolysosome via lysosome efflux transporters for reuse, for example, in the production of new proteins. The release of nutrients from the autophagolysosome reactivates mTOR, which triggers autophagy termination and the formation of nascent lysosomes. This process is called autophagic lysosome reformation. Under conditions of low nutrition, the cycle is repeated.
Figure 2 Molecular signaling pathway involved in autophagy.
Abbreviation: HCC, hepatocellular carcinoma.
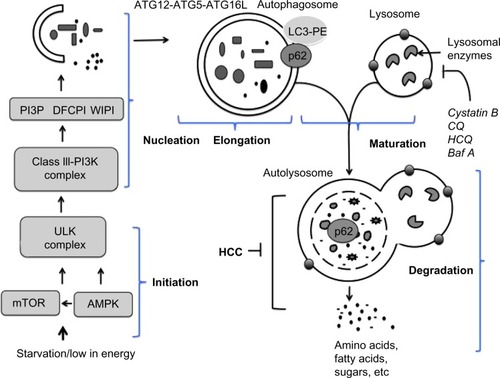
The fusion of an amphisome with a lysosome requires Lamp2a and a small amount of GTPase Rab7.Citation15,Citation16 The retrieval of ATG proteins from the mature autophagosome is mediated by ATG2, ATG9, and ATG18.Citation17 The ER is the most common site for the initiation of the autophagy membrane source, but autophagy initiation can involve Golgi apparati, endosomes, plasma membrane, and mitochondria. Different forms of autophagy can be induced by nutrient starvation, stress related to virus infection, and chemotherapy. Increased autophagic degradation can lead to cell death, which may be important in tissue homeostasis. A decreased autophagy response has also been linked to the development of cancer and neurological diseases. We here describe the important role of autophagy in chronic liver disease in humans.
Autophagy in chronic liver diseases
Mechanisms of acute and chronic liver injury in humans have been linked mainly to infection by hepatitis viruses, alcohol abuse, and fat deposition. These agents target mostly hepatocytes, as hepatocytes are the predominant cell type in the liver subject to acute and chronic injury. One of the host-related factors responsible for the evolution of chronic liver disease to liver cirrhosis and HCC is the degree of hepatocellular injury. An increase in serum aminotransferase has been used as a surrogate marker for assessing the extent of hepatic injury in patients with chronic liver diseases of both viral and nonviral etiology.
Liver injury is caused by three distinct types of cell death: apoptosis, necrosis, and autophagy. Apoptosis is a form of cell death mediated by an intracellular proteolytic cascade, in which cells die neatly. Apoptotic cells are usually phagocytosed either by neighboring cells or by a macrophage. Necrosis is a form of cell death mediated by acute injury in which the cell swells, bursts, and spills its contents into surrounding areas, causing an inflammatory response. Apoptosis and autophagy are interrelated biological processes important for maintaining tissue homeostasis and carcinogenesis. Hepatocellular apoptosis acts as a tumor suppressor or (prodeath) in the liver. It has been shown that the genes controlling apoptosis (Bcl-2) are involved in carcinogenesis. Oncogenic mutations in the Bcl-2 gene that inhibit apoptosis can lead to tumor initiation, progression, or metastasis. Alternatively, oncogenes that promote cellular apoptosis can initiate selective pressure to override apoptosis during multistage carcinogenesis. A number of excellent reviews have described the role of hepatocyte apoptosis in prodeath (tumor suppression) during chronic liver injury and carcinogenesis.Citation18,Citation19 Similarly, hepatocellular autophagy can also cause tumor suppression in chronic liver disease, and an impaired autophagy response can lead to malignant transformation and HCC. It is well known that HCC develops in the background of liver cirrhosis after many years of chronic liver disease due to hepatitis virus infection and alcoholic and nonalcoholic liver diseases. Available evidence suggests that the autophagy response is deregulated in chronic liver disease and liver cirrhosis, which can lead to HCC. To date, a consistent autophagic tumor suppressor mechanism causing the development of chronic liver disease, cirrhosis, and HCC has not been identified.
Infection with hepatitis B virus (HBV) and hepatitis C virus (HCV) has been shown to induce an autophagy response both in vitro and in vivo in chronically infected liver.Citation20 It has been observed that the autophagy response promotes HBV and HCV replication, whereas autophagy suppression inhibits replication. These results suggest that viral infection induces the autophagy response to degrade organelles and long-lived proteins needed to generate energy and sustain virus replication in hepatocytes, and, if uncontrolled, the induced autophagy response could lead to autophagic cell death and the elimination of infected hepatocytes in the liver. Actually, both viruses lead to chronic infection.
The prosurvival function of HBV and HCV infection has not been well established, with the exception of several studies that have used hepatoma cell lines to show that HBV or HCV infection inhibits autophagic degradation.Citation21–Citation23 The autophagy response is decreased in chronic liver disease on account of both alcoholic and nonalcoholic liver diseases. This conclusion is supported by the fact that suppression of autophagy by pharmacological agents or siRNA against ATG7 significantly exacerbates liver injury, whereas autophagy induction improves chronic liver disease caused by alcoholic and nonalcoholic fatty liver disease.Citation24 The decreased autophagy response in alcoholic and nonalcoholic liver disease can lead to the accumulation of misfolded protein aggregates, which can increase oxidative stress, DNA damage, and genomic instability, all of which favor carcinogenesis. These results are consistent with human data showing that chronic liver disease and HCC develop in the presence of these factors. Since autophagy induction also improves alcoholic and nonalcoholic liver diseases, it is expected that autophagy induction should also improve liver cirrhosis related to alcoholic and nonalcoholic fatty liver disease. It appears that autophagy modulation (induction or inhibition) may be a potential therapeutic strategy for treatment of liver cirrhosis, but so far this strategy has produced mixed results, with the exception that autophagy induction using rapamycin has been shown to be beneficial for the treatment of hepatic fibrosis due to alpha-1 antitrypsin deficiency.Citation25 Additional investigations to understand the role of autophagy in liver cirrhosis should guide whether autophagy inhibition or induction strategies will be beneficial for the treatment of liver fibrosis.
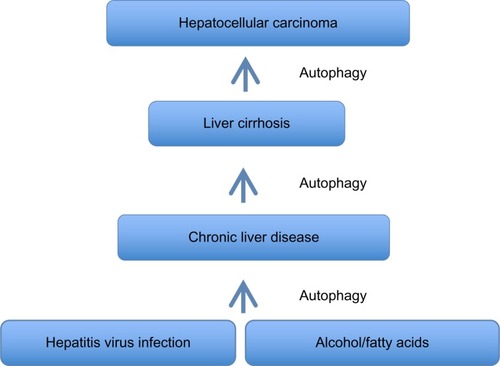
Taken together, the evidence indicates that the autophagy response increases in chronic liver disease, and persists in the stage of liver cirrhosis (). An understanding of whether autophagy acts as a cell death pathway or a prosurvival pathway should allow the development of novel therapeutic strategies for treatment of liver cirrhosis and HCC.
Figure 3 Summary of autophagy response in chronic liver disease, liver cirrhosis, and hepatocellular carcinomas.
Autophagy as a tumor suppressor mechanism in HCC
HCC accounts for more than 500,000 to 600,000 deaths per year worldwide.Citation26 During the last 2 decades, significant progress has been made in understanding the role of autophagy in cancer development, including the development of HCC.Citation27–Citation29 Available evidence suggests that autophagy may serve as a tumor suppressor in cases of chronic liver disease and liver cirrhosis, and that autophagy deficiency may lead to HCC.
The following studies support the idea that HCC develops in the absence of autophagy. Abnormal expression of the autophagy gene Beclin 1 has been found associated with the development of a variety of cancers, including ovarian, breast, prostate, melanoma, colon, and brain.Citation30–Citation37 Heterozygous deletion of Beclin 1 increases susceptibility to spontaneous malignancies and accelerates HBV-related HCC.Citation38 Mice lacking one copy of the gene encoding for the Beclin 1 regulator protein (called AMBRA1) also develop tumors.Citation39 The role of other autophagy genes (UVRAG, Bif1, ATG4C, ATG5, and ATG7) in tumor suppression has been confirmed in mouse models.Citation40–Citation44
Studies by Takamura et alCitation43 show that deletion of either the ATG5 or ATG7 gene in mice results in the development of hepatomegaly and hepatocellular adenoma at age 6–9 months. Takamura et al also found that accumulations of ubiquitinated proteins/aggregates are present in the hepatocytes of tumor-bearing mice but not in the hepatocytes of nontumor mice. They concluded that the tumor cells originated from autophagy-deficient hepatocytes, in association with mitochondrial swelling, p62 accumulation, oxidative stress, and an increased DNA damage response. Specific deletion of p62 expression in the hepatocytes of ATG7-deficient mice decreased tumor size, thus supporting the observation that hepatic p62 expression contributes to tumorigenesis.
Another study addressing p62 expression showed that p62 accumulation leads to hepatocellular adenoma through activation of Nrf2 target genes. The authors showed that p62 competed with the binding between Nrf2 and Keap1, resulting in enhanced transcriptional activation of Nrf2-specific genes in the autophagy-deficient hepatocytes.Citation44 Liver-specific autophagy-deficient mice contained adenomas linked to the formation of p62- and Keap1-positive cellular aggregates, and activation of Nrf2 target genes.
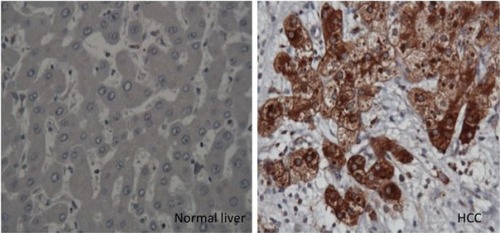
To verify whether this mechanism operates in humans, we showed that high accumulations of autophagy flux protein (p62) are present in paraffin-embedded HCC tissues (). Thus, p62 expression was found in the HCC samples but not in the surrounding nontumorous hepatocytes.Citation45 The deposition of p62/ubiquitin/keratin-like protein aggregates observed in ATG7 knockout mouse is consistent with Mallory body detection in the hepatocytes of patients with alcoholic and nonalcoholic liver diseases.Citation46 Decreased p62 expression, as determined by Western blot analysis, has been used as a reliable marker of autophagy flux. The p62 protein has been found to contribute to carcinogenesis through multiple signaling pathways, including NF-kB, Nrf2, Wnt/β-catenin, and mTOR.Citation47–Citation53 All of these reports provide evidence that autophagy plays a role in tumor suppression, and that autophagy deficiency could lead to HCC. Mechanistically, autophagy deficiency leads to the accumulation of misfolded proteins, dysfunctional mitochondria, the generation of reactive oxygen species, oxidative stress leading to increased DNA damage, the accumulation of double-stranded DNA breaks, increased DNA content, chromosomal instability leading to the cell transformation stage, and carcinogenesis (). If this hypothesis is correct, then the mechanism by which autophagy deficiency is created in cirrhotic livers should be an interesting area of investigation.
Figure 4 Immunohistochemical staining of hepatocytes in HCC and cirrhotic liver.
Autophagy as a prosurvival (oncogenic) mechanism in HCC
Cancer cells need autophagy to generate the energy required to sustain growth and survival under various metabolic as well as therapeutic stress conditions.Citation54 Autophagy plays a prosurvival role during HCC development. Increased autophagy flux has been reported in advanced HCC,Citation55,Citation56 and an increased autophagy response has been found to correlate with malignant progression and poor prognosis of HCC.Citation57 Autophagy was demonstrated to promote HCC invasion through activation of the epithelial–mesenchymal transition.Citation58 In pancreatic cancer, autophagy promotes cell growth, survival, invasion, and metastasis.Citation59–Citation63 It has also been shown that in cells expressing oncogenic RAS, autophagy is required for promotion of cancer, as it maintains oxidative metabolism and facilitates glycolysis.Citation59 Autophagy promotes tumor growth in a mouse model of RAS-driven pancreatic cancer by suppressing p53 activation.Citation64 A similar observation has been made in HCC, indicating that the autophagy response is also needed to inactivate tumor suppressors to promote tumor development. Administration of dethylnitrosamine to wild-type mice inactivated p53-developed HCC, whereas liver-specific ATG5-knockout mice developed only benign hepatic adenoma due to induction of multiple tumor suppressors, including p53.Citation65 Autophagy induction promotes growth of cancerous stem cell–derived mammary tumors.Citation66 Available evidence suggests that 50%–60% of tumors grown under hypoxic conditions show an increased autophagy response.Citation67 Tumor microenvironments, which are clearly different from those of normal tissue, have limited blood supply and are hypoxic, low in energy due to high mitotic activity, acidic, and inflammatory. These conditions induce autophagy by activating various pathways.Citation68,Citation69 All these lines of evidence support the hypothesis that autophagy induction is required for tumor progression, which could also explain why HCC develops more frequently in cirrhotic than in normal liver.
Altered autophagy signaling in HCC
The role of autophagy as a tumor suppressor or oncogenic inducer is unclear, as an increase or decrease in the autophagy response in cancer is often regulated by overactivation or inactivation of oncogenic signaling. This topic is highly complex, and this review therefore considers only selected pathways that are relevant to autophagic regulation in HCC. The interactions between antiapoptotic protein family Bcl-2 and autophagy protein Beclin 1 are of particular importance in the regulation of autophagy.Citation70,Citation71 The interaction of Bcl-2 with wild-type Beclin 1 inhibits the autophagy response, whereas mutant Beclin 1, which is defective in the Bcl-2 binding domain, can induce autophagy. Apoptosis and autophagic cell death are the two mechanisms of cell death that are controlled at the level of interactions between antiapoptotic protein Bcl-2 and autophagic protein Beclin 1. A minimal interaction between these two proteins (Bcl-2 and Beclin 1) favors an increased autophagy response, whereas a maximal interaction leads to autophagy inhibition. The dissociation of Bcl-2 from Beclin 1 is important for activation of autophagy, whereas their association inhibits autophagy. Therefore, the presence of mutant Beclin 1 protein or mutant multidomain protein members of the Bcl-2 family (Bcl-XL) could inhibit this interaction and induce the autophagy response. The interaction between Bcl-2 and Beclin 1 can also be affected by EGFR/mTOR signaling.
A significant number of cancers show high activations of receptor tyrosine kinases, such as epidermal growth factor (EGF). Epidermal growth factor receptor (EGFR) signaling is of key importance in liver injury, inflammation, fibrogenesis, and neoplastic transformation. The EGFR, a receptor tyrosine kinase in the ErbB family, consists of four members: EGFR (ErbB1, HER1), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4). The EGFR pathway becomes deregulated in HCC by a number of mechanisms, including overproduction of ligands, overproduction of receptors, and activation of receptors.Citation72,Citation73 During liver injury and regeneration, hepatocytes express high levels of ErbB1, and the expression of most ligands (eg, EGF and TGF alpha) is increased.Citation74 The EGFR signaling is triggered when the ligand binds to the extracellular ligand-binding domain of the EGFR, which initiates receptor homo-/hetero-dimerization and autophosphorylation in specific tyrosine residues in the intracellular kinase domain of the receptor. Multiple phosphorylation at the kinase domain generates docking sites for a variety of signaling proteins, such as Shc, GRb2, Grb7, Crk, PLC, the kinase Src, PI3K, protein phosphatases SHP1 and SHP2, as well as the ubiquitin ligase Cbl E3.Citation75 The EGFR signaling engages other signaling pathways generated from growth factors, cytokines, and inflammatory mediators to support growth and survival functions.
Studies supporting the role of EGFR-modulated autophagy have shown that overexpression of the EGFR represses autophagy, whereas silencing the EGFR in cancer cells leads to induction of autophagy.Citation76 The EGFR signaling that is relevant to autophagy regulation includes the P13K–AKT–mTOR pathway and the EGFR–Beclin 1 axis. It has been well established that mTOR is the master regulator of autophagy, that mTOR is directly controlled by EGFR signaling,Citation77 and that activation of EGFR inhibits autophagy by reducing Beclin 1 levels.Citation78–Citation80 Reports indicate that EGFR signaling also induces the autophagy response in some cancers, with the response being due to ligand-independent EGFR signaling via the truncated receptor. The EGFR mutant vIII, a naturally occurring EGFR mutant lacking 801 base pairs ligand-binding domain (exons 2–7), can stimulate ligand-independent activity in glycolytic tumors in the brain.Citation81 Tumor-expressing EGFR vIII shows higher activation of the autophagy response due to upregulation of genes involved in cell metabolism, such as glucose transporters (GLUT1 and GLUT3), hexokinase 2 (HK2), and pyruvate dehydrogenase kinase (PDK1).Citation78 The autophagy response is also induced in various models of RAS-induced lung and pancreatic tumors.Citation72
Another explanation for the induced autophagy response in HCC is related to ERK signaling. Among the four classes of MAPK signaling in cancer cells, ERK signaling is activated in response to proliferation signals, while p38 and JNK are activated in response to various stresses.Citation82 The EGFR signaling activates membrane-bound RAS, which interacts with RAF. The RAF phosphorylates two serine residues on the kinase mitogen protein kinase-1 and -2 (MEK1/2); MEK then activates ERK1/2 by phosphorylation of threonine and tyrosine residues, which are separated by one amino acid (threonine-183 and tyrosine-185 of ERK1/2). The activated ERKs phosphorylate numerous cytoplasmic and nuclear proteins to induce cell proliferation; ERK1/2 promotes cell proliferation, being overexpressed in human HCC. It has been reported that direct activation of ERK by MEK can promote autophagy without other signals.Citation83 All of these lines of evidence indicate that altered EGF signaling could activate or inhibit the autophagy response in diverse cancers, including in HCC.
The p53 tumor suppressor, which is inactivated in more than 50% of human cancers, coordinates a wide varieties of responses, including DNA damage, transactivation of cell cycle arresting proteins, metabolism, proapoptotic function, and autophagy.Citation84 Accumulating evidence indicates that p53 can modulate the autophagy response in cancer cells in a dual fashion, depending on its subcellular localization. Nuclear p53 induces the autophagy response by inducing transcription of two modulators: sestrin 1/2 and damage-regulated autophagy modulator (DRAM). Sestrin 1/2 activates autophagy by inhibiting mTOR signaling.Citation85 Cytoplasmic p53 represses autophagy through inactivation of AMPK, which activates mTOR signaling.Citation86 Cytoplasmic p53 can operate at the mitochondrial level to promote cell death and repress the autophagy response.
Autophagy induction in HCC therapy
Significant interest has emerged in the use of autophagy-inducing agents for the treatment and prevention of chronic liver diseases and HCC. As discussed earlier, autophagy plays a dual role in the pathophysiology of chronic liver disease, liver cirrhosis, and HCC, and it is therefore important to define the liver diseases appropriate for autophagy induction or inhibition therapy. The liver diseases that are expected to benefit from autophagy induction include alcoholic fatty liver disease and alpha-1 antitrypsin deficiency.Citation87 Based on a “proof–principle approach”, autophagy induction using carbamazepine has been proven to benefit patients with alpha-1 antitrypsin deficiency.Citation25 However, autophagy induction therapy may be problematic in the treatment of chronic viral hepatitis, as autophagy induction enhances the replication of HBV and HCV. Autophagy induction might also impair the host’s innate immunity and capacity to clear infection.Citation88 Moreover, the autophagy induction therapy approach may not be applicable to those patients with alcoholic or fatty liver disease who are chronically infected with HBV or HCV.
Autophagy induction can be accomplished by the use of both pharmacological and nonpharmacological agents. The best nonpharmacological approach for autophagy induction may be calorie restriction and regular exercise. This approach provides protection against high fat diet–induced diabetes in mice.Citation89 In addition to calorie restriction, other nutritional factors such as coffee and vitamin D intake may be used to improve health through autophagy induction.Citation90–Citation97 Consumption of caffeine induces autophagy, which reduces hepatic steatosis in mice with nonalcoholic fatty liver disease.Citation93 Based on these reports, it is expected that calorie restriction, exercise, coffee consumption, and vitamin D can be adopted in the treatment of chronic liver disease due to alcoholic and nonalcoholic fatty liver disease in humans.
An alternative approach to inducing autophagy in a tissue-specific manner is by gene delivery of vectors that express autophagy genes. A study demonstrated that TFEB gene delivery improves the outcome of a variety of diseases, including obesity/diabetes and alpha-1 antitrypsin deficiency.Citation98 The small-sized molecules currently approved by the US FDA to induce autophagy include carbamazepine, clonidine, lithium, metformin, rapamycin, rilmenidine, sodium valproate, verapamil, trifluoperazine, statin, and tyrosine kinase inhibitors.Citation99,Citation100 These agents can be used for the treatment of liver disease in the context of whether or not autophagy induction would be beneficial. Available evidence suggests that autophagy acts as a tumor suppressor, but is insufficient in HCCs. Therefore, induction of autophagy should help to reverse the malignant phenotype and improve chemotherapeutic treatment of HCC. Based on this reasoning, autophagy-inducing agents can be used along with the FDA-approved drug sorafenib for the treatment of liver cancer. Sorafenib is a multitargeted receptor tyrosine kinase inhibitor that has been approved as a standard therapy for advanced HCC.Citation101 Sorafenib alone has only modest effects in prolonging the survival of HCC patients.Citation102 Studies have shown that sorafenib itself induces the autophagy response and accumulates autophagosomes in HCC cells through inhibition of the mTOR pathway.Citation103 Sorafenib also induces the expression of ER stress response genes (such as IRE-1 and CHOP), eIF2alpha phosphorylation, and the autophagy response in HCC cells.Citation104 Whether chemotherapy drugs that induce autophagic cell death can be used in combination with sorafenib to improve the therapeutic response in HCC patients is currently under investigation, as the concept has been supported by several studies showing that small-molecule drugs inhibit HCC growth through autophagy induction.Citation105–Citation108 A number of chemotherapy drugs known to induce autophagic cell death (such as tamoxifen, etoposide, temozolomide, varinostat, arsenic trioxide, sodium selenite, and metformin) could be used in combination with sorafenib to inhibit HCC cells, especially in cases of defects in the apoptosis pathway. The success of combination therapies using sorafenib and other autophagy inducers needs further validation.
Autophagy inhibition in HCC therapy
Autophagy is required for tumor cell survival, and therefore autophagy inhibition could be explored as a potential therapeutic strategy for cancer treatment. A wide variety of pharmaceutical inhibitors that block different steps of the autophagy process are commercially available. Pharmaceutical inhibitors of HCC growth include 3MA, wortmannin, spautin-1, thapsigargin, vorinostat, chloroquine (CQ), hydroxychloroquine (HCQ), monensin, lucanthone, matrine, xanthohumol, azithromycin, bafilomycin A1, and concanamycin A.Citation99 Among these, CQ and HCQ, which are used to treat malaria, are FDA-approved drugs commonly used as autophagy inhibitors in various experimental tumor models.Citation109–Citation111 Both CQ and HCQ are lysosomal lumen alkalizers that inhibit the activity of lysosomal hydrolases by neutralizing acidic pH in the lumen of lysosomal vesicles. Alkalization of lysosomal vesicles leads to the accumulation of autophagosomes by blocking lysosomal degradation.Citation112 Based on this mechanism, CQ and HCQ have been used as anticancer drug candidates in humans.Citation113
Another lysosomal inhibitor that has been developed, Lys05 (a dimeric CQ), accumulates in the lysosome and shows antitumor activity more potent than that of HCQ.Citation114 A number of new potent autophagy inhibitors have been developed that inhibit autophagy by preventing fusion of autophagosomes with lysosomes, thus causing acidification of the lysosome and lysosomal degradation.Citation115–Citation117 At present, several ongoing cancer clinical trials include autophagy inhibitors along with other chemotherapy agents (http://www.clinical-trials.gov). Autophagy inhibitors can enhance the effectiveness of oxaliplatin, cisplatin, 5-fluorouracil, and sorafenib in HCC models.Citation103,Citation118,Citation119 Coadministration of sorafenib and CQ decreases tumor growth more significantly than administration of either agent alone. It has been demonstrated in experimental animal model that autophagy as inhibitors interact synergistically with either proteasome inhibitor or angiogenesis inhibitor to inhibit HCC growth.Citation119,Citation120
Cancer-initiating cells (ie, cancer stem cells) have been identified in a variety of cancers, but in only a small subpopulation of tumors.Citation121 However, such cells in tumors can differentiate into multiple heterogeneous lineages of cancer cells. Available evidence indicates that current cancer treatments are ineffective in the elimination of the cancer stem cell population, thus resulting in tumor relapse and chemoresistance. Inhibition of autophagy by CQ was found to decrease the viability of liver cancer stem cells under conditions of hypoxia and nutritional starvation.Citation122 It is anticipated that future research will clarify whether autophagy inhibition or induction will have a clinical benefit in the management of HCC chemotherapy.
Currently, there are more than 30 ongoing cancer treatment clinical trials using autophagy inhibitors (HCQ or CQ) in spite of the fact that many tumors, including HCC, show insufficiency in autophagy response ().Citation123 The mechanisms by which the autophagy inhibitors show strong antitumor response in the clinic are not well established. It has been reported that the anticancer mechanisms exhibited by HCQ or CQ are complex and involve more than one mechanism. Some studies reported that CQ sensitizes cancer cells to chemotherapy by inhibiting autophagy,Citation124 inhibiting anticancer drug extrusion by blocking transporter P-glycoprotein,Citation125 promoting apoptosis through lysosomal membrane permeabilization,Citation126 and impairing DNA repair.Citation127 The anticancer mechanism of CQ has been reported to be independent of autophagy inhibition.Citation128 All these results indicate that further understanding of the anticancer mechanisms should establish the therapeutic potential of CQ in cancer.
Conclusion
Autophagy has been recognized as a tumor suppressor mechanism in the liver, and increased autophagy levels have been observed in cases of chronic liver disease, liver cirrhosis, and HCC. The hepatic autophagy response impairs the innate and adaptive immune response. Recent studies have shown that HCC may be associated with an insufficient autophagy response, and available evidence suggests that an insufficient response in HCC could be related to either impaired expression of autophagy genes or altered autophagy signaling. Future research will address whether an increased or decreased autophagy response is associated with the development of HCC related to liver cirrhosis. Autophagy inhibitors as chemotherapeutic agents have shown promising results in the treatment of HCC, by reducing cancer stem cell evolution and improving the immune response against HCC. In summary, autophagy modulation provides new prospects for anti-HCC therapies. We propose that more basic research is needed to further understand the detailed mechanisms of autophagy modulation, and to explore future applications of autophagy modulation to the treatment of liver disease.
Acknowledgments
We thank Samantha Hoekst for critically reviewing this manuscript and Troy Taliancich in the Pathology Department for assistance with image generation. This work was supported by NIH grants CA127481, CA089121, and AI103106.
Disclosure
The authors report no conflicts of interest in this work.
References
- HeCKlionskyDJRegulation mechanisms and signaling pathways of autophagyAnnu Rev Gsssenet2009436793
- YinXMDingWXGaoWAutophagy in the liverHepatology2008471773178518393362
- CuervoAMKnechtETerleckySRDiceJFActivation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvationAm J Physiol199526912001208
- KomatsuMWaguriSUenoTImpairment of starvation-induced and constitutive autophagy in Atg7-deficient miceJ Cell Biol200516942543415866887
- EfeyanACombWCSabatiniDMNutrient-sensing mechanisms and pathwaysNature201551730231025592535
- LambCAYoshimoriTToozeSAThe autophagosome: origins unknown, biogenesis complexNat Rev Mol Cell Biol20131475977424201109
- MizushimaNLevineBCuervoAMKlionskyDJAutophagy fights disease through cellular self-digestionNature20084511069107518305538
- AriasECuervoAMChaperone-mediated autophagy in protein quality controlCurr Opin Cell Biol20112318418921094035
- JewellJLRussellRCGuanKLAmino acid signaling upstream of mTORNat Rev Mol Cell Biol20131413313923361334
- MizushimaNThe role of the ATG1/ULK1 complex in autophagy regulationCurr Opin Cell Biol20102213213920056399
- JankuFMcConkeyDJHongDSKurzrockRAutophagy as a target for anticancer therapyNat Rev Clin Oncol2011852853921587219
- MatsunagaKSaitohTTabataKTwo Beclin 1-binding proteins, ATG14L and Rubicon, reciprocally regulate autophagy at different stagesNat Cell Biol20091138539619270696
- KirkinVMcEwanDGNovakIDikicIA role for ubiquitin in selective autophagyMol Cell20093425926919450525
- PuissantAFenouilleNAubergerPWhen autophagy meets cancer through p62/SQSTM1Am J Cancer Res2012239741322860231
- JagarSBucciCTanidaIRole of Rab7 in maturation of late autophagic vacuolesJ Cell Sci20041174837484815340014
- TanakaYGuhdeGSuterAAccumulation of autophagic vacuoles and cardiomyopathy in LAMP-2 -deficient miceNature200040690290610972293
- YangZKlionskyDJMammalian autophagy: core molecular machinery and signaling regulationCurr Opin Cell Biol20102212413120034776
- MalhiHGuicciardiMEGoresGJHepatocyte Death: a clear and present dangerPhysiol Rev2010901165119420664081
- CanbayAFriedmanSGoresGJApoptosis: the nexus of liver injury and fibrosisHepatology20043927327814767974
- RautouPECazals-HatemDFeldmannGChanges in autophagic response in patients with chronic hepatitis C virus infectionAm J Pathol20111782708271521641393
- TaguwaSkambaraHFujitaNDysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virusJ Virol201185131851319421994453
- SirDChenWLChoiJWakitaTyenTSOuJInduction of incomplete autophagic response by hepatitis C virus via the unfolded protein responseHepatology2008481054106118688877
- LiuBFangMHuYHuangBLiNChangCHepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturationAutophagy20141041643024401568
- LinCWZhangHLiMPharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in miceJ Hepatol20135899399923339953
- HidvegiTEwingMHalePAn autophagy-enhancing drug promotes degradation of mutant alpha-1-antitrypsin Z and reduces hepatic fibrosisScience201032922923220522742
- El-SeragHBHepatocellular CarcinomaN Eng J Med201136511181127
- ZhiXZhongQAutophagy in cancerF1000 Prime Rep2015718
- WhiteEThe role for autophagy in cancerJ Clin Invest2015125424625654549
- GalluzziLPietrocolaFBravo-San PedroJMAutophagy in malignant transformation and cancer progressionEMBO J20153485688025712477
- LiangXHJacksonSSeamanMInduction of autophagy and inhibition of tumorigenesis by beclin 1Nature199940267267610604474
- SaitoHInazawaJSaitoSDetailed deletion mapping of chromosome 17q in ovarian and breast cancers: 2-cM region on 17q21.3 often and commonly deleted in tumorsCancer Res199353338233858100738
- GaoXZacharekASalkowskiALoss of heterozygosity of the BRCA1 and other loci on chromosome 17q in human prostate cancerCancer Res199555100210057866981
- AitaVMLiangXHMurtyVVCloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21Genomics199959596510395800
- ShenYLiDDwangLLDengRZhuXFDecreased expression of autophagy related proteins in mammalian epithelial ovarian cancerAutophagy200841067106818776739
- PirtoliLCeveniniGTiniPThe prognostic role of beclin 1 protein expression in high grade gliomaAutophagy2009593093619556884
- LiBXLiCYPengRQThe expression of beclin 1 is associated with favorable prognosis in stage IIIB colon cancersAutophagy2009530330619066461
- MiraccoCCosciEOliveriGProtein and mRNA expression of autophagy gene Beclin 1 in human brain tumorsInt J Oncol20073042943617203225
- QuXYuJBhagatGFuruyaNPromotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy geneJ Clin Invest20031121809182014638851
- CianfanelliVFuocoCLorenteMAMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradationNat Cell Biol201517203025438055
- LiangCFengPKuBAutophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAGNat Cell Biol2006868869916799551
- TakahashiYCoppolaDMatsushitaNBif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesisNat Cell Biol200791142115117891140
- MarinoGSalvador-MontoliuNFueyoAKnechtEMizushimaNLopez-OtinCTissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3J Biol Chem2007282185731858317442669
- TakamuraAKomatsuMHaraTAutophagy-deficient mice develop multiple liver tumorsGenes Dev20112579580021498569
- InamiYWaguriSSakamotoAPersistent activation of Nrf2 through p62 in hepatocellular carcinoma cellsJ Cell Biol201119327528421482715
- BaoLChandraPKMorozKImpaired autophagy response in human hepatocellular carcinomaExp Mol Path20149614915424369267
- ZatloukalKFrenchSWStumptnerCFrom Mallory to Mallory-Denk bodies: what, how and whyExp Cell Res20073132033204917531973
- MathewRKarpCMBeaudoinBAutophagy suppresses tumorigenesis through elimination of p62Cell20091371062107519524509
- KomatsuMKurokawaHWaguriSThe selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1Nat Cell Biol20101221322320173742
- MoscatJDiaz-MecoMTp62 at the crossroads of autophagy, apoptosis, and cancerCell20091371001100419524504
- JinZLiYPittiRCullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signalingCell200913772173519427028
- GaoCCaoWBaoLAutophagy negatively regulates Wnt signaling by promoting Disheveled degradationNat Cell Biol20101278179020639871
- DuranAAmanchyRLinaresJFp62 is a key regulator of nutrient sensing in the mTORC1 pathwayMol Cell20114413414621981924
- ManleySWilliamsJADingW-XRole of p62/SQSTM1 in liver physiology and pathogenesisExp Mol Med2013238525538
- DegenhardtKMathewRBeaudoinBAutophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesisCancer Cell200610516416843265
- LazovaRCampRLKlumpVSiddiquiSFAmaravadiRKPawelekJMPunctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis and poor outcomeClin Cancer Res20121837037922080440
- MikhaylovaOStrattonYHallDVHL-regulated MiR-204 suppresses tumor growth inhibition of LC3B-mediated autophagy in renal clear cell carcinomaCancer Cell20122153254622516261
- WuDHJiaCCChenJAutophagic LC3B overexpression correlates with malignant progression and predicts a poor prognosis in hepatocellular carcinomaTumor Biol2014351222512233
- LiJYangBZhouQAutophagy promotes HCC cell invasion through activation of epithelial-mesenchymal transitionCarcinogenesis2013341343135123430956
- GuoJYChenHYMathewRFanJActivated Ras requires autophagy to maintain oxidative metabolism and tumorigenesisGenes Dev20112546047021317241
- LockRRoySKenificCMAutophagy facilitates glycolysis during Ras-mediated oncogenic transformationMol Biol Cell20112216517821119005
- YangSWangXContinoGPancreatic cancers require autophagy for tumor growthGenes Dev20112571772921406549
- LockRKenificCMLeidalAMAutophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasionCancer Discov2014446647924513958
- GuoJYXiaBWhiteEAutophagy-mediated tumor promotionCell20131551216121924315093
- RosenfeldtMTO’PretJMortonJPp53 status determined the role of autophagy in pancreatic tumor developmentNature201350429630024305049
- TianYKuoCFWangLGovindrajanSPetrovicLMOuJHAutophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesisCell Death Differentiation2015221025103425526090
- GongCBauvyCTonelliGBeclin 1 and Autophagy are required for the tumorigenicity of breast cancer stem-like progenitor cellsOncogene2013322261227222733132
- YangXYuDDYanFThe role of autophagy induced by tumor microenvironment in different cells and stages of cancerCell Biosci201551425844158
- LiuEYRyanKMAutophagy and cancer-issues we need to digestJ Cell Sci20121252349235822641689
- AmaravadiRKLippincott-SchwartzJYinXMPrinciple and current strategies for targeting autophagy for cancer treatmentClin Cancer Res20111765466621325294
- ShimizuSKanasekiTMizushimaNRole of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genesNat Cell Biol200461221122815558033
- PattingreSTessaAQuXBcl-2 antiapoptotic proteins inhibit beclin 1 dependent autophagyCell200512292793916179260
- KenificCMDebnathJCellular and metabolic functions for autophagy in cancer cellsTrends Cell Biol201525374525278333
- YardenYSliwkowskiMXUntangling the ErbB signaling networkNat Rev Mol Cell Biol2001212713711252954
- ChoiKJBaikIHYeSKLeeYhMolecular targeted therapy for hepatocellular carcinoma: present status and future directionsBiol Pharm Bull20153898699126133708
- McDonellLMKernohanKDBoycottKMSawyerSLReceptor tyrosine kinase mutations in developmental syndrome and cancer: two sides of the same coinHum Mol Genet201524R1R60R6626152202
- CarverRSStevensonMCSchevingLARussellWEDiverse expression of ErbB receptors proteins during rat liver development and regenerationGastroenterology20021232017202712454858
- BerasainCAvilaMAThe EGFR signaling system in the liver: from hepatoprotection to hepatocarcinogenesisJ Gastroenterol20144992324318021
- JuttenBRouschopMAEGFR signaling and autophagy dependence for growth, survival and therapy resistanceCell Cycle201413425124335351
- LiXLuYPanTFanZRole of autophagy in cetuximab-mediated cancer therapy against EGFRAutophagy201061066107720864811
- WeiYZouZBeckerNAndersonMEGFR-mediated Beclin1 phosphorylation in autophagy suppression, tumor promotion and chemoresistanceCell20131541269128424034250
- BabicIAndersonESTanakaKEGFR mutation induced alternative splicing of Max contribute to growth of glycolytic tumors in brain cancerCell Metab2013171000100823707073
- HazzalinCAMahadevanLCMAPK-regulated transcription: a continuously variable gene switch?Nat Rev Mol Cell Biol20023304011823796
- CorcelleENeboutMBekriSDisruption of autophagy at maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activityCancer Res2006666861687016818664
- SoussiTp53 alterations in human cancers: more questions than answersOncogene2007262145215617401423
- BudanovAVKarinMp53 target genes sestrin 1 and senstrin 2 connect genotoxic stress and mTOR signalingCell200813445146018692468
- TasdemirEMaiuriMCGalluziLRegulation of autophagy by cytoplasmic p53Nat Cell Biol20081067668718454141
- LevineBPackerMCodognoPDevelopment of autophagy inducers in clinical medicineJ Clin Invest2015125142425654546
- DereticVSaitohTAkiraSAutophagy in infection, inflammation and immunityNat Rev Immunol20131372273724064518
- HeCBassikMCMoresiVExercise-induced BCL2 regulated autophagy is required for muscle glucose homeostasisNature201248151151522258505
- HandschinCSpiegelmanBMThe role of exercise and PGC1α in inflammation and chronic diseaseNature200845446346918650917
- JiaKLevineBAutophagy is required for dietary restriction-mediated life span extension in C. elegansAutophagy2007359759917912023
- WatanabeTTakemuraGKanamoriHRestriction of food intake prevents postinfarction heart failure by enhancing autophagy in the surviving cardiomyocytesAm J Pathol20141841384139424641899
- SinhaRAFarahBLSinghBKCaffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in miceHepatology2014591366138023929677
- MoonJHLeeJHParkJYCaffeine prevents human prion protein-mediated neurotoxicity through the induction of autophagyInt J Mol Med20143455355824938171
- FreedmanNDParkYAbnetCCHollenbeckARSinhaRAssociation of coffee drinking with total and cause-specific mortalityN Engl J Med20123661891190422591295
- Hoyer-HansenMNordbrandtSPJaattelaMAutophagy as a basis for the health-promoting effects of vitamin DTrends Mol Med20101629530220488750
- Hoyer-HansenMBastholmLMathiasenISEllingFJaattelaMVitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell deathCell Death Differ2005121297130915905882
- PastoreNBlomenkampKAnnunziataFGene transfer of master regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha 1-anti-trypsin deficiencyEMBO Mol Med2013539741223381957
- Vakifahmetoglu-NorbergHXiaHYuanJPharmacologic agents targeting autophagyJ Clin Invest201512551325654545
- ZhangMZWangYPaueksakonPHarrisRCEGFR inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagyDiabetes2014632063207224705402
- LlovetJMRicciSMazzaferroVSorafenib in advanced hepatocellular carcinomaN Eng J Med2008359378390
- WilhelmSCarterCLynchMDiscovery and development of sorafenib: a multikinase inhibitor for treating cancerNat Rev Drug Discov2006583584417016424
- ShimizuSTakeharaTHikitaHInhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinomaInt J Cancer201213154855721858812
- ShiYHDingZBZhouJTargeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosisAutophagy201171159117221691147
- TaiWTShiauCWChenHLMcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cellsCell Death Dis20134e48523392173
- GaoMYehPYLuYSOSU-03012, a novel celecoxib derivative induces reactive oxygen species-related autophagy in hepatocellular carcinomaCancer Res2008689348935719010909
- YuHCLinCSTaiWTNilotinib induces autophagy in hepatocellular carcinoma through AMPK activationJ Biol Chem2013288182491825923677989
- BarefordMDParkMAYacoubASorafenib enhances pemetrexed cytotoxicity through an autophagy-dependent mechanism in cancer cellsCancer Res2011714955496721622715
- AvalosYCanalesJBravo-SaguaRTumor suppression and promoting by autophagyBiomed Res Int2014201460398025328887
- JiangPMuzushimaNAutophagy and human diseasesCell Res201424697924323045
- MahalingamDMiraMSarantopousJCombined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic and pharmacodynamics analysis of hydroxychloroquine in combination with the HDAC inhibitor varinostat in patients with advanced solid tumorsAutophagy2014101403141424991835
- SchneiderPKorolenkoTABuschUA review of drug-induced lysosomal disorders of the liver in man and laboratory animalsMicrosc Res Tech1997362532759140926
- GunjaNRobertDMcCoubrieDSurvival after massive hydroxychloroquine overdosesAnaesth Intensive Care20093713013319157361
- AmaravadiRKWinklerJDLys05: a new lysosomal autophagy inhibitorAutophagy201281383138422878685
- JuhászGInterpretation of bafilomycin, pH neutralizing or protease inhibitor treatments in autophagic flux experiments: novel considerationsAutophagy201281875187622874642
- CarewJSEspitiaCMEsquivelJA2ndLucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosisJ Biol Chem20112866602661321148553
- WangZZhangJWangYMatrine, a novel autophagy inhibitor, blocks trafficking and the proteolytic activation of lysosomal proteasesCarcinogenesis20133412813823002236
- GuoXLLiDHuFTargeting autophagy potentiates chemotherapy-induced apoptosis and proliferation inhibition in hepatocarcinoma cellsCancer Lett201232017117922406827
- HuiBShiYHDingZBProteasome inhibitor interacts synergistically with autophagy inhibitor to suppress proliferation and induce apoptosis in hepatocellular carcinomaCancer20121185560557122517429
- GuoXLLiDSunKInhibition of autophagy enhances anticancer effects of bevacizumab in hepatocarcinomaJ Mol Med (Berl)20139147348323052483
- LinY-HHuangY-CChenL-HChuP-MAutophagy in cancer stem/progenitor cellsCancer Chemther Pharmacol201557879886
- SongYJZhangSSGuoXLAutophagy contributes to the survival of Cd133+liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironmentCancer Lett2013339708123879969
- SiuXChenRWangZAutophagy and chemotherapy resistance: a promising therapeutic target for cancer treatmentCell Death and Disease20134e83824113172
- VlahopoulosSCritselisEVoutsasIFNew use for old drugs? Prospective targets of chloroquines in cancer therapyCurr Drug Targets201415984385125023646
- YamagishiTSahniSSharpDMArvindAJansonPJRichardsonDRP-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestrationJ Biol Chem201328844317613177124062304
- EnzemullerSGonzalezPDebatinKMFuldaSChloroquine overcomes resistance of lung carcinoma cells to the dual PI3K/mTOR inhibitor PI103 by lysosome mediated apoptosisAnticancer Drugs2013241141923111416
- LiuEYXuNO’PreyJLoss of autophagy causes a synthetic lethal deficiency in DNA repairProc Natl Acad Sci201511277377825568088
- MaycottePAryalSCummingsCTThorbumJMorganMJThorburnAChloroquine sensitizes breast cancer cells to chemotherapy independent of autophagyAutophagy20128220021222252008