
Abstract
Zika virus (ZIKV) is an arbovirus that is transmitted by Aedes mosquitos. Its prototype was isolated in 1947 from serum of a sentinel Rhesus monkey (Macaca mulatta) in the Zika forest of Uganda. As a member of the genus Flavivirus, family Flaviviridae, ZIKV is enveloped and icosahedral and possesses a single-stranded, positive-sense RNA genome of approximately 10.7 kb. Epidemiologically, infection by ZIKV has become a global health concern in recent years because of the occurrence of epidemics, its speed of dissemination, routes of transmission, and the sequelae it can cause especially in newborns. At the neural level, there are still many gaps in our understanding of the mechanisms that induce ZIKV infection-associated microcephaly. However, some studies already demonstrated that underlying cell death is determinant to induce the congenital malformation. In this report, we reviewed the various mechanisms of cell injury involved in the immunopathogenesis of ZIKV infection and discussed its relationship with the death of neuronal and glial cells development and microcephaly.
Perspectives
Zika virus (ZIKV) is a member of the family Flaviviridae and the genus Flavivirus; it was originally isolated in 1947 from a febrile sentinel rhesus monkey in the Zika forest (Uganda).Citation1 In 2015, the World Health Organization (WHO) declared the Zika epidemic a public health emergency in Brazil owing to the outbreak and exponential growth of reported cases of microcephaly and other congenital defects.Citation1,Citation2
ZIKV exhibits extreme versatility and modulates mechanisms of immune evasion; recent experimental advances have shown that ZIKV infects different cell types in several organs and tissues, confirming that transmission occurs not only by bites of infected mosquitos (Aedes), but also by sexual and vertical routes).Citation3 In this context, it is believed that receptors belonging to the TIM and TAM families, particularly AXL, facilitate ZIKV entry in cells.Citation4
Considering the concept of immune privilege and the high selectivity of the blood-placental, blood-testis, and blood–brain barrier to control protein and immune cell entry in the organs, the immune responses mediated by dendritic cells, M1/M2 macrophages, endothelial cells, and CD4 and CD8 lymphocytes reflect the complexity of the host defense system and its organization to combat infectious agents.Citation5–Citation7
Recent experimental studies in vitro and in vivo have shown that several mechanisms of cell injury are involved in the immunopathogenesis of ZIKV infection. These studies suggest that there is a link between microcephaly and cell death resulting from the neuroinflammatory response, whereby apoptosis, necrosis, necroptosis, pyroptosis, paraptosis, and autophagy increase the death of neuronal and glial cells and, consequently, result in brain atrophy and ZIKV-induced microcephaly.
Apoptosis, Necrosis, And Necroptosis
The development of microcephaly presents a great challenge owing to the lack of a comprehensive understanding of the evolutionary mechanisms and dynamics of the process. Accordingly, further studies are needed to clarify these issues and to determine the mechanisms underlying cell injury. A loss of tissue homeostasis and thereby a cell functional imbalance can result in the induction of necrosis or apoptosis pathways.Citation8,Citation9 Both necrosis and apoptosis are observed in cases of ZIKV-induced microcephaly.Citation8,Citation9
More recent investigations showed the involvement of components of the necroptotic cell death pathway to limit neuronal Zika virus infection.Citation10,Citation11 In cortical neurons, the activation of Z-DNA-binding protein 1 (ZBP1) and RIPK1/RIPK3 can trigger the formation of intracellular cascade which modulate the expression of immunoresponsive gene 1 (IRG1), as well as itaconate metabolite. In this process, itaconate inhibits the formation of succinate dehydrogenase (SDH) and consequently suppresses the replication of viral genomes.Citation11
With respect to apoptosis, extrinsic and intrinsic pathways act jointly to enhance cell damage. In the extrinsic pathway, cytokines, growth factors, and death ligands induce the activation of death receptors responsible for initiate the intrinsic cascade. Activation of the intrinsic cascade results in mitochondrial damage that alters the electron transportation chain, provokes the formation of pores in the membrane where cytochrome C is released to bind with Apaf1, and forms the apoptosome complex responsible for activating a series of caspases that direct the cell to apoptosis.Citation12,Citation13 This process involves the caspases 6, 7, 8, 9, and, in particular, caspase 3, which mediate programmed cell death induced by ZIKV.Citation12,Citation14,Citation15 Classic apoptosis leads to a process of natural cell death in which the host defense system induces various mechanisms to maintain homeostasis, without causing major damage; in microcephaly cases, apoptosis induces the uncontrolled death of neurons and glia cells at several stages.Citation7,Citation12,Citation15 This has direct relationship with cellular cycle as evidenced in study cell death showing that in the ventricular zone, progression and mitotic abnormalities of progenitor neural cells in the brain are associated with the intense production of caspase 3, 7, 8, and 9.Citation15
Pyroptosis
Dynamic, interacting processes are linked to the immune response, and recent study of cell death mechanisms has shown that endoplasmic reticulum stress directly contributes to the activation of signaling proteins that modulate excitotoxicity, increase intracellular calcium production, and enhance cell damage.Citation16 In the CNS, an intense neuroinflammatory response and loss of mitochondrial homeostasis results in the development of a new cell death mechanism that is extremely harmful to neurons and glial cells, i.e., pyroptosis in microcephaly cases ZIKV-induced.Citation17–Citation20 Interestingly, pyroptosis differs from apoptosis and necrosis since it is dependent on the activation of the inflammasome, a multiprotein complex characterized by the activation of NLRP3 and AIM2, receptors that recognize pathogen-associated molecular patterns (PAMPs) of the ZIKV and induce the release of caspase 1 to activate pyroptosis and trigger the maturation of IL-1β, IL-18, and IL-33.Citation17–Citation20 Studies of these cytokines are necessary to understand the neuroinflammatory mechanisms involved in the development of microcephaly; IL-1β, IL-18, and IL-33 belong to the IL-1 family and modulate the activities of the M1 (IL-1β and IL-18) and M2 (IL-33) macrophages/microglia as well as the Th1 (IL-1β and IL-18) and Th2 (IL-33) lymphocytes.Citation7,Citation20 Recently in experimental in vitro was showed that ZIKV enhance proinflammatory response to promote NLRP3 inflammasome activation by stabilizing caspase‐1 to suppress cGAS‐mediated type I IFN signaling. In this context, NS1 block the proteasomal degradation of the caspase 1 recruiting USP8 to cleave K11‐linked poly‐ubiquitin chains from caspase‐1 at Lys134. The implications of this process enhance inflammasome activation and inhibit type I IFN signaling.Citation21
Paraptosis
As an immune escape mechanism and adaptation, ZIKV uses the cellular machinery to transform the cell into a setting for viral replication, inducing massive cytoplasmic vacuolization in human epithelial cells, primary human fibroblasts, and human astrocytes. This process is associated with a new form of cell death that does not result in apoptotic corpuscles, known as paraptosis.Citation22 The mechanisms underlying paraptosis in cells of the CNS involve PIK3, AKt, PERK, IRE-1, and ATF-6, as well as vesicle transport orchestrated by the endoplasmic reticulum and Golgi apparatus.Citation16,Citation22 Interesting that ZIKV‐induced vacuoles are dependent on viral translocation into ER through Sec61 and on PI3K/Akt signaling. Therefore, the paraptosis makes the cell a dangerous ZIKV reservoir via the massive cytoplasmic vacuolization, functional alterations of the endoplasmic reticulum, delayed phagolysosome maturation, and the excessive production of ROS; accordingly, it can contribute to cell destruction and ZIKV dissemination.
Autophagy
In addition to paraptosis, another cell death pathway implicated in the pathogenesis of ZIKV-infection is autophagy.Citation23 Autophagy regulation in the brain is associated with ZIKV tropism by several CNS cells. In vitro the infection of human fetal neural stem cells (fNSCs) with ZIKV leads to impaired neurosphere formation and elevated autophagy. Infection with ZIKV strains MR766 and IbH30656 efficiently induced LC3-I to LC3-II conversion and LC3 puncta formation of fNSCs in the presence or absence of lysosome inhibitor bafilomycin A1. Interesting that in this process two ZIKV proteins (NS4A and NS4B) inhibit AKt and consequently the PIK3 cascade and mTOR production causing the deregulation of neurogenesis aberrant activation of autophagy.Citation23 Curiously this both proteins produced by dengue virus do not have the same effect on neurogenesis. Therefore, the identification of this mechanism suggests a strong actuation of NS4A and NS4B to modulate the immune evasion of ZIKV in the viral pathogenesis.
Conclusion And Future Direction
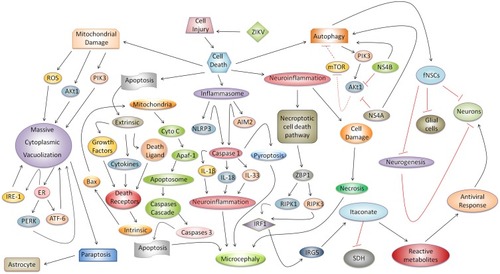
Many questions about microcephaly induced by ZIKV remain. However, investigations of several mechanisms of cell injury involved in the immunopathogenesis of ZIKV infection suggest that there is a link between microcephaly and cell death in which the neuroinflammatory response in situ can induce apoptosis, necrosis, and pyroptosis and consequently result in brain atrophy. In vitro triggering of the necroptosis, paraptosis, and autophagy enhance death of neuronal and glial cells. Therefore, identification of these mechanisms enables the discovery of several immune-targets for anti-ZIKV therapeutic intervention. However, we emphasize that studies in vivo need to be performed to confirm this hypothesis. In summary, we propose an integrated model that synthesizes the relationships among these underlying mechanisms ().
Figure 1 Possible integrated mechanism of cell injury and its relationship with microcephaly ZIKV-induced and cell death. Cell injury is determined by a complex physiopathological response, in which apoptosis, necrosis, necroptotic cell death pathway, pyroptosis, paraptosis, and autophagy can directly impact on the development of microcephaly during ZIKV congenital infection or in the death of glial cells or neurons. Classic apoptosis, associated with the progression of extrinsic and intrinsic pathways, results in the activation of caspase 3, which is responsible for programmed cell death in cells of the neural parenchyma and thereby aggravates microcephaly. The cell stress and neuroinflammatory responses promote activation of the necroptotic cell death pathway which triggers an antiviral response in neurons regulated by actuation of ZBP1, RIPK1/RIPK3, IRF1, IRG5, and itaconate. In the neural parenchyma, with a loss of tissue architecture, intense cellular damage aggravated by neuroinflammation, and extravasation of the enzymatic content, can result in the destruction of neurons and glial cells by necrosis induced by ZIKV in microcephaly cases. Another type of cell death associated with microcephaly, which is dependent on inflammasome activation, is pyroptosis. In the inflammasome, receptors (e.g., NLRP3 and AIM2) recognize ZIKV PAMPs and release caspase 1, which, once activated, promotes IL-1β, IL-18, and IL-33 maturation; these cytokines enhance the neuroinflammatory response and promote the development of microcephaly. In addition, caspase 1 provokes pyroptosis and cell death in the neural parenchyma during ZIKV-induced microcephaly. Regarding mitochondrial damage, a new type of cell death known as paraptosis causes massive cytoplasmic vacuolization in neuronal and glial cells mainly astrocytes. This relationship intensifies the response to PIK3, IRE1, PERK, and ATF-6, endoplasmic reticulum stress, and ROS. Finally, during autophagy, two ZIKV proteins (NS4A and NS4B) modulate the PIK3/AKt1 pathway, inhibiting neurogenesis and AKt1 and mTOR responses, increasing autophagic activity, and resulting in deleterious effects in vitro in neurosphere leading human fetal neural stem cells (fNSCs) to cell death.
Author contributions
All authors contributed towards data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Funding
This work was supported by the Ministry of Science, Technology and Innovation/National Council for Scientific and Technological Development CNPQ/Brazil (grant numbers: 303999/2016-0, 439971/2016-0, and 440405/2016-5), and CAPES (Zika Fast-track).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Screaton G, Mongkolsapaya J. Evolution of neurovirulent Zika virus. Science. 2017;358:863–864. doi:10.1126/science.aaq129729146794
- Possas C, Brasil P, Marzochi MC, et al. Zika puzzle in Brazil: peculiar conditions of viral introduction and dissemination - a review. Mem Inst Oswaldo Cruz. 2017;112:319–327. doi:10.1590/0074-0276016051028443985
- Miner JJ, Diamond MS. Zika virus pathogenesis and tissue tropism. Cell Host Microbe. 2017;21:134–142. doi:10.1016/j.chom.2017.01.00428182948
- Meertens L, Labeau A, Dejarnac O, et al. Axl mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep. 2017;18:324–333. doi:10.1016/j.celrep.2016.12.04528076778
- Quicke KM, Bowen JR, Johnson EL, et al. Zika virus infects human placental macrophages. Cell Host Microbe. 2016;20:83–90. doi:10.1016/j.chom.2016.05.01527247001
- Siemann DN, Strange DP, Maharaj PN, Shi PY, Verma S. Zika virus infects human sertoli cells and modulates the integrity of the in vitro blood-testis barrier model. J Virol. 2017;91:e00623–17. doi:10.1128/JVI.00623-1728878076
- Azevedo RSS, de Sousa JR, Araujo MTF, et al. In situ immune response and mechanisms of cell damage in central nervous system of fatal cases microcephaly by Zika virus. Sci Rep. 2018;1:1. doi:10.1038/s41598-017-17765-5
- Solomon IH, Milner DA, Folkerth RD. Neuropathology of Zika virus. J Neuroinfect Dis. 2016;7:220. doi:10.4172/2314-7326.100022027525286
- Chimelli L, Avvad-Portari E. Congenital Zika virus infection: a neuropathological review. Childs Nerv Syst. 2018;34:95–99. doi:10.1007/s00381-017-3651-329167994
- Chaudhary V, Yuen KS, Chan JF, et al. Selective activation of type II interferon signaling by Zika virus NS5 protein. J Virol. 2017;91:e00163–17. doi:10.1128/JVI.00163-1728468880
- Daniels BP, Kofman SB, Smith JR, et al. The nucleotide sensor ZBP1 and kinase RIPK3 induce the enzyme IRG1 to promote an antiviral metabolic state in neurons. Immunity. 2019;50:64–76.e4. doi:10.1016/j.immuni.2018.11.01730635240
- de Sousa JR, Azevedo RSS, Martins Filho AJ, et al. Correlation between apoptosis and in situ immune response in fatal cases of microcephaly caused by Zika virus. Am J Pathol. 2018;9440:30153–30156.
- Ghosh Roy S, Sadigh B, Datan E, Lockshin RA, Zakeri Z. Regulation of cell survival and death during Flavivirus infections. World J Biol Chem. 2014;5:93–105. doi:10.4331/wjbc.v5.i2.9324921001
- Cugola FR, Fernandes IR, Russo FB, et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature. 2016;534:267–271. doi:10.1038/nature1829627279226
- Souza BS, Sampaio GL, Pereira CS, et al. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci Rep. 2016;23:39775. doi:10.1038/srep39775
- Blázquez AB, Escribano-Romero E, Merino-Ramos T, Saiz JC, Martín-Acebes MA. Stress responses in flavivirus-infected cells: activation of unfolded proten response and autophagy. Front Microbiol. 2014;5:266. doi:10.3389/fmicb.2014.0054724917859
- He Z, Chen J, An S, et al. The NLRP3 inflammasome activation mediates Zika virus associated inflammation. J Infect Dis. 2018;217:1942–1951. doi:10.1093/infdis/jiy12929518228
- Wang W, Li G, De Wu LZ, et al. Zika virus infection induces host inflammatory responses by facilitating NLRP3 inflammasome assembly and interleukin-1β secretion. Nat Commun. 2018;9:106. doi:10.1038/s41467-017-02645-329317641
- Hamel R, Dejarnac O, Wichit S, et al. Biology of Zika virus infection in human skin cells. J Virol. 2015;89:8880–8896. doi:10.1128/JVI.00354-1526085147
- de Sousa JR, Azevedo RDSDS, Martins Filho AJ, et al. In situ inflammasome activation results in severe damage to the central nervous system in fatal Zika virus microcephaly cases. Cytokine. 2018;111:255–264. doi:10.1016/j.cyto.2018.08.00830199767
- Zheng Y, Liu Q, Wu Y, et al. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1‐caspase‐1 axis. Embo J. 2018;37:e99347. doi:10.15252/embj.20189934730065070
- Monel B, Compton AA, Bruel T, et al. Zika virus induces massive cytoplasmic vacuolization and paraptosis-like death in infected cells. Embo J. 2017;36:1653–1668. doi:10.15252/embj.20169559728473450
- Liang Q, Luo Z, Zeng J, et al. Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell. 2016;19:663–671. doi:10.1016/j.stem.2016.07.01927524440