
Abstract
Nonalcoholic fatty liver disease (NAFLD) has gained importance in recent decades due to drastic changes in diet, especially in Western countries. NAFLD occurs as a spectrum from simple hepatic steatosis, steatohepatitis to cirrhosis, and even hepatocellular carcinoma. Although the molecular mechanisms underlying the development of NAFLD have been intensively investigated, many issues remain to be resolved. Autophagy is a cell survival mechanism for disposing of excess or defective organelles, and has become a hot spot for research. Recent studies have revealed that autophagy is linked to the development of NAFLD and regulation of autophagy has therapeutic potential. Autophagy reduces intracellular lipid droplets by enclosing them and fusing with lysosomes for degradation. Furthermore, autophagy is involved in attenuating inflammation and liver injury. However, autophagy is regarded as a double-edged sword, as it may also affect adipogenesis and adipocyte differentiation. Moreover, it is unclear as to whether autophagy protects the body from injury or causes diseases and even death, and the association between autophagy and NAFLD remains controversial. This review is intended to discuss, comment, and outline the progress made in this field and establish the possible molecular mechanism involved.
Introduction
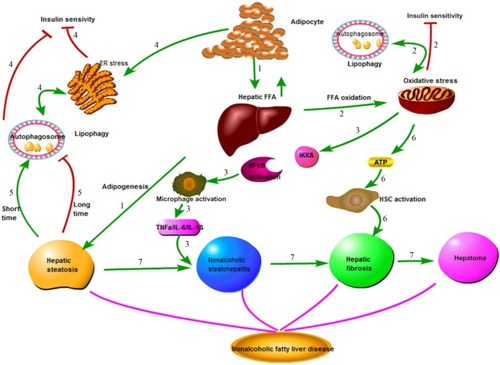
Nonalcoholic fatty liver disease (NAFLD) is the most common hepatic disease in Western countries, and is defined as evidence of hepatic steatosis, either by imaging or histology, with no cause for secondary hepatic fat accumulation such as significant alcohol consumption, use of steatogenic medication, or hereditary disorders.Citation1 The prevalence of NAFLD is 20%–30% in Western countries,Citation2 which causes considerable concern with its progression from steatosis to steatohepatitis, cirrhosis, and even hepatocellular carcinoma (HCC; ). NAFLD is associated with hepatic insulin resistance and type 2 diabetes.Citation3 Recent studies have revealed that patients with NAFLD are at a higher risk of developing coronary atherosclerosis, which might be an independent risk factor.Citation4 The mechanisms involved in the onset of NAFLD are complicated and are not entirely clear at present. Recent advances in the understanding of the molecular processes contributing to autophagy have provided insight into the relationship between autophagy and NAFLD. Autophagy might stimulate lipid metabolism,Citation5 and thus has therapeutic potential in NAFLD.Citation6
Figure 1 Pathophysiology of nonalcoholic fatty liver disease (NAFLD).
Abbreviations: FFA, free fatty acid; ER, endoplasmic reticulum; HSC, hepatic stellate cells.
Autophagy
Autophagy is a critical intracellular pathway that targets defective organelles to the lysosomes for degradation, and this process is highly preserved in eukaryotic cells. As an initial protective mechanism, autophagy is usually triggered by starvation or damage to maintain survival.
Three different types of autophagy are defined based on how the target substrates are delivered to the lysosomes for degradation: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA).Citation7 Macroautophagy is the major type of autophagy. Initially, a small vascular sac called the isolation membrane is formed to enclose a portion of the cytoplasm or organelles, which results in the formation of a double-membrane structure named the autophagosomeCitation8 () The autophagosome fuses with the lysosome to form the autolysosome, which is involved in the degradation of defective or excessive organelles for energy recyclingCitation9 ().
Figure 2 Electron micrograph showing ultrastructure of hepatocytes from a NAFLD mouse model.
Abbreviation: NAFLD, nonalcoholic fatty liver disease.

Figure 3 Molecular mechanisms involved in autophagy and NAFLD.
Abbreviations: NAFLD, nonalcoholic fatty liver disease; FFA, free fatty acid; ROS, reactive oxygen species; ER, endoplasmic reticulum; WAT, white adipose tissue; BAT, brown adipose tissue.
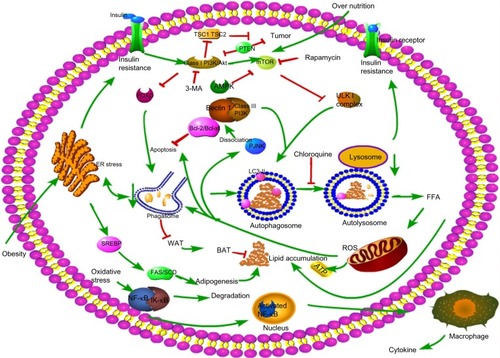
More than 30 autophagy-related genes (Atgs) have been discovered in yeast and play important roles in autophagy, many of which have mammalian homologs.Citation10 Many molecules are involved in the formation and working process of autophagy. Atg8/LC3, Atg7, and Atg6/Beclin-1 with the class III phosphoinositide 3-kinase (PI3K) complex are the best characterized. The process of autophagosome formation involves three major steps: 1) uncoordinated 51-like kinase (ULK) 1 helps in the double-membrane formation in the initiation phase; 2) nucleation with the Beclin-1/class III PI3K complex; and 3) elongation of the isolation membrane with the help of LC3 lipidation.Citation8,Citation11 Conjugation of yeast Atg8 or mammalian LC3 with phosphatidylethanolamine (PE) during autophagy results in the formation of an insoluble form of Atg8 (Atg8-PE) or LC3 (LC3-II), which contributes to autophagosome formation. Atg8/LC3 stays on the membrane until it is degraded by lysosomes. Therefore, Atg8/LC3 is widely used as a marker for monitoring autophagy.Citation12
Several signaling pathways are involved in the regulation of autophagy, among which, the class I PI3K/Akt/mTOR signaling pathway is the most classic one. PI3K promotes the phosphorylation of Akt, which subsequently leads to the activation of mTOR and the inhibition of autophagy. Conversely, inhibiting the activation of Akt induces a high level of autophagy, suggesting that class I PI3K/Akt/mTOR inversely regulates autophagy.Citation13 When cells are under normal nutrient conditions, mTOR will be activated and phosphorylates ULK1, which subsequently combines with Atg13 and FIP200 and results in the inhibition of autophagosome formation (). Rapamycin or nutrient deprivation-induced inhibition of mTOR leads to dephosphorylation of ULK1, ULK2, and Atg13, and activates ULK to phosphorylate FIP200, resulting in the stimulation of autophagy.Citation14,Citation15 Another drug named 3-methyladenine (3-MA) inhibits autophagy by suppressing the class III PI3K/Beclin-1 signaling pathway, which positively regulates autophagy.Citation12 Furthermore, AMP-activated protein kinase (AMPK) plays a role in the regulation of autophagy. AMPK is activated by elevation of the AMP/ATP ratioCitation12 in accordance with upregulation of autophagy under conditions of starvation, as described in . Initial studies also found that AMPK is a positive regulator of autophagy both in the yeastCitation16 and mammalian cells.Citation17
Autophagy and steatosis
Autophagy reduces lipid droplets via lipophagy
Several studies have demonstrated that autophagy is closely associated with metabolism, and this has caused considerable concern recently. Autophagy is characterized by digesting damaged organelles that are used to generate energy and materials for cell recycling.Citation18 Degradation of cellular lipid stores through autophagy (lipophagy) is another way to degrade lipid besides cytosolic lipases, which opens up a new understanding of lipid degradation.Citation19 Increased levels of fatty acids sustain the rates of mitochondrial β-oxidation for generation of ATP.Citation20 However, excessive fatty acids will cause damage to mitochondria if they surpass the oxidation capacity of mitochondria. The increase in autophagy observed in a fatty liver can be confirmed by the presence of lipids enclosed within autophagic vacuoles and elevated expression of LC3-II, a bioprotein marker of autophagy.
Singh et al demonstrated that hepatocyte triglyceride content was significantly increased when challenged with fatty acid supplementation or cultured in methionine and choline-deficient medium with exogenous lipid supplementation treated with 3-MA, a pharmacological inhibitor of autophagy.Citation19 Conversely, treatment with rapamycin, a pharmacological stimulator of autophagy, significantly decreases lipid droplets.Citation19 It was reported recently that thyroid hormones, known to be important in promoting metabolism, probably stimulate fatty acid oxidation by induction of hepatic autophagy, which could be blocked by Atg5 siRNA.Citation21 Another study showed that treatment with caffeine reduced cellular lipid accumulation and the serum level of β-hydroxybutyrate, a product of lipolysis, and is accompanied with the increase of LC3-II. However, Atg5 knockdown in hepatocytes significantly increased intracellular lipids and reduced β-hydroxybutyrate levels.Citation22 Another study demonstrated that pharmacological intervention with rapamycin or carbamazepine (mTOR-independent inducer of autophagy) in high-fat diet (HFD)-fed mice obviously reduced hepatic and blood triglyceride, blood glucose and plasma insulin levels,Citation23 indicating that modulation of autophagy alleviates NAFLD via an mTOR-dependent or -independent pathway.Citation24 Autophagy removes lipid droplets not only in NAFLD but also in other factor-induced fatty liver diseases such as ethanol-induced steatosis.Citation25
All the studiesCitation5,Citation12,Citation19,Citation20 mentioned earlier showed similar results that intracellular reduction in lipid droplets was associated with a concomitant increase in autophagy flux, suggesting that autophagy plays essential roles in lipolysis, called lipophagy, which may provide a new way to eliminate fat in addition to lipase-mediated lipolysis.Citation26,Citation27 In fact, the effect of autophagy on lipid clearance may not be restricted to macrolipophagy, and indirect autophagy of organelles such as mitochondria and peroxisomes are probably involved; however, more investigations are needed to determine the independent effect of macrolipophagy.Citation26
Autophagy regulates adipogenesis and adipose tissue differentiation
Contrary to the lipolysis effect, autophagy may also work on adipogenesis and lipocyte differentiation. It has been found that Atg7 knockdown of white adipose tissue (WAT) in mice showed a remarkable phenotype. The mutant mice were smaller than their wild-type counterparts, with larger numbers of smaller adipocytes filled with multilocular lipid droplets, as observed by light microscopy, and higher numbers of mitochondria, as observed by electron microscopy. All the results demonstrate that autophagy is critical for WAT adipogenesis, especially for the formation of the unilocular lipid droplet structure and for the mitochondria homeostasis control.Citation28
Likewise, Ma et alCitation29 discovered that deletion of FIP200, a core subunit of the mammalian autophagy related 1 complex, reduced both HFD- and starvation-induced steatosis and downregulated the gene expression of enzymes involved in de novo lipogenesis, which include ATP-citrate lyase (Acly), acetyl-CoA carboxylase (Acaca), fatty acid synthase (Fasn), and stearoyl-CoA desaturase-1 (Scd1). This was consistent with another studyCitation30 which reported that knockdown of Atg7 in pre-adipocytes inhibited lipid accumulation. Moreover, adipocyte-specific knockout of Atg7 generated lean mice with decreased WAT and increased brown adipocytes.Citation30 Similar results were demonstrated by other studies where autophagy deficiency exerted a protective role against obesity by inhibiting adipogenesis.Citation31–Citation33 In addition to adipocytes, researchers using hepatocytes also demonstrated that intracellular lipid droplets were suppressed by Atg7 knockdown.Citation34
Autophagy plays essential roles not only in adipogenesis but also in the process of WAT differentiation into mature tissue. Mouse embryonic fibroblasts with Atg5 deletion failed to complete the differentiation process, with the main cells developing multiple small lipid droplets and eventually dying.Citation35 Although mitochondria are mostly responsible for fatty acid oxidation, their contribution to adipogenesis cannot be neglected. The number of mitochondria is increased in the early stage of adipogenesis and during maturation of WAT, due to the requirement of both energy and the key substrates for lipogenesis such as acetyl-CoA and glycerol 3-phosphate. However, the number of mitochondria was decreased by autophagy, called mitophagy, which may reflect the decreased need for lipogenesis in mature white adipocytes. The most significant features of adipose-specific Atg knockout in mice were an increased number of mitochondria and multiple small lipid droplets, compared with mature white adipocytes.Citation36
Autophagy is also involved in the cell-type differentiation of adipocytes. WAT and brown adipose tissue (BAT) are two different types of tissue. In WAT, lipid storage prevents free fatty acid (FFA) lipotoxicity and provides energy when nutrients are restricted. BAT has a reduced capacity for lipid storage but higher rate of oxidation to provide energy.Citation20 Tissue Atg7 knockout in mice had decreased WAT but increased BAT, along with an increased rate of oxidation and lean body mass, suggesting an inhibitory effect of autophagy on differentiation of WAT to BATCitation30,Citation37 ().
Collectively, autophagy is involved in lipolysis, lipogenesis, and differentiation of adipocytes. The two mechanisms are not contradictory, but coexist and coact on lipid regulation. The imbalance between lipogenesis and lipolysis may result in steatosis, and studies focusing on expanding one side, such as lipolysis, may be of interest for NAFLD therapy.
Molecular mechanisms linked with autophagy and steatosis
Effect of lipotoxicity on autophagy varies with time
The effect of autophagy varies with time as well as nutritional status. Autophagy is activated by short-term stimulation of lipid availability as a self-protective mechanism to degrade lipid droplets.Citation19 Both ob/ob mice and chronic HFD mice show markedly impaired hepatic autophagy, particularly expression of Atg7.Citation38 Furthermore, chronic exposure to lipid challenge has an inhibitory effect on CMA, which is probably associated with accelerated degradation of LAMP-2A.Citation39 Liu et al found that long-term insulin resistance inhibited autophagy by inhibition of the constitutive nuclear form of FOXO1, which controls the transcription of autophagy-related genes, while short-term insulin resistance activates autophagy via the mTOR complexCitation40 (). Long-term lipid load may change membrane lipid composition and reduce autophagosome/lysosome fusion both in vitro and in vivo, which may partly explain why excessive lipid reduces autophagy and accelerates lipid accumulation.Citation41
Autophagy and aging
Autophagy declines with aging in parallel with increased lipid accumulation in various organs, which may be associated with the increased prevalence of NAFLD, type II diabetes, and other metabolic disorders. The age-related changes in liver macroautophagic proteolysis have been extensively studied in vitro and in vivo.Citation7 Both macroautophagy and CMA decrease with age.Citation39,Citation42 What is responsible for the decline in autophagy upon aging? Insulin/IGF-1 is the best characterized pathway that is highly conserved in humans.Citation43 Alterations in the IGF-1 signaling pathway may lead to extension of life and affect the process of autophagy.Citation44 IGF-1 may inhibit macroautophagic proteolysis and degradation of mitochondria harboring deleterious mtDNA mutations.Citation45 The IGF-1 signaling cascade includes PI3K/Akt pathway and is downstream of mTOR. Downregulation of PI3K/Akt stimulates autophagy, so autophagy may partly account for extension of life.Citation13
Autophagy and insulin resistance
The pathogenesis of NAFLD involves a two-hit hypothesis.Citation46 The first hit is insulin resistance, which is closely associated with the metabolic syndrome. Insulin resistance enhances lipolysis, increasing serum levels of FFA and decreasing very-low-density lipoprotein, which delivers triglycerides and cholesterol from the liver to peripheral organs, and promotes the resynthesis of lipid, thus leading to lipid storage in the liver. At the same time, fatty acid oxidation is increased and impairs mitochondrial oxidation, contributing to oxidative stressCitation47 (). As previously shown, insulin has an inhibitory effect on autophagy in the liver, but the mechanism remains unknown. This result is in agreement with a former study in which autophagy flux in rats treated with HFD for 2 weeks was upregulated, but was nearly inhibited after giving HFD for 10 weeks.Citation48 FOXO transcription factors are regulators of longevity downstream of insulin signaling, which is involved in promoting expression of autophagy genes and the ubiquitin–proteasome system, known as two cell clearance processs.Citation49 Another signaling pathway related to the inhibition of autophagy in the presence of insulin is class I PI3K/AKT that leads to the activation of mTOR and phosphorylation of its downstream p70S6k and autophagy inhibition, which can be blocked by rapamycin, an inhibitor of mTOR ().Citation50 Furthermore, Atg7 mutant mice showed impaired glucose tolerance, abnormal levels of insulin, and reduced β-cell mass.Citation51 Defective hepatic autophagy promotes endoplasmic reticulum stress and causes insulin resistance.Citation38
In contrast, autophagy deficiency in skeletal muscles leads to impaired activity of mitochondrial oxidative phosphorylation and subsequent induction of FGF-21 through ATF4 activation. As a result, autophagy deficiency enhanced oxidation and energy expenditure through browning of WAT and increased insulin sensitivity.Citation32 In conclusion, the relationship between autophagy and insulin resistance is controversial. On the one hand, insulin resistance-induced steatosis reduces autophagy, which in turn aggravates insulin resistance and ER stress (), and on the other hand, autophagy deficiency is involved in attenuating insulin resistance.
Autophagy and steatohepatitis
Benign hepatic steatosis can progress to nonalcoholic steatohepatitis (NASH), the state of inflammation and hepato-cellular injury that is characterized by elevation of alanine aminotransferase, aspartate aminotransferase, and several adipokines and cytokines. Adiponectin, leptin, TNF-α, and IL-6 play critical roles in the process of inflammation and liver injuryCitation52 ().
Several studies have demonstrated that autophagy exhibits cellular protective effects, leading cells to survival from death stimuli.Citation53–Citation56 Cell damage is increased in long-term steatosis and aging in cells and tissues, suggesting that cell damage is correlated with autophagy reduction. Wang et al have discovered that autophagy inhibition by shRNA-mediated knockdown of Atg5 sensitizes cells to death from menadione by overactivation of JNK and the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) 1/2 signaling pathway.Citation56
However, autophagy is a double-edged sword that mediates cell proliferation as well as cell death. Reactive oxygen species (ROS) produced by excessive FFA oxidation in mitochondria activate the JNK signaling pathway, promoting the dissociation of Bcl-2/Beclin-1 complex and autophagy induction, which is accompanied by apoptosis ().Citation57,Citation58 The effect of autophagy on life and death of a cell is controversial.
Immune activation and related signaling pathways
Immune activation is a prerequisite for the development of NASH, which exacerbates steatosis, including innate immune pathways.Citation59 Salminen et al have discovered that inhibition of autophagic capacity with aging accelerates inflammation via activation of NLR P3.Citation60
Activation of JNK and IKKB promotes the synthesis of AP-1 and NF-κB, leading to an inflammatory reaction.Citation59 Insulin resistance and oxidative stress stimulate phosphorylation and subsequent degradation of IKKB, accompanied by trans-location of activated NF-κB to the nucleus, causing inflammatory cascades, which in turn aggravate NF-κB activation (). Extracellular lipid can activate IKKB by engaging TLRs. Mice deficient in hepatocyte TLR4 attenuated severity of steatosis after challenge with an HFD, indicating that TLR4 is an important proinflammatory mediator.Citation61 TLR may play an important role in pathogen-induced autophagy via a TRIF-dependent, MyD88-independent TLR4 signaling pathway, which has not been recognized before.Citation62
Saitoh et al have shown that Atg16L1 deficiency disrupts the formation of autophagosome, resulting in more severe damage and higher amounts of inflammatory cytokines such as IL-1β and IL-18, under stimulation with lipopolysaccha-ride.Citation63 The mechanism by which autophagy regulates non-pathogen immunity, such as NASH, has not been elucidated and needs further investigation.
Apoptosis
Hepatocyte apoptosis due to FFA toxicity, called lipoapoptosis, is an important mechanism in the progression of NAFLD or NASH.Citation64–Citation66 Natarajan et al discovered that hepatocyte and cholangiocyte lipoapoptosis is induced by FFAs.Citation66 Activation of p38-MAPK, ERK, and JNK signaling pathways is involved in the mechanism of lipoapoptosis,Citation65,Citation66 but the exact mechanism involved in the regulation of lipoapoptosis remains controversial.
Autophagy and apoptosis are two major ways of programmed cell death. There is significant cross talk between autophagy and apoptosis, as they can cooperate, antagonize, or assist each other under different conditions.Citation67 MCL-1, an antiapoptotic Bcl-2 homolog, is a stress sensor that controls autophagy and apoptosis, and MCL-1 degradation not only induces apoptosis but also activates autophagy.Citation68 It is established that the cross talk between apoptosis and autophagy is mediated, at least partially, by interaction of the Beclin-1 and Bcl-2 complexes.Citation69 Bcl-2 functions as an antiapoptotic protein as well as an anti-autophagy protein by inhibitory interaction with Beclin-1, to help maintain autophagy at levels that are compatible with cell survival rather than cell death.Citation69 The connection between Bcl-2 and Beclin-1 or Bax is disrupted to induce autophagy and apoptosis, respectively, under conditions of stress.Citation57 Localization of Bcl-2 to the ER prevents Beclin-1-mediated, starvation-regulated autophagy by binding to NAF-1, which stabilizes the interaction between Bcl-2 and Beclin-1 at the ER surface.Citation70 Proapoptotic Bcl-2 homology (BH)3-only proteins have been described in previous studies to bind with the BH3-binding pocket of Bcl-2 or Bcl-XL, leading to the disassociation of Bcl-2/XL and Beclin-1, resulting in autophagy or apoptosis, respectively.Citation71,Citation72 Activation of the JNK1 signaling pathway stimulates the disassociation of Bcl-2 from Beclin-1 via phosphorylating multiple residues at the Bcl-2 regulatory position, leading to inactivated Bcl-2 and autophagy induction Citation65 ().
There is another type of protein called death-associated protein kinase (DAPK) that is reported to promote disassociation of Bcl-2 from Beclin-1 by phosphorylating Thr119 of Beclin-1.Citation73 HMGB1 is also suggested to be involved in the disassociation of Bcl-2 from Beclin-1.Citation74,Citation75 Autophagy is regarded as a cell survival mechanism; however, the interaction of Bcl-2 – an antiapoptotic protein –with Beclin-1 results in the inactivation of Beclin-1 and maintains autophagy to a certain extent. The disassociation of the complex leads to apoptosis and autophagy. The cross talk between autophagy and apoptosis is controversial and needs further investigation.
Autophagy and liver fibrosis
NAFLD is characterized by its course from simple steatosis to steatohepatitis, fibrosis, and cirrhosis. Hepatic fibrosis is the progressive accumulation of extracellular matrix that occurs in various chronic liver diseases,Citation76 including NAFLD. Hepatic stellate cells (HSCs) function well in hepatic fibrosis, which may be a possible link with autophagy. HSC activation is critical in liver fibrosis, as it leads to the production of excessive extracellular matrixCitation77 (). According to a recent study, autophagy was increased in mice treated with CCl4 or thioacetamide, accompanied by lipid droplet loss and HSC activation. On the contrary, blockade of autophagy with siRNA, resulting in Atg5 knockdown, reduces fibrosis and matrix accumulation and increases the number and size of lipid droplets.Citation78 Autophagy fuels tissue fibrogenesis not only in liver but also in other organs such as the lungs and kidneys,Citation79 and may be a new factor in HSC activation.Citation80 Autophagy affects the activation of HSCs not only due to the energy supply by autophagy, but also due to the effect of autophagy on cell growth cycle. 3-MA-induced autophagy inhibition is reported to block the G2 phase of HSC-T6 cells, a rat HSC line, leading to inhibition of cell proliferation.Citation81 However, the relationship between autophagy and HSC proliferation remains controversial. An early study found that rapamycin – an mTOR target inhibitor and autophagy stimulator – reduced fibrosis,Citation82 and that this was due to its inhibition of cell proliferation or its indirect effect on autophagy induction; however, the mechanisms involved remain unclear.
Although autophagy plays essential roles in HSC activation and fibrosis promotion, its effect on antifibrosis and collagen degradation should not be neglected. In α-1-antitrypsin deficiency, a disease in which the α-1-antitrypsin mutant Z protein results in protein aggregates and chronic liver injury, an autophagy-enhancing drug was demonstrated to reduce the hepatic load and reversed fibrosis.Citation83 Both genetic and pharmacological inhibition of autophagy in mice resulted in increased levels of type I collagen in mouse kidneys and primary mesangial cells, suggesting that autophagy promotes intracellular degradation of type I collagen.Citation84 However, whether it exhibits the same effect in liver remains unclear. Moreover, autophagy is regarded as a cell survival mechanism; it reduces oxidative stress and ER stress, and inhibits inflammation, thereby leading to fibrosis reduction.
We suggest that if autophagy could be selectively inhibited in HSCs and other fibrotic cells, a special blocker for autophagy would be an attractive candidate for antifibrotic strategies. However, this is challenging, as the tissue of the whole body is a complex of various cell types. Furthermore, since fibrosis is the result of an imbalance between collagen synthesis and degradation, the role of autophagy in collagen degradation is of great interest to the study of its antifibrotic mechanism.
Autophagy and hepatic carcinogenesis
NAFLD has the potential to progress into hepatic carcinoma. The role of autophagy in hepatic carcinogenesis is complex and is regarded as a double-edged sword.Citation85 Controversy exists as to whether autophagy inhibits or promotes carcinogenesis. Liang et al first reported that autophagy-related gene Beclin-1 was associated with cancer suppression.Citation86 Several tumor suppressor genes such as PTEN, TSC1, and TSC2 stimulate autophagy by inhibiting mTOR pathway (). On the contrary, class I PI3K/Akt activates oncogenes and inhibits autophagy by activating mTOR.Citation11 Qu et alCitation87 found that Beclin-1, a gene closely related to autophagy, was deleted in 40%–75% of the cases of human sporadic breast, ovarian, and prostate cancers. Beclin-1 gene knockout in mice increased the frequency of spontaneous hepatic malignancies and reduced autophagy.Citation87 Similarly, it has been shown that autophagy may function in tumor suppression by mitigating metabolic stress and, in concert with apoptosis, by preventing death by necrosis.Citation88 This evidence suggests that defective autophagy is positively correlated with carcinoma development.
However, autophagy is also a cell survival mechanism to help cells live through the environment of stress or nutrient restriction, thus leading to proliferation of tumor cells under stress, and may promote metastasis by enhancing tumor cell survival.Citation89,Citation90 Autophagy may suppress the p53 response by limiting DNA damage, oxidative stress, oncogene activation, and tumor cells less sensitive to starvation-induced cell death, thereby protecting cells from stress and promoting tumorigenesis.Citation91 Furthermore, a recent study demonstrated that autophagy promoted HCC cell invasion by activation of epithelial–mesenchymal transition through a TGF-β/Smad3 signaling-dependent manner.Citation92 In addition, LC3 is highly expressed in hepatoma compared with normal tissues, which is regarded as an independent predictor of hepatoma recurrence after surgery.Citation93
As yet, it is undetermined as to whether autophagy promotes or suppresses carcinoma. The double-edged sword effect of autophagy on tumors makes it important as a potential therapeutic option for cancer suppression, if properly controlled. However, several important questions are of concern. How long does it take for autophagy inhibition or activation to suppress an established tumor? Does the inhibition or activation effect depend on the tumor type or the drug concentration? How does autophagy function in tumors deficient in autophagy-related genes? In summary, activation of autophagy may represent a promising and effective strategy for liver cancer therapy, while the inhibition of autophagy may lead to the death of malignant cells due to energy restriction.Citation94 Further investigations are needed to determine the dual role of autophagy in carcinoma.
Future perspectives and conclusion
Autophagy is associated with NAFLD. Much work is needed to determine the exact relationship between autophagy and NAFLD, and the results from various studies are controversial. Most studies have provided evidence that autophagy deficiency leads to steatosis, since autophagy digests lipid droplets, and drugs that increase autophagy may be a potential therapeutic approach in NAFLD.
However, several studies have demonstrated that autophagy is essential in adipogenesis and differentiation. Whether the difference in the results is related to the extent of autophagy or activation of other signaling pathways needs further investigation. It may also be significant to focus on other cell types such as HSCs and Kupffer cells, which are also involved in the progression of NAFLDCitation95 in addition to hepatocytes. More attention should be paid to the contribution of other types of autophagy to the pathogenesis of NAFLD; for example, CMA, though macroautophagy is regarded as the major type of autophagy.
There are some other questions that deserve consideration. Is the mechanism of lipid regulation tissue specific in the liver? Upregulation of autophagy protects hepatocytes against lipotoxicity, but cancer may also be promoted since autophagy is regarded as a cellular survival mechanism and helps cells to live through the conditions of stress and nutrient restriction. The answer to whether autophagy is a friend or foe in cancer is controversial. Moreover, the cross talk between apoptosis and autophagy, and the extent autophagy provides a protective effect in cells, need to be investigated. Autophagy induces HSC activation and stimulates hepatic fibrosis; however, autophagy fuels HSC activation and works on collagen degradation and inflammation inhibition, which seems to inhibit fibrosis. It is unclear as to whether the different effects of autophagy on fibrosis are cell type- or tissue-specific. The selective inhibitor of autophagy may be an ideal candidate for antifibrotic medicines.
In conclusion, a better understanding of the bidirectional characters of autophagy and its regulatory mechanisms may allow the practical use of autophagy for therapeutic purposes in the future.
Acknowledgments
Supported by Research Grant for Key Clinical Discipline Construction of Shanghai Municipality, People’s Republic of China (grant number: ZK2012B20).
Disclosure
The authors report no conflicts of interest in this work.
References
- ChalasaniNYounossiZLavineJEThe diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological AssociationHepatology2012552005202322488764
- MilicSStimacDNonalcoholic fatty liver disease/steatohepatitis: epidemiology, pathogenesis, clinical presentation and treatmentDig Dis20123015816222722431
- BirkenfeldALShulmanGINon alcoholic fatty liver disease, hepatic insulin resistance and type 2 diabetesHepatology20135971372323929732
- KimDChoiSYParkEHNonalcoholic fatty liver disease is associated with coronary artery calcificationHepatology20125660561322271511
- SinhaRAFarahBLSinghBKCaffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in miceHepatology2014591366138023929677
- AmirMCzajaMJAutophagy in nonalcoholic steatohepatitisExpert Rev Gastroenterol Hepatol2011515916621476911
- CuervoAMBergaminiEBrunkUTDrogeWFrenchMTermanAAutophagy and aging: the importance of maintaining “clean” cellsAutophagy2005113114016874025
- RautouPEMansouriALebrecDDurandFVallaDMoreauRAutophagy in liver diseasesJ Hepatol2010531123113420810185
- MizushimaNYoshimoriTOhsumiYThe role of Atg proteins in autophagosome formationAnnu Rev Cell Dev Biol20112710713221801009
- MizushimaNYoshimoriTLevineBMethods in mammalian autophagy researchCell201014031332620144757
- LevineBKroemerGAutophagy in the pathogenesis of diseaseCell2008132274218191218
- YinXMDingWXGaoWAutophagy in the liverHepatology2008471773178518393362
- ShintaniTKlionskyDJAutophagy in health and disease: a double-edged swordScience200430699099515528435
- MurrowLDebnathJAutophagy as a stress-response and quality-control mechanism: implications for cell injury and human diseaseAnnu Rev Pathol2013810513723072311
- JungCHJunCBRoSHULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machineryMol Biol Cell2009201992200319225151
- WangZWilsonWAFujinoMARoachPJAntagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85pMol Cell Biol2001215742575211486014
- TakagiHMatsuiYHirotaniSSakodaHAsanoTSadoshimaJAMPK mediates autophagy during myocardial ischemia in vivoAutophagy2007340540717471015
- RabinowitzJDWhiteEAutophagy and metabolismScience20103301344134821127245
- SinghRKaushikSWangYAutophagy regulates lipid metabolismNature20094581131113519339967
- LiuKCzajaMJRegulation of lipid stores and metabolism by lipophagyCell Death Differ20132031122595754
- SinhaRAYouSHZhouJThyroid hormone stimulates hepatic lipid catabolism via activation of autophagyJ Clin Invest20121222428243822684107
- SinhaRAFarahBLSinghBKCaffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in miceHepatology2014591366138023929677
- LinCWZhangHLiMPharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in miceJ Hepatol20135899399923339953
- Gracia-SanchoJGuixe-MuntetSHideDBoschJModulation of autophagy for the treatment of liver diseasesExpert Opin Investig Drugs201423965977
- DingWXLiM ChenXAutophagy reduces acute ethanol-induced hepatotoxicity and steatosis in miceGastroenterology20101391740175220659474
- ZechnerRMadeoFCell biology: another way to get rid of fatNature20094581118111919407787
- MaoYChengJYuFLiHGuoCFanXGhrelin attenuated lipotoxicity via autophagy induction and nuclear factor-kappaB inhibitionCell Physiol Biochem20153756357626329041
- GoldmanSZhangYJinSAutophagy and adipogenesis: implications in obesity and type II diabetesAutophagy2010617918120110772
- MaDMoluskyMMSongJAutophagy deficiency by hepatic FIP200 deletion uncouples steatosis from liver injury in NAFLDMol Endocrinol2013271643165423960084
- SinghRXiangYWangYAutophagy regulates adipose mass and differentiation in miceJ Clin Invest20091193329333919855132
- ZhangYGoldmanSBaergaRZhaoYKomatsuMJinSAdipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesisProc Natl Acad Sci U S A2009106198601986519910529
- KimKHJeongYTOhHAutophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokineNat Med201319839223202295
- LapierreLRSilvestriniMJNunezLAutophagy genes are required for normal lipid levels in C. elegansAutophagy2013927828623321914
- ShibataMYoshimuraKFuruyaNThe MAP1-LC3 conjugation system is involved in lipid droplet formationBiochem Biophys Res Commun200938241942319285958
- BaergaRZhangYChenPHGoldmanSJinSTargeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in miceAutophagy200951118113019844159
- GoldmanSJZhangYJinSAutophagic degradation of mitochondria in white adipose tissue differentiationAntioxid Redox Signal2011141971197821126221
- SinghRAutophagy and regulation of lipid metabolismResults Probl Cell Differ201052354620865370
- YangLLiPFuSCalayESHotamisligilGSDefective hepatic autophagy in obesity promotes ER stress and causes insulin resistanceCell Metab20101146747820519119
- Rodriguez-NavarroJAKaushikSKogaHInhibitory effect of dietary lipids on chaperone-mediated autophagyProc Natl Acad Sci U S A2012109E705E71422331875
- LiuHYHanJCaoSYHepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulinJ Biol Chem2009284314843149219758991
- KogaHKaushikSCuervoAMAltered lipid content inhibits autophagic vesicular fusionFaseb J2010243052306520375270
- CuervoAMDiceJFAge-related decline in chaperone-mediated autophagyJ Biol Chem2000275315053151310806201
- LongoVDFinchCEEvolutionary medicine: from dwarf model systems to healthy centenarians?Science20032991342134612610293
- Brown-BorgHMBorgKEMeliskaCJBartkeADwarf mice and the ageing processNature1996384338900272
- GuYWangCCohenAEffect of IGF-1 on the balance between autophagy of dysfunctional mitochondria and apoptosisFebs Lett200457735736015556609
- DayCPJamesOFSteatohepatitis: a tale of two “hits”?Gastroenterology19981148428459547102
- KawanoYCohenDEMechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver diseaseJ Gastroenterol20134843444123397118
- PapackovaZDankovaHPalenickovaEKazdovaLCahovaMEffect of short- and long-term high-fat feeding on autophagy flux and lysosomal activity in rat liverPhysiol Res201261Suppl 2S67S7623130905
- WebbAEBrunetAFOXO transcription factors: key regulators of cellular quality controlTrends Biochem Sci20143915916924630600
- KanazawaTTaneikeIAkaishiRAmino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytesJ Biol Chem20042798452845914610086
- EbatoCUchidaTArakawaMAutophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat dietCell Metab2008832533218840363
- PinzaniMPathophysiology of non-alcoholic steatohepatitis and basis for treatmentDig Dis20112924324821734391
- PuaHHDzhagalovIChuckMMizushimaNHeYWA critical role for the autophagy gene Atg5 in T cell survival and proliferationJ Exp Med2007204253117190837
- FukuoYYamashinaSSonoueHAbnormality of autophagic function and cathepsin expression in the liver from patients with nonalcoholic fatty liver diseaseHepatol Res20144491026103624299564
- WuDCederbaumAIInhibition of autophagy promotes CYP2E1-dependent toxicity in HepG2 cells via elevated oxidative stress, mitochondria dysfunction and activation of p38 and JNK MAPKRedox Biol2013155256524273738
- WangYSinghRXiangYCzajaMJMacroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stressHepatology20105226627720578144
- MukhopadhyaySPandaPKSinhaNDasDNBhutiaSKAutophagy and apoptosis: where do they meet?Apoptosis20141955556624415198
- YouleRJNarendraDPMechanisms of mitophagyNat Rev Mol Cell Biol20111291421179058
- MaherJJLeonPRyanJCBeyond insulin resistance: Innate immunity in nonalcoholic steatohepatitisHepatology20084867067818666225
- SalminenAKaarnirantaKKauppinenAInflammaging: disturbed interplay between autophagy and inflammasomesAging (Albany NY)2012416617522411934
- JiaLViannaCRFukudaMHepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistanceNat Commun20145387824815961
- XuYJagannathCLiuXDSharafkhanehAKolodziejskaKEEissaNTToll-like receptor 4 is a sensor for autophagy associated with innate immunityImmunity20072713514417658277
- SaitohTFujitaNJangMHLoss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta productionNature200845626426818849965
- MeiSNiHMManleySDifferential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytesJ Pharmacol Exp Ther201133948749821856859
- KomiyaKUchidaTUenoTFree fatty acids stimulate autophagy in pancreatic beta-cells via JNK pathwayBiochem Biophys Res Commun201040156156720888798
- NatarajanSKInghamSAMohrAMSaturated free fatty acids induce cholangiocyte lipoapoptosisHepatology20146061942195624753158
- NikoletopoulouVMarkakiMPalikarasKTavernarakisNCrosstalk between apoptosis, necrosis and autophagyBiochim Biophys Acta201318333448345923770045
- GermainMNguyenAPLe GrandJNMCL-1 is a stress sensor that regulates autophagy in a developmentally regulated mannerEmbo J20113039540721139567
- PattingreSTassaAQuXBcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagyCell200512292793916179260
- ChangNCNguyenMGermainMShoreGCAntagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1Embo J20102960661820010695
- ObersteinAJeffreyPDShiYCrystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only proteinJ Biol Chem2007282131231313217337444
- FengWHuangSWuHZhangMMolecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1J Mol Biol200737222323517659302
- ZalckvarEBerissiHMizrachyLDAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagyEmbo Rep20091028529219180116
- KangRLiveseyKMZehHJLozeMTTangDHMGB1: a novel Beclin 1-binding protein active in autophagyAutophagy201061209121120935509
- ShenMLuJDaiWEthyl pyruvate ameliorates hepatic ischemia-reperfusion injury by inhibiting intrinsic pathway of apoptosis and autophagyMediators Inflamm20132013461536
- MorenoMChavesJFSancho-BruPGhrelin attenuates hepato-cellular injury and liver fibrogenesis in rodents and influences fibrosis progression in humansHepatology20105197498520077562
- FriedmanSLMechanisms of hepatic fibrogenesisGastroenterology20081341655166918471545
- Hernandez-GeaVGhiassi-NejadZRozenfeldRAutophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissuesGastroenterology201214293894622240484
- Hernandez-GeaVFriedmanSLAutophagy fuels tissue fibrogenesisAutophagy2102884985022617442
- ThoenLFGuimaraesELGrunsvenLAAutophagy: a new player in hepatic stellate cell activationAutophagy2102812612822082960
- LiuMHeYZhangJEffect of autophagy inhibitor 3-methyladenine on proliferation and activation of hepatic stellate cellsXi Bao Yu Fen Zi Mian Yi Xue Za Zhi210329809812 Chinese23948405
- ZhuJWuJFrizellERapamycin inhibits hepatic stellate cell proliferation in vitro and limits fibrogenesis in an in vivo model of liver fibrosisGastroenterology19991171198120410535884
- HidvegiTEwingMHalePAn autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosisScience201032922923220522742
- KimSINaHJDingYWangZLeeSJChoiMEAutophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-beta1J Biol Chem2012287116771168822351764
- WhiteEDiPaolaRSThe double-edged sword of autophagy modulation in cancerClin Cancer Res2009155308531619706824
- LiangXHJacksonSSeamanMInduction of autophagy and inhibition of tumorigenesis by beclin 1Nature199940267267610604474
- QuXYuJBhagatGPromotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy geneJ Clin Invest20031121809182014638851
- DegenhardtKMathewRBeaudoinBAutophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesisCancer Cell200610516416843265
- BhutiaSKMukhopadhyaySSinhaNAutophagy: cancer’s friend or foe?Adv Cancer Res2013118619523768510
- YangSWangXContinoGPancreatic cancers require autophagy for tumor growthGenes De201125717729
- GuoJYXiaBWhiteEAutophagy-mediated tumor promotionCell20131551216121924315093
- LiJYangBZhouQAutophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transitionCarcinogenesis2013341343135123430956
- YangJDSeolSYLeemSHGenes associated with recurrence of hepatocellular carcinoma: integrated analysis by gene expression and methylation profilingJ Korean Med Sci2011261428143822065898
- LavallardVJGualPAutophagy and non-alcoholic fatty liver diseaseBiomed Res Int20142014120179
- BarattaJLNgoALopezBKasabwallaNLongmuirKJRobertsonRTCellular organization of normal mouse liver: a histological, quantitative immunocytochemical, and fine structural analysisHistochem Cell Biol200913171372619255771