Abstract
Over the last ten years, genome sequencing capabilities have expanded exponentially. There have been tremendous advances in sequencing technology, DNA sample preparation, genome assembly, and data analysis. This has led to advances in a number of facets of bacterial genomics, including metagenomics, clinical medicine, bacterial archaeology, and bacterial evolution. This review examines the strengths and weaknesses of techniques in bacterial genome sequencing, upcoming technologies, and assembly techniques, as well as highlighting recent studies that highlight new applications for bacterial genomics.
History of bacterial genome sequencing
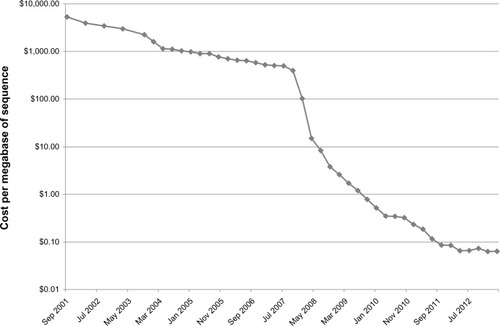
The first sequenced bacterial genome was Haemophilus influenzaeCitation1 in 1995. Since then, the Genomes Online DatabaseCitation2 lists 2,264 finished bacterial genomes and 4,067 permanent draft genomes (genomes that are sequenced but not completely closed). The majority of these have been deposited since 2008, after the commercial introduction of high-throughput sequencing. A number of sequencing techniques have been subsequently introduced making bacterial genome sequencing significantly cheaper and easier. This has decreased the cost per megabase of sequence by five logs (see ), which has allowed for sequencing of large numbers of genomes. These advances have allowed movement from sequencing individual genomes to sequencing multiple strains. However, the general workflow of bacterial sequencing remains generally unchanged – sample preparation, DNA sequencing, sequence assembly, and bioinformatic analysis. This review will examine each of these, as well as examining some of the current applications of these technologies.
Figure 1 Cost per megabase of sequencing, from 2001 to 2012.
Adapted from the NIH NHGRI Genome Sequencing Program website (http://www.genome.gov/sequencingcosts/).
Abbreviations: NIH, National Institutes of Health, NHGRI, National Human Genome Research Institute.
Sample preparation
The major advance in sample preparation is enabling more effective isolation of small amounts of DNA, allowing genome sequencing from limited or degraded initial samples. This includes the development of isothermal amplification for multiple displacement amplification (MDA). This technique uses the phi29 DNA polymerase combined with random hexamers to produce DNA fragments in the multiple-kilobase range.Citation3 This allows genomic-scale sequencing from small starting samples of DNA. Based on studies in Anaplasma, it appears that sequencing after phi29 amplification provides similar genomic coverage and single’nucleotide’polymorphism’(SNP) rates as traditional sample preparation.Citation4 While additional chimeric sequences (a single sequence derived from two separate pieces of DNA) were generated, these did not interfere with genome assembly. MDA has been used to sequence the genome from a single unculturable intracytoplasmic symbiont of Draeculacephala minerva.Citation5 This may provide a significant amount of information on genome sequences from unculturable bacteria, allowing whole genome sequencing rather than the limited information from metagenomic studies.
Sequencing technologies
The biggest revolution in genomics the last several years has been the emergence of new sequencing technologies. These have shifted the bottleneck in genome sequencing from generation of raw sequence to bioinformatic processing of samples. Each sequencing technology has specific strengths and weaknesses, making selection of the appropriate technique important to obtaining the desired experimental results. 1 and give an overview of different sequencing technologies and some relative strengths and weaknesses. Individual techniques are described below. However, these technologies are continuously revised; the mean read length of pyrosequencing, for example, has grown from approximately 150 bpCitation6 to approximately 700 bpCitation7 in the last five years. Consultation with the sequencing center early in the planning stage of an experiment is helpful in obtaining the best results, as they can provide updates on the technologies in use and tailor the sequencing runs to the needs of the experiment.
Table 1 An overview of current sequencing technologies
Table 2 Relative strengths and weaknesses of current sequencing technologies
Current technologies
Pyrosequencing (454)
Pyrosequencing (454) (Roche Inc., Branford, CT, USA) uses a “sequencing by synthesis” approach. Deoxynucleotides are added one at a time and incorporation is detected by converting the amount of phosphorus released in deoxynucleotide incorporation into a light signal that is read by the sequencer. Because of this, it tends to have difficulty with homopolymeric tracts, as the difference in light intensity between progressively longer nucleotide repeats is relatively less. In general, the strengths of pyrosequencing are its relatively long read lengths and rapid turnaround time, which make it especially useful for de novo sequencing projects and organisms with large numbers of repeats or long repetitive regions.
Sequencing by Oligo Ligation Detection
Sequencing by Oligo Ligation Detection (SOLiD) (Life Technologies Corporation, Grand Island, NY, USA) uses a “sequencing by ligation” approach. Numerous degenerate 8-mers are ligated to the single stranded DNA (ssDNA) template, with two nucleotides specific for the strand being sequenced and the remaining six bases degenerate. As the probes are ligated to the template, fluorescent dyes are cleaved off and detected by the sequencer. Every nucleotide participates in two ligation reactions, which allows for error checking of each read. This gives SOLiD an advantage for SNP detection, as it tends to have high reliability in SNP sequencing. However, since each nucleotide sequence is based off of a combination of two reads (termed “colorspace”), rather than a nucleotide sequence with a quality score, fully utilizing these data require tools designed for SOLiD sequences.
MySeq and HiSeq
MySeq and HiSeq (Illumina Inc., San Diego, CA, USA), machines use a “sequencing by synthesis” technique, where individual DNA molecules are attached to the surface of flow cells and isothermal ‘bridging’ amplification is used to amplify signals. These are then sequenced using reversible fluorophore-labeled nucleotides, which are optically read from each flow cell. While these have high accuracies and produce large amounts of raw data, the individual read lengths tend to be shorter, which can be problematic for genomes with large repeats. Illumina’s Nextera sample preparation kit can allow for template amounts as low as 50 ng, which can be useful for organisms that are difficult to culture.
The Ion Personal Genome Machine® (PGM™)
Ion Torrent Personal Genome Machine® (PGM™) (Life Technologies Corporation, Grand Island, NY, USA) uses a “sequencing by synthesis” approach, measuring the hydrogen ions released during deoxynucleotide incorporation. This is measured by semiconductors in the disposable chips used by the machine for sequencing. As there is no optical component of the sequencer, machine throughput can be increased by modifications to the chips used without additional sequenced modification, which has led to a tremendous increase in the throughput since the initial release. This also allows selection of chips giving the appropriate sequencing coverage for the desired application, which can make sequencing more cost-effective. While there is generally high accuracy, there are difficulties with high adenine-thymine (AT)-rich sequences, which can lead to gaps in coverage.Citation8 In addition to the PGM, Life Technologies has also released the Proton system, which allows for larger chips with more sequence per run.
PacBio RS II Single Molecule Real-time Sequencing
PacBio RS II Single Molecule Real-time Sequencing (SMRT) (Pacific Biosciences Inc., Menlo Park, CA 94025) uses a variation of “sequencing by synthesis”, using fluorescent-labeled deoxynucleotides added to a zero-mode waveguide (ZMW) with a DNA polymerase embedded in the bottom. As deoxynucleotides are added to the template, the fluorescent signals are read in real-time by the sequencer. While the accuracy of individual reads tends to be low (∼85% or so), errors tend to be random, rather than due to specific DNA features, so increased coverage allows for high cumulative accuracy rates.Citation9 The main advantage of PacBio is its long read length; while mean read lengths tend to be approximately 5 kb, reads of more than 10 kb are not uncommon. Also, as the machine observes the reaction in real time, it can detect some base modifications, such as methylationCitation10, without additional reagents due to alterations in the deoxynucleotide incorporation time. In addition, experiments have been made to sequence DNA without the initial amplification step in library preparation.Citation11
Future technologies
GnuBio
GnuBio (GnuBio Inc., Cambridge, MA USA) sequencer uses a sequencing by amplification approach, using microfluidics to combine target selection, DNA amplification, library preparation, and sequencing into one instrument.Citation12 While this is targeted at clinical applications, this has a number of applications for microbial sequencing. Beta testing began in April of 2013.
GridION/MiniION
GridION/MiniION (Oxford Nanopore Technologies Ltd, Oxford, UK) systems use nanopore technology and disposable cartridges to perform a number of possible experiments, including DNA sequencing.Citation13 The nanopores use voltage variation produced when ssDNA is fed through the nanopore via an enzyme. No amplification step is necessary, allowing examination of DNA sequence modifications (such as methylation) directly. The GridION is meant to be an expandable, reusable system for core laboratories, while the MiniION is a single-use device for individual laboratories.
Genome assemblers
While raw sequence data is useful, it is significantly more valuable after assembly into contiguous DNA sequences (contigs). There are a number of strategies for assembly, and sequences can be assembled either de novo or assembled against a reference sequence. A number of assemblers have been used on bacterial genome sequences; some of the more common options are discussed below in alphabetical order.
ABySS
ABySS (Assembly By Short Sequences) is a de novo parallel paired-end assembler that works with Illumina, SOLiD, pyrosequencing, and Sanger reads.Citation14 It also works with combinations of technologies by calculating the distribution of read sizes for each, so an accurate empirical distribution can be obtained. In addition, it has been adapted for transcriptome assembly with RNA seq data.
Celera Assembler
CABOG (Celera Assembler with the Best Overlap Graph)Citation15 is a de novo assembler that was first developed for the original human genome project. It has subsequently been modified to assemble pyrosequencingCitation16, Illumina, and PacBio reads. While it is primarily geared toward mammalian sequences, it can also be utilized for microbial sequences.Citation17
Edena
Edena (Exact DE Novo Assembler) is a de novo overlaps graph-based short reads assembler.Citation18 It requires reads to be a similar length, as it is designed for Illumina-based sequences; therefore, pyrosequencing and Sanger-based reads would need to be trimmed to a similar length to be processed. This program is specifically designed for bacterial genome assemblies.
EULER-SR
EULER-SR is a de novo assembler that uses an A-Bruijn graph technique to assemble Sanger, pyrosequencing, and Illumina reads.Citation19 This is geared toward assembly of DNA sequences from individual organisms, as well as clustering of sequences from metagenomic analyses.
MaSuRCA
MaSuRCA (Maryland Super Read Cabog Assembler) is a new de novo genome assembler that combines de Bruijn graphs and Overlap-layout-consensus approaches to increase efficiency.Citation20 It can use a combination of short (Illumina and SOLiD) and longer (pyrosequencing) reads. This assembler performed best on a recent comparison of several modern assemblers with a number of bacterial genomic data sets.Citation21
MIRA
MIRA (Mimicking Intelligent Read Assembly) is a whole genome shotgun and expressed sequence tag (EST) assemblerCitation22 for Sanger and pyrosequencing reads, as well as Illumina, Life Technologies, and PacBio reads with the development version.Citation23 It can perform both de novo and reference-based assemblies. It features sequence editors, allowing repair of sequencing errors and use of quality data in generating assemblies. It also will assemble to a reference sequence and call SNPs and other mutations.
SOAP suite
SOAPdenovo2 (Short Oligonucleotide Analysis Package) is made up of multiple modules that perform error correction, assembly, paired end mapping, and scaffold constructionCitation24, and is specifically designed for de novo assembly of Illumina reads. While this was designed for large genomes, it has been tested and works well on microbial genomes as well. There is a separate program, SOAP2 and SOAP3, that align reads to reference genomes. In addition to the assembler, there are additional tools for SNP and indel detection.
SOPRA
SOPRA (Statistical Optimization of Paired Read Assembly) is a de novo assembler that attempts to compensate for inaccuracies in the high throughput reads.Citation25 It accepts pyrosequencing, Illumina, and SOLiD reads, and can use data on mate pair distances to create scaffolds. It can convert SOLiD colorspace to base-space, and use that for quality checking. However, SOPRA requires contigs as input; the developers recommend Velvet as a contig assembler, but the program can use FASTA contigs generated by any program.
Velvet
Velvet is a De Bruijn graph-based de novo assemblerCitation26 that can assemble Illumina, SOLiD, pyrosequencing, and Sanger reads.Citation27 In addition, if compiled to support colorspace, it can use colorspace assembly as well as base-space assembly. Velvet is one of the first De Bruijn graph assemblers, and has continued to be updated, including updates to allow for mixed-length assembly and paired-end assembly.Citation28
Optical mapping
In addition to traditional assembly of sequence reads into contigs, high-resolution optical mapping has been combined with contig assembly to allow more rapid assembly of contigs and determination of gap locations.Citation29 Software is able to take the optical map and arrange contigs, either with the assistance of a reference sequence or de novo. Whole genome mapping has been used as a scaffold to perform the initial assembly of pyrosequencing reads to better identify gaps in sequence coverage, allowing complete genome assembly without paired-end sequencing.Citation30
Accessory programs
In addition to sequence assembly, there are a number of other computer programs that can be helpful in further processing sequence data.
Trimmomatic
Trimmomatic is useful for processing Illumina data, screening libraries for a number of quality parameters, including adapter trimming, cropping, trimming based on a minimum length, and converting quality scores.Citation31 Sequences that do not meet quality guidelines are automatically trimmed out. However, unlike a number of other methods for sequence trimming, Trimmomatic is aware of paired end data, and maintains the paired end links.
CGView Comparison Tool
CGView Comparison Tool (CCT) is a program to visually compare multiple circular genomesCitation32 that takes sequence alignment output and uses it to visualize the results against a reference genome. One strength over many other tools is the ability to compare thousands of genomes in the same map.
Artemis Comparison Tool
The Artemis Comparison Tool (ACT) is another tool to visualize multiple genome comparisons.Citation33 For people who use Artemis for genome annotation, the user interface for ACT is almost identical, making it easy to use. The interactive user interface makes it useful for examination of genomes and SNP detection. In addition to the stand-alone program, a web tool (WebACT) has also been developed for online work.Citation34
Galaxy
Galaxy is a web application that can use a variety of bioinformatics tools.Citation35 It is also extensible, so programmers can add support for nearly any desired bioinformatics tool. While it started as primarily a method for working with text-based data, such as DNA sequences, recent developments have added data visualization tools as well.Citation36 The main strength of Galaxy is the ability for multiple researchers to work on data sets together via web browsers. In addition to sharing datasets, researchers can also share workflows, allowing others to replicate their results and allowing editing and saving of workflows for future use. There are public servers for Galaxy, but it can also be downloaded and run locally or in the cloud to use additional storage and computing resources.
Applications of genome sequencing
Clinical medicine
One recent development has been the application of high-throughput DNA sequencing to clinical applications. First, as requirements for DNA template concentration and purity for genome sequencing decrease, clinical samples can be directly sequenced, allowing for organism identification and possible identification of traits such as antibiotic resistance.Citation37 While complete genome assembly is still time consuming, high-throughput sequencing and assembly can reveal a tremendous amount of information about target organisms without obtaining the complete genome sequence.Citation38 This may also make large numbers of clinical samples available for use in research studies, as complete genome sequences of Chlamydia trachomatis were isolated from discarded swabs after testing.Citation39
In addition to rapidly determining genotype/phenotype association, whole genome sequencing (WGS) techniques have been used in several public health surveys, analyzing nosocomial infections in hospitals and differentiating them from non-outbreak isolatesCitation40 and in retrospective analysis to track the spread of infections through hospitals.Citation41 Whole genome sequencing gives the ability to determine where an infection was acquired from, and has, in some cases, revealed previously unknown bacterial reservoirs.Citation42 This may be aided by the continued sequencing of multiple bacterial strains, as evidenced by the determination of a minimum core genome for Streptococcus suis, which allowed determination of genes unique to animal versus human strains. Other genomic analyses have detected infection with multiple strains of the same organism, revealing previously unknown transmission events.Citation43
Another related activity is using microbial genomics and metagenomics for forensics. This has been done for cases of bioterrorism, such as the anthrax letter attack investigation, where the isolates from the letters were linked and were different from those previously suspected in the investigation.Citation44 Microbial sequencing may also be used in the future for criminal investigation, as skin microbial populations are relatively unique and can be used to identify items handled by people up to two weeks previously.Citation45
However, the abundance of sequence information makes bioinformatics the bottleneck in utilization of sequences in clinical samples. Future developments may help automate sequence assembly and annotationCitation46, as well as automating bacterial typing from whole genome sequences,Citation47 speeding analysis.
Genomic archaeology
In addition to clinical medicine, the reduction in DNA template requirements for sequencing have produced profound developments in genomic archaeology. Medieval isolates of Yersinia pestis from victims of the black death were sequenced using the Illumina platform, yielding 93% genome coverage.Citation48 This has revealed that current isolates of Y. pestis appear to be descended from the medieval strain, and that the virulence of the Black Death organism does not appear to be due to bacterial genotype. In another study, multiple ancient isolates of Mycobacterium leprae were sequenced from bone lesions and compared to modern isolates.Citation49 This is the first study to assemble a complete genome de novo from ancient sequences, rather than use a modern reference sequence for scaffolding. This has allowed tracking of the spread of leprosy from ancient times to modern day, as well as drawing conclusions about why leprosy disappeared from Europe but persists in many developing countries today. Other studies have examined the bacterial composition of ancient dental calculus,Citation50 allowing for comparisons of historical bacterial populations compared with modern day oral flora, and using that to examine environmental factors associated with dental disease. Finally, another study examined the bacterial populations in waterlogged, preserved wood,Citation51 which will aid in preserving historic wrecks and establishing underwater archaeological parks.
Metagenomics studies
While sequencing advances have caused a huge growth in the field of metagenomics, metagenomic studies have started to be exploited as sources of raw sequence for genome projects. The increases in DNA sequencing throughput have allowed shifting metagenomics studies from amplification of 16S to shotgun sequencing of the entire sample DNA population.Citation52 Generally, this depends on having a predominance of small numbers of microbial genomes in the population, allowing for assembly into complete or near-complete genomes.Citation53,Citation54 However, one study combined multiple metagenomics studies from a population to assemble twelve near-complete or complete genome sequencesCitation55 from low-prevalence populations.
Bacterial evolution
With the large numbers of sequenced genomes, a variety of techniques from other organisms have subsequently been applied to bacteria. Genome-wide association studies have begun to be applied to bacteria. In one study, Campylobacter strains from a variety of hosts were examined to determine factors involved in host specificity.Citation56 While most lineages were able to switch hosts, some lineages were associated with specific hosts. These were linked with vitamin B5 biosynthesis genes, and cattle isolates were able to grow better in vitamin B5-depleted media. Another study examined the microbiota in patients with and without type 2 diabetes, finding a significant decrease in butyrate-producing bacteria and an increase in opportunistic pathogens.Citation57
Other studies have examined a number of bacteria to determine changes associated with the development of pathogenicity. One study found that pathogenic bacteria have smaller genomes, with less ribosomal RNA, less transcriptional regulators, and more genes for toxins and DNA replication.Citation58 Similar reductions are detected in experimental populations with multiple generations.Citation59 Other studies have examined multiple strains to correlate phenotypic differences with polymorphisms and transcriptional differences in bacteria that are unable to be cultured.Citation60 Other studies have examined the rate of polymorphism formation in multiple species, finding that SNPs can occur in non-random locations depending on the nature of the mutation.Citation61
Future work will likely involve correlating genomic data with transcriptional regulatory data, metabolic pathway reconstruction, and proteomics data.Citation62 While the ultimate goal would be to establish whole-cell models of bacterial systems, the raw data to drive these models will still be complete, edited bacterial genomes.
Conclusion
Advances in sample preparation, DNA sequencing, and assembly technology have caused an explosion in the number of sequenced bacterial genomes, and are enabling new uses for bacterial genome sequencing. As technology improves, the number of applications will only increase, making understanding the spectrum of technology more important. Further, collaboration will be more important, making web tools for manipulation of genomic data more useful.
Disclosure
The author has no conflicts of interest to report.
References
- FleischmannRDAdamsMDWhiteOWhole-genome random sequencing and assembly of Haemophilus influenzae RdScience199526952234965127542800
- PaganiILioliosKJanssonJThe Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadataNucleic Acids Res201240Database issueD571D57922135293
- AlsmadiOAlkayalFMoniesDMeyerBFSpecific and complete human genome amplification with improved yield achieved by phi29 DNA polymerase and a novel primer at elevated temperatureBMC Res Notes200924819309528
- DarkMJLundgrenAMBarbetAFDetermining the repertoire of immunodominant proteins via whole-genome amplification of intracellular pathogensPLoS One201274e3645622558468
- WoykeTTigheDMavromatisKOne bacterial cell, one complete genomePLoS One201054e1031420428247
- LiuLLiYLiSComparison of next-generation sequencing systemsJ Biomed Biotechnol2012201225136422829749
- GS FLX+ SystemRoche Corporation 454 Sequencing home page2013Accessed July 26, 2013
- QuailMASmithMCouplandPA tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencersBMC Genomics20121334122827831
- KorenSSchatzMCWalenzBPHybrid error correction and de novo assembly of single-molecule sequencing readsNat Biotechnol201230769370022750884
- MurrayIAClarkTAMorganRDThe methylomes of six bacteriaNucleic Acids Res20124022114501146223034806
- CouplandPChandraTQuailMReikWSwerdlowHDirect sequencing of small genomes on the Pacific Biosciences RS without library preparationBiotechniques201253636537223227987
- GnuBIOGnuBIO home page2013http://gnubio.com/Accessed July 29, 2013
- Oxford Nanopore TechnologiesOxford Nanopore Technologies home page2013https://www.nanoporetech.com/Accessed July 29, 2013
- SimpsonJTWongKJackmanSDScheinJEJonesSJMBirolƒnABySS: A parallel assembler for short read sequence dataGenome Research20091961117112319251739
- Celera AssemblerSourceforge: Celera Assembler home page2013http://sourceforge.net/apps/mediawiki/wgs-assembler/index.php?title=Main_PageAccessed July 29, 2013
- MillerJRDelcherALKorenSAggressive assembly of pyrosequencing reads with matesBioinformatics200824242818282418952627
- GillespieJJJoardarVWilliamsKPA Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyleJ Bacteriol2012194237639422056929
- HernandezDFrancoisPFarinelliLOsterasMSchrenzelJDe novo bacterial genome sequencing: millions of very short reads assembled on a desktop computerGenome Res200818580280918332092
- ChaissonMJPevznerPAShort read fragment assembly of bacterial genomesGenome Res200818232433018083777
- Developing Methods for Improving Genome AssemblyThe University of Maryland Genome Assembly Group home page2013http://www.genome.umd.edu/masurca.htmlAccessed August 2, 2013
- MagocTPabingerSCanzarSGAGE-B: an evaluation of genome assemblers for bacterial organismsBioinformatics201329141718172523665771
- ChevreuxBPfistererTDrescherBUsing the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTsGenome Res20041461147115915140833
- MIRASourceforge MIRA home page2013http://sourceforge.net/apps/mediawiki/mira-assembler/index.php?title=Main_PageAccessed July 30, 2013
- LuoRLiuBXieYSOAPdenovo2: an empirically improved memory-efficient short-read de novo assemblerGigascience2012111823587118
- DayarianAMichaelTPSenguptaAMSOPRA: Scaffolding algorithm for paired reads via statistical optimizationBMC Bioinformatics20101134520576136
- ZerbinoDRBirneyEVelvet: algorithms for de novo short read assembly using de Bruijn graphsGenome Res200818582182918349386
- NagarajanNPopMSequence assembly demystifiedNat Rev Genet201314315716723358380
- ZerbinoDRMcEwenGKMarguliesEHBirneyEPebble and rock band: heuristic resolution of repeats and scaffolding in the velvet short-read de novo assemblerPLoS One2009412e840720027311
- MillerJMWhole-genome mapping: a new paradigm in strain-typing technologyJ Clin Microbiol20135141066107023363821
- Onmus-LeoneFHangJCliffordRJEnhanced de novo assembly of high throughput pyrosequencing data using whole genome mappingPLoS One201384e6176223613926
- LohseMBolgerAMNagelARobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomicsNucleic Acids Res201240Web Server issueW622W62722684630
- GrantJRArantesASStothardPComparing thousands of circular genomes using the CGView Comparison ToolBMC Genomics20121320222621371
- CarverTJRutherfordKMBerrimanMRajandreamMABarrellBGParkhillJACT: the Artemis Comparison ToolBioinformatics200521163422342315976072
- AbbottJCAanensenDMBentleySDWebACT: an online genome comparison suiteMethods Mol Biol2007395577417993667
- GoecksJNekrutenkoATaylorJGalaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciencesGenome Biol2010118R8620738864
- GoecksJEberhardCTooTNekrutenkoATaylorJWeb-based visual analysis for high-throughput genomicsBMC Genomics20131439723758618
- TorokMEPeacockSJRapid whole-genome sequencing of bacterial pathogens in the clinical microbiology laboratory – pipe dream or reality?J Antimicrob Chemother201267102307230822729921
- BertelliCGreubGRapid bacterial genome sequencing: methods and applications in clinical microbiologyClin Microbiol Infect20131998031323601179
- Seth-SmithHMHarrisSRSkiltonRJWhole-genome sequences of Chlamydia trachomatis directly from clinical samples without cultureGenome Res201323585586623525359
- ReuterSEllingtonMJCartwrightEJRapid Bacterial Whole-Genome Sequencing to Enhance Diagnostic and Public Health MicrobiologyJAMA Intern Med2013 (in print)
- SnitkinESZelaznyAMThomasPJTracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencingSci Transl Med20124148148ra116
- NubelUNachtnebelMFalkenhorstGMRSA transmission on a neonatal intensive care unit: epidemiological and genome-based phylogenetic analysesPLoS One201381e5489823382995
- EyreDWCuleMLGriffithsDDetection of mixed infection from bacterial whole genome sequence data allows assessment of its role in Clostridium difficile transmissionPLoS Comput Biol201395e100305923658511
- RaskoDAWorshamPLAbshireTGBacillus anthracis comparative genome analysis in support of the Amerithrax investigationProc Natl Acad Sci U S A2011108125027503221383169
- FiererNLauberCLZhouNMcDonaldDCostelloEKKnightRForensic identification using skin bacterial communitiesProc Natl Acad Sci U S A2010107146477648120231444
- RichardsonEJWatsonMThe automatic annotation of bacterial genomesBrief Bioinform201314111222408191
- JolleyKAMaidenMCAutomated extraction of typing information for bacterial pathogens from whole genome sequence data: Neisseria meningitidis as an exemplarEuro Surveill20131842037923369391
- BosKISchuenemannVJGoldingGBA draft genome of Yersinia pestis from victims of the Black DeathNature2011478737050651021993626
- SchuenemannVJSinghPMendumTAGenome-wide comparison of medieval and modern Mycobacterium lepraeScience2013341614217918323765279
- De La FuenteCFloresSMoragaMDNA from human ancient bacteria: a novel source of genetic evidence from archaeological dental calculusArchaeometry2013554767778
- PallaFMancusoFPBilleciNMultiple approaches to identify bacteria in archaeological waterlogged woodJournal of Cultural Heritage201314Suppl 3e61e64
- TringeSGvon MeringCKobayashiAComparative metagenomics of microbial communitiesScience2005308572155455715845853
- Garcia MartinHIvanovaNKuninVMetagenomic analysis of two enhanced biological phosphorus removal (EBPR) sludge communitiesNat Biotechnol200624101263126916998472
- TysonGWChapmanJHugenholtzPCommunity structure and metabolism through reconstruction of microbial genomes from the environmentNature20044286978374314961025
- AlbertsenMHugenholtzPSkarshewskiANielsenKLTysonGWNielsenPHGenome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomesNat Biotechnol201331653353823707974
- SheppardSKDidelotXMericGGenome-wide association study identifies vitamin B5 biosynthesis as a host specificity factor in CampylobacterProc Natl Acad Sci U S A201311029119231192723818615
- QinJLiYCaiZA metagenome-wide association study of gut microbiota in type 2 diabetesNature20124907418556023023125
- MerhejVGeorgiadesKRaoultDPostgenomic analysis of bacterial pathogens repertoire reveals genome reduction rather than virulence factorsBrief Funct Genomics201312429130423814139
- LeeMCMarxCJRepeated, selection-driven genome reduction of accessory genes in experimental populationsPLoS Genet201285e100265122589730
- PierleSADarkMJDahmenDPalmerGHBraytonKAComparative genomics and transcriptomics of trait-gene associationBMC Genomics20121366923181781
- BryantJChewapreechaCBentleySDDeveloping insights into the mechanisms of evolution of bacterial pathogens from whole-genome sequencesFuture Microbiol20127111283129623075447
- FariaJPOverbeekRXiaFRochaMRochaIHenryCSGenomescale bacterial transcriptional regulatory networks: reconstruction and integrated analysis with metabolic modelsBrief Bioinform2013