Abstract
Raltegravir, the only integrase (IN) inhibitor approved for use in HIV therapy, has recently been licensed. Raltegravir inhibits HIV-1 replication by blocking the IN strand transfer reaction. More than 30 mutations have been associated with resistance to raltegravir and other IN strand transfer inhibitors (INSTIs). The majority of the mutations are located in the vicinity of the IN active site within the catalytic core domain which is also the binding pocket for INSTIs. High-level resistance to INSTIs primarily involves three independent mutations at residues Q148, N155, and Y143. The mutations significantly affect replication capacity of the virus and are often accompanied by other mutations that either improve replication fitness and/or increase resistance to the inhibitors. The pattern of development of INSTI resistance mutations has been extensively studied in vitro and in vivo. This has been augmented by cell-based phenotypic studies and investigation of the mechanisms of resistance using biochemical assays. The recent elucidation of the structure of the prototype foamy virus IN, which is closely related to HIV-1, in complex with INSTIs has greatly enhanced our understanding of the evolution and mechanisms of IN drug resistance.
The management of HIV-1 infection and drug resistance
Over 20 antiretroviral drugs (ARVs) have been licensed for the treatment of HIV-1 infection in the past 25 years.Citation1 This has significantly improved the prognosis of HIV-infected individuals and reduced the mortality and morbidity rates worldwide.Citation2,Citation3 The initial class of drugs approved for HIV-1 therapy were the nucleoside reverse transcriptase inhibitors (NRTIs) which are nucleoside analogs that inhibit DNA synthesis by acting as chain terminators.Citation4 Subsequently, further classes of ARVs with different inhibitory mechanisms and/or targets were approved for use against HIV-1, these being non-NRTIs and protease inhibitors (PIs).Citation5,Citation6 All three classes target viral enzymes required for HIV-1 viral replication, namely reverse transcriptase and protease. Therefore, it is not surprising that drug-resistant variants eventually emerge against these agents due to HIV’s high mutation rate and lack of a proofreading mechanism. The occurrence of drug resistance was a particular problem in the early years of ARV therapy when the drugs were used separately. This led to the idea of using no less than three different drugs belonging to at least two different classes to increase the genetic barrier to resistance development in what is termed highly active antiretroviral therapy (HAART).
Nonetheless, the emergence of drug-resistant HIV-1 variants in patients undergoing HAART remains a major reason for treatment failure in HIV-1 therapy. Several factors contribute to treatment failure in the era of HAART, and these include poor adherence due to adverse effects associated with the drugs, high pill burden, or irregular supply of the ARVs particularly in resource-poor countries. In addition, the transmission of drug-resistant viral strains can compromise and limit the effectiveness of first-line treatment regimens. The past few years have seen the introduction of three new classes of ARVs and second-generation drugs or variants of the original classes, such as the NRTI-variant tenofovir, which possess different inhibitory mechanisms and/or fewer adverse effects. The three new classes are fusion inhibitors, entry inhibitors, and integrase (IN) inhibitors. This review focuses on the pattern of resistance development to IN inhibitors as currently understood at the molecular and structural level.
HIV-1 IN and integration
Following entry of HIV into a host cell, the virus synthesizes a double-stranded (ds) DNA copy of its RNA genome. The viral DNA is then irreversibly inserted into the host genome in a process called integration.Citation7,Citation8 This is a defining step in the virus life cycle as it establishes a perpetual source of viral progeny for the lifetime of the cell. Integration is mediated by the virally encoded enzyme IN, and targeting this protein or its actions is a useful antiviral strategy.Citation9–Citation13 IN performs two main enzymatic reactions to facilitate the integration of HIV DNA into the host genome. The first reaction, termed 3′ processing, prepares the viral DNA ends for insertion into target DNA by removing a pair of nucleotides at the 3′ end of both viral DNA strands (). This exposes a 3′ hydroxyl group on the terminal adenosine of the conserved CA dinucleotide. The 3′ processing occurs in the cytoplasm within a high-molecular-weight preintegration complex (PIC) made up of viral DNA together with viral and cellular proteins.Citation14–Citation18 The 3′ processing step is followed by the active transfer of the PIC into the nucleus, a move facilitated by the interaction of the PIC with nucleopore complex proteins.Citation19
Figure 1 HIV-1 DNA integration. HIV-1 virus synthesizes a dsDNA (red) copy of its RNA genome following entry of the virus into host cell cytoplasm. HIV-1 integrase removes 3′ end GT dinucleotides on both viral DNA ends to expose a 3′ hydroxyl group on terminal adenosines by 3′ processing. The 3′ processed viral DNA is then imported into the nucleus where strand transfer occurs resulting in the integration of the two viral DNA ends into host DNA (black) at positions five base pairs (bp) apart. Host DNA repair enzymes then cleave unpaired viral CA dinucleotides, fill in the five bp gaps (green), and ligate the DNA ends.
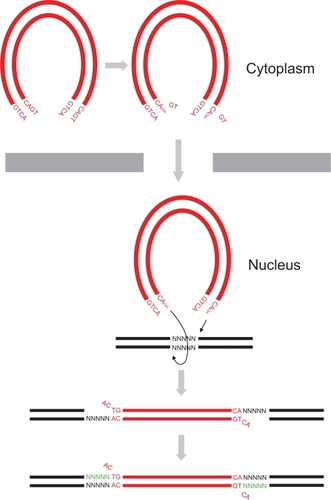
Following nuclear transfer, IN performs the second of its enzymatic reactions, namely strand transfer (). The exposed 3′ hydroxyl groups at the viral DNA ends are used to attack a pair of phosphodiester bonds in the target DNA at sites that are five base pairs (bp) apart, resulting in ligation of the viral DNA strands to host cell genomic DNA. Finally, the unpaired viral CA dinucleotides on the nonligated viral DNA strands are removed, and the resulting five bp gaps on either end of the newly formed junctions are repaired and ligated. These reactions are believed to be performed by host DNA repair enzymes.Citation20 No specific DNA sequence motif has been shown to be a preferred target for HIV-1 integration, but HIV-1 has been shown to selectively insert its DNA within transcriptional units of actively expressed host genes, whereas gamma-retroviruses, such as MLV, prefer to integrate their DNA upstream of host genes in the promoter region.Citation21–Citation24
IN is encoded by the C-terminal region of the HIV-1 pol gene and is a 288 amino acid-long protein that is conventionally divided into three structural and functional domains, namely N-terminal domain (NTD), catalytic core domain (CCD), and C-terminal domain (CTD). The structure of the entire HIV-1 IN has proved elusive; however, the structures of its three domains have been determined separately as well as two-domain fragments.Citation25–Citation27 These studies have revealed that the NTD encompasses IN residues 1–49 and is made up of a triplet of α-helices, containing a double histidine/cysteine (H12-H16-C40-C43) zinc-binding motif. The motif plays a role in the dimerization of IN monomers and the binding of cellular factors.Citation28 IN residues 50–212 make up the CCD which contains the IN active site composed of a triad of acidic residues D64, D116, and E152, also called the DDE motif. The motif is essential for the coordination of divalent metal ions (Mg2+ or Mn2+) that are essential for IN enzymatic functions.Citation28 The CTD, residues 213–288, contains SH3 domains that nonspecifically bind to DNA.Citation29 It is believed that the functional entity of HIV-1 IN is a tetramer assembled from two symmetrical dimers each bound to one of the viral DNA ends.Citation30 Elucidation of the structure of full-length HIV-1 IN and its mode of action has recently benefited from the determination of the crystal structure of prototype foamy virus (PFV) IN tetramer in complex with 3′ processed viral DNA ends.Citation30,Citation31 This IN–DNA complex is called an intasome and is the minimal structure required for integration into target DNA. The structural model of HIV-1 intasome based on the PFV intasome shows the two viral DNA ends engaged by the active site to be ∼18Å apart which is equivalent to a five bp distance, thus validating the intasome as the integration functional unit.Citation30 The intasome is also the preferred target for inhibitors of IN, rather than the unbound enzyme, indicating a direct or indirect involvement of the viral DNA ends in inhibitor binding.
Inhibitors of HIV-1 IN
To date, only one IN inhibitor has been licensed for use in HIV-1 treatment; this being raltegravir which is marketed under the brand name Isentress® (Merck & Co., Inc., White house station, NJ) and was also formerly called MK-0518. Raltegravir, which was approved for use by the US Food and Drug Administration in 2007, is a diketo acid (DKA) analog. A signature feature of DKAs is a β-hydroxy ketone moiety (), and the compounds were the first molecules to be reported as potent and specific IN strand transfer inhibitors (INSTIs). The first two DKA compounds to enter clinical trials were S-1360 and L-870,810, but these agents demonstrated poor efficacy and toxicity, respectively, and were not developed further.Citation32 However, another INSTI, namely elvitegravir, is in the late stages of clinical development and is expected to be approved for clinical use soon, with several others at different stages of development. Elvitegravir is structurally similar to quinolone antibiotics, but like raltegravir, it contains a β-hydroxy ketone moiety (). The crystal structures of PFV in complex with the inhibitors, as well as the structural models of the HIV intasome, show that the oxygen atoms of the β-hydroxy ketone moiety chelate the divalent metal ions that are coordinated by the DDE motif of the IN active site, thereby impeding their participation in the DNA strand transfer reaction (). At the same time, the halobenzyl moieties of the inhibitors end stacked up against the cytosine of the CA dinucleotide which forces the reactive 3′ hydroxyl group of the terminal adenosine away from the active site (). The drugs also make contact with residues Q146 and R231.Citation30 In addition, raltegravir interacts with N117, Y143, N144, and P145, while elvitegravir makes only one additional contact with C65.Citation30
Figure 2 Structures of raltegravir and elvitegravir. A) Raltegravir. B) Elvitegravir. The β-hydroxy ketone and halobenzyl moieties are indicated. The atoms are indicated and/or represented by different colored spheres: C, gray; O, red; N, blue; Cl, green; F, brown. Hydrogen atoms are not shown. The chemical structures were created using MarvinSketch software (ChemAxon, Budapest, Hungary).
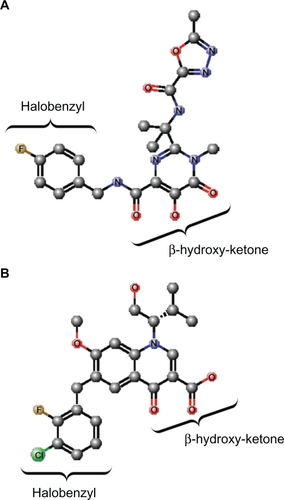
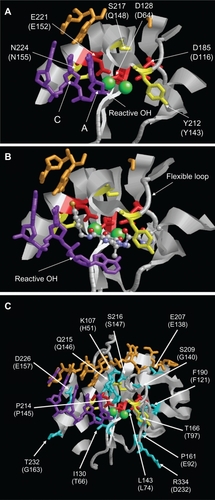
Figure 3 Structure of the PFV IN active site. A) Structure of PFV IN active site within 14Å of Mn2+ ions showing location of the three active site residues (red sticks), three residues where primary resistance mutations occur (yellow sticks), and Mn2+ ions (green spheres). B) Structure of PFV IN active site in complex with raltegravir showing the three oxygen atoms (red spheres) of the β-hydroxy ketone moiety chelating the Mn2+ ions. The halobenzyl moiety (with brown fluoride atom) is seen stacked close to the cytosine (C) of the CA dinucleotide of the donor DNA strand (purple sticks) which results in the displacement of the terminal adenosine (A) and its 3′ hydroxyl group from the active site. C) Structure of PFV IN active site within 20Å of Mn2+ ions showing location of some of the residues where secondary resistance mutations occur (cyan sticks). PFV residues are indicated, and the corresponding HIV-1 residues are in brackets. The nontransferred DNA strand is shown as brown sticks. Protein data bank codes are 3OY9 and 3L2V,Citation31 and the diagrams were created using RasMol software (University of Massachusetts, Amherst, MA, USA).
Raltegravir and elvitegravir are very potent inhibitors of the IN strand transfer reaction with 50% inhibitory concentrations in the nanomolar range.Citation33 Both drugs possess good safety profiles and are rarely associated with severe adverse events. A significant Achilles’ heel for both drugs, as discussed in the next section, is a relatively low genetic barrier to the development of resistance such that a single mutation is enough to cause a major reduction in susceptibility. However, raltegravir and elvitegravir differ in their metabolic pathways, raltegravir being metabolized mainly by glucuronidation mediated by uridine-diphosphate-glucoronosyltransferase 1A1, whereas elvitegravir is metabolized by cytochrome P450 (CYP3A4/5) and secondarily by glucuronidation.Citation34 As a consequence, the half-life of elvitegravir can be boosted by the coadministration of an inhibitor of CYP3A4 such as ritonavir.
Resistance patterns of IN inhibitors
Multiple mutations conferring resistance to raltegravir and elvitegravir have been described in vitro and in vivo in viruses from patients failing on raltegravir therapy. These mutations mostly involve amino acid substitutions that occur in the vicinity of the IN inhibitor-binding pocket in the CCD.Citation31,Citation32 Considerable cross-resistance has been demonstrated between the two inhibitors which is not surprising as structural models have shown a similar binding mechanism for raltegravir and elvitegravir at the active site of IN.Citation30,Citation31 Primarily, mutations causing high-level resistance to raltegravir arise independently at three residues, namely Q148 (to H, R, or K), N155 (to H), and less frequently Y143 (to C, H, or R).Citation12 However, mutations at T66 and E92 have also been shown to significantly affect susceptibility to both drugs when present alone in vitro and in vivo, respectively.Citation35,Citation36 Predictably, these mutations negatively affect IN enzymatic activity, and therefore, viral replication capacity such that on the cessation of raltegravir treatment, the resistance mutations are quick to disappear, demonstrating their impact on fitness of the virus.Citation37,Citation38 As a consequence, the primary mutations are often accompanied by one or more specific secondary mutations that either increase resistance or restore viral fitness or both (–). Some of the accessory mutations, such as L74I, T97A, E138K, V151I, G163R, V165I, V201I, I203M, and T206S, are natural polymorphisms which could influence the rate of development of resistance in viruses that contain these mutations at initiation of IN inhibitor therapy.Citation39–Citation43 The majority of these secondary mutations are also located in the vicinity of the IN active site (). Interestingly, only three IN residues where drug resistance mutations occur, namely Y143, P145, and Q146, were shown to contact raltegravir and/or elvitegravir in the structural models of the HIV intasome in complex with the inhibitors.Citation30 Mutations occurring at the other sites are hypothesized to affect the conformation of the active site indirectly, so perturbing the binding of the inhibitors while preserving IN enzymatic activity. IN resistance accessory mutations have also been described to occur in the CTD at residue S230, suggesting a direct role for the other domains in IN enzymatic functions. Indeed, the new HIV intasome model shows that IN–viral DNA interactions not only involve residues in CCD, but also the other two domains.
Table 1 Secondary resistance mutation patterns associated with Q148HRK
Table 2 Secondary resistance mutation patterns associated with N155H
Table 3 Secondary resistance mutation patterns associated with Y143CHRK
The Q148R/H/K resistance pathway
Q148 is located on the active site flexible loop, consisting of residues 140–148 which is involved in CCD–DNA contacts (). Arginine, histidine, or lysine substitutions at position 148 have been shown to reduce both the susceptibility to raltegravir and elvitegravir as well as the replication capacity of the virus.Citation42,Citation44 Consequently, substitutions at Q148 often occur with the secondary mutations, such as G140S/A or E138K, which improve viral replication capacity (). These secondary mutations appear after the establishment of the codon 148 primary mutation, and the nature of the amino acid substitution at position 148 dictates the effect secondary mutations have on drug susceptibility or replication fitness.Citation42,Citation45,Citation46 Fransen et al showed that on addition of G140S to a Q148R/H mutant, susceptibility to raltegravir was reduced further compared to Q148R/H alone.Citation42 In contrast, addition of the G140S mutation to Q148K increased susceptibility to raltegravir. This study also showed that the addition of G140S improved the replication capacity of Q148H/K mutants but not Q148R.Citation42 Another study went further to reveal that the G140S/A mutants alone impaired IN strand transfer activity without affecting the 3′ processing reaction.Citation47 On the other hand, the Q148R/H/K mutants showed defective activities in both strand transfer and 3′ processing. Thus, it was hypothesized that substituting Q with R, H, or K (all of which have longer and bulkier side chains) alters the binding of the viral DNA to IN, therefore affecting both the strand transfer and 3′ processing activities. However, complementation studies have demonstrated that only the G140S/Q148H double mutant was capable of restoring IN activities to almost wild-type levels and that this complementation only operates in cis. This suggests that this double mutant is the only mutant able to restore an active site for viral DNA, allowing 3′ processing and strand transfer activities, while not re-establishing the raltegravir binding site.Citation47 Hence, this may explain the evolution toward this replication efficient double mutant virus in patients failing on raltegravir treatment.
The N155H resistance pathway
The N155 residue is located on α-helix 4, close to active site acidic residue E152 involved in chelation of metal ions (). The mutation N155H is generally associated with lower raltegravir resistance than Q148 mutations, which may explain its eventual disappearance and replacement with either the Q148R/H/K or Y143C/H/R mutations during raltegravir treatment failure ().Citation42,Citation48 This mutant has been shown to reduce the replication capacity of the virus by impairing strand transfer activity and to some extent 3′ processing activity.Citation37,Citation49 A mechanism by which the N155H mutant causes resistance to raltegravir has been reported by Grobler et al. They proposed that the N155 residue in the IN active site interacts with the residues responsible for binding the magnesium cations required for IN activity. As raltegravir binds to IN through interactions with these metal ions, mutation of this residue may prevent raltegravir from binding by disrupting the metal ion active site arrangement.Citation50
Figure 4 Schematic representation of the evolution of raltegravir primary resistance mutations. Initially, mutations conferring resistance to raltegravir have been shown to primarily occur at residues Q148 and N155. Subsequently, switches from 148 or 155 pathways to 148 or 143 pathways have been observed.

Interestingly, studies have shown that the N155H pathway has a broad range of reductions in raltegravir susceptibility which is dependent on the type of secondary mutations that have also been accumulated (). However, it has been suggested that the 155 pathway does not confer a stable state of raltegravir resistance regardless of the accumulated secondary mutations since the type of secondary mutations in a viral population containing the N155H primary mutation vary greatly over time.Citation45 Among the INSTI resistance-associated secondary mutations, the E92Q mutation has been shown to preferentially occur with the N155H mutation by clonal analysis and also that the addition of the E92Q mutation to N155H results in further decreases in both replication capacity and raltegravir susceptibility.Citation42 It was thus hypothesized that selection of E92Q after the establishment of N155H in the virus is probably driven by the increase in raltegravir resistance rather than to compensate for replication capacity.Citation42 However, a recent study by Fun and colleagues has shown that subsequent selection of the Q95K mutation by the N155H-containing virus resulted in increased raltegravir resistance as well as a partially restored replication capacity.Citation37 Further addition of the V151I mutation resulted in reduction in replication capacity but an additional increase in raltegravir resistance, generating a virus with an overall higher level of fitness in the presence of raltegravir. Indeed, this triple mutant virus was shown to rapidly dominate the viral population in one patient during raltegravir treatment failure.Citation37
The Y143C/H/R resistance pathway
Similar to Q148, Y143 is located on the active site flexible loop (). As mentioned earlier, residue Y143 is one of a few residues that directly make contact with raltegravir when the inhibitor is bound at the active site.Citation30 Therefore, development of IN inhibitor resistance via mutation of Y143 can be explained by a direct effect on inhibitor binding. As with the other resistance pathways, mutations at position 143 in the viral IN gene have been shown to increase resistance to raltegravir while reducing the replication capacity of the virus.Citation40,Citation48,Citation51 The level of raltegravir resistance associated with this pathway, however, is believed to be higher than that of the Q148H/G140S double mutant and other primary mutations.Citation51 In vitro studies show that the Y143R/C mutants have impaired 3′ processing and strand transfer activities. The 3′ processing activity was more affected in the R mutant than the C mutant, whereas the strand transfer activity was more efficient in the R mutant. Regardless, both mutants had similar levels of resistance to raltegravir leading the authors to conclude that perhaps the Y143C mutant was a transient form of the Y143R mutant, as a Y to C change involves a single bp change, whereas a Y to R change requires two bp changes. Structural modeling analysis of the Y143R/C mutants suggested that these mutants may allow for an alternative recognition site for DNA binding, in particular, the post-3′ processing contact with the 5′AC overhang, while preventing the binding of raltegravir to the enzyme.Citation51 In another study by Reigadas et al, the strand transfer activity of the Y143R/C mutants was shown to be severely impaired, but the 3′ processing activity was only moderately impaired. They showed that this mutant was highly resistant to ralte-gravir but had a much lower replication capacity than wild-type virus. Drawing on conclusions from unpublished data from Fransen et al,Citation42 Reigadas and colleagues suggested that perhaps the protease and/or reverse transcriptase of the virus help to rescue this replication capacity deficit.Citation48 Studies have shown that Y143R/C mutants can be present either alone or in association with other secondary mutations (). Little is yet known about the effects of these secondary mutations on the raltegravir resistance levels and replication capacity of the viruses expressing the Y143R/C mutations. Nonetheless, it is widely considered that these mutations play a positive role in IN activity and/or resistance to raltegravir.Citation40,Citation48,Citation51
Several other IN resistance mutation patterns have been described that do not involve mutations at Y143, Q148, or N155 (). However, the three main resistance pathways described above have been shown to be mutually exclusive. Several studies have shown that the different pathways can be present in the same viral population within patients undergoing virological failure albeit on different genomes.Citation38,Citation42,Citation45,Citation48,Citation52 This suggests that a virus containing more than one of the primary mutations is probably severely impaired and replicatively compromised. Nonetheless, it has been suggested that the evolution of raltegravir resistance mutations is mostly driven by the need to increase levels of resistance during drug-selective pressure rather than replication fitness.Citation40,Citation42 Indeed, a switch from the 155 to the 148 pathway is often observed during treatment failure () and is believed possibly to be linked to higher levels of raltegravir resistance seen with the 148 pathway.Citation45 Switches from either the 148 or 155 pathways to the 143 pathway have also been observed during treatment failure.Citation40,Citation48,Citation53 However, in this case, it is believed that the eventual emergence of the 143 pathway may be associated with its replicative advantage in the presence of raltegravir rather than an increase in resistance levels.Citation44
Table 4 Other integrase resistance mutation patterns
Two second-generation INSTIs are in late stages of development by GlaxoSmithKline (GSK) and Shionogi, namely S/GSK1349572 and S/GSK1265744.Citation54 Of interest, the pattern of resistance mutations associated with S/GSK1349572 in cell culture passages was shown to be different from that seen with raltegravir or elvitegravir. Resistance mutation patterns associated with S/GSK1349572 included T124A, S153Y, T124A/S153Y, T124A/S153F, and L101I/T124A/S153Y. S/GSK1349572 has been shown to either completely or partially inhibit raltegravir- and elvitegravir-resistant viruses in vitro.Citation55–Citation57 However, combination mutations involving primary mutations at Q148 and N155 have been shown to still cause a significant loss of activity although less than that observed with raltegravir. This suggests that these compounds have a similar mode of action to raltegravir and elvitegravir.
Use of raltegravir in clinical practice
Initially, raltegravir was approved for use in patients who had experienced treatment failure on previous regimens; however, it is now being considered for use in first-line regimens.Citation58 Several studies have looked at the effectiveness of raltegravir in treatment-experienced and treatment-naïve patients.
Treatment-experienced patients
The BENCHMRK trial showed that raltegravir was highly effective in the treatment of patients when combined with an optimized background regimen. A total of 57% of patients receiving raltegravir had undetectable viral load (VL) (<50 RNA copies/mL) at 96 weeks, compared with 26% of placebo recipients who only received optimized therapy. The best results were seen when raltegravir was combined with two new active agents.Citation59 Significantly, greater rises in CD4 counts were also seen in the raltegravir recipients. Thus, overall, these studies showed that raltegravir could contribute to long-term viral suppression in patients with multidrug-resistant HIV, provided that other drugs in the regimen were also effective.
Treatment-naïve patients
Raltegravir has been shown to be noninferior to efavirenz when combined with tenofovir and emtricitabine in the STARTMRK trial and was associated with faster virological responses.Citation60 There have also been studies to examine the effectiveness of NRTI-sparing combinations where ralte-gravir has been used with ritonavir-boosted lopinavir or unboosted atazanavir. The first showed similar outcomes to use of raltegravir with tenofovir and emtricitabine,Citation61 whereas the latter was discontinued due to adverse effects and a higher number of virological failures.Citation62 Thus, the combination of raltegravir with tenofovir and emtricitabine is now regarded as a useful possible alternative first-line therapy, although long-term experience with potential toxicities due to raltegravir have yet to be determined.Citation63 A coformulation pill known as Quad, comprising of elvitegravir with a novel pharmacoenhancer called cobicistat (GS-9350) plus emtricitabine and tenofovir, is also undergoing clinical trials in ART-naïve HIV-infected individuals. In phase II trials, Quad has been shown to have similar efficacy and to be associated with less adverse effects compared to Atripla (efavirenz + emtricitabine + tenofovir).Citation64
Switching therapies
Two trials, SWITCHMRK and SPIRAL, have looked at replacing PIs with raltegravir in suppressed patients, but only in SPIRAL was the raltegravir switch found to be noninferior.Citation65,Citation66 The reasons for the different outcomes are believed to lie in the previous treatment histories of the patients and whether they had experienced virological failure before.
Future research and optimal use of existing and emerging agents
The development of HIV-1 IN inhibitors has been ongoing for nearly two decades, but success has been long time coming, in terms of drugs approved for clinical use, with only raltegravir licensed for clinical use to date. In part, this is because the knowledge of how IN inhibitors work and the mechanism of IN drug resistance has been incomplete due to the lack of a crystal structure of HIV-1 IN alone or in complex with inhibitors. The recent elucidation of the structure of the PFV intasome in complex with first-generation INSTIs has helped produce better structural models of the HIV-1 intasome–INSTI complex.Citation30,Citation31 This is a significant event that will advance the strategic design and development of new active site inhibitors against both wild-type IN and drug-resistant IN mutants or spur improvements of existing INSTIs. Despite the close relation of PFV and HIV-1 IN proteins, structural differences do exist between the two, such as the presence of the N-terminal extension domain in PFV, which is involved in nonspecific DNA binding of viral DNA ends.Citation31 Therefore, HIV IN drug design will benefit even more from the elucidation of a crystal structure of HIV-1 IN in complex with inhibitors, and that still remains a major goal for IN inhibitor drug design and development.
In addition to understanding the mechanism of inhibition and drug resistance profiles of available INSTIs, an HIV-1 intasome model or crystal structure will enable the development of inhibitors that can target other IN regions apart from the active site, akin to the development of RTIs. Zidovudine was the first RTI developed as a competitive nucleoside analog inhibitor that blocked HIV-1 DNA synthesis by binding at the polymerase active site.Citation4 Following its discovery, structural modeling played a prominent role in the design and development of non-nucleoside inhibitors that bound outside the polymerase active site.Citation5 This new class of inhibitors blocked HIV-1 DNA synthesis by affecting the conformation of the active site, a feat better understood with the help of structural models.Citation5 As mentioned earlier, the discovery of this new class of ARVs prompted the use of combination therapy as the resistance mutation profiles for the two classes are mutually exclusive. It is, therefore, envisaged that understanding the structure of the HIV-1 intasome could fuel the search for agents that inhibit HIV-1 integration by binding outside of the IN active site and/or by a different mechanism, which can then be used in combination with first-generation INSTIs, raltegravir or elvitegravir.
With that goal in mind, several avenues for the discovery of IN inhibitors are already being explored and could produce future drugs in addition to the first- and second-generation INSTIs.Citation32 Other reactions or interactions involving IN, apart from the strand transfer reaction, could provide potential targets for blocking HIV-1 integration. These include the 3′ processing reaction, the interaction of IN with both viral and target DNA, the dimerization or oligomerization of IN, and the interaction of IN with host or viral cofactors. Several such agents have been discovered in the past few years, but some of these have not been developed further partly due to a lack of an understanding of their mechanism of action. Structural models of these inhibitors in complex with an HIV-1 intasome can lead to improvements in their design and alternative means of inhibiting HIV-1 DNA integration.
The holy grail for HIV therapy is the complete eradication of the virus in infected individuals; however, this still remains elusive. Studies on viral eradication have shown that the reduction of VL in infected individuals during effective HAART occurs rapidly during the early phases. However, the later phase involves a further gradual and long-term reduction in VL with estimates of more than 60 years for complete eradication of the virus from the infected individual to occur.Citation67,Citation68 It is believed that the latency of HIV-1 infection and probably the persistent low-level expression of nascent virions from long-lived cells infected prior to commencement of HAART could be significant bottlenecks to achieving eradication. One strategy, which has been attempted, is ARV treatment intensification with or without the use of selective inducers of HIV-1 expression. Raltegravir has been one of the ARVs chosen for intensification therapy partially because it has been documented to produce an accelerated decay of HIV-1 RNA in infected individuals. This is believed to result from improved distribution of raltegravir to less accessible sites and cells capable of producing HIV-1 particles, such as in the cerebrospinal fluid.Citation69 However, preliminary results have proved disappointing with unsustainable reductions of residual HIV-1 viremia and no eradication of the virus.Citation70,Citation71 In the future, intensification treatments could involve the use of raltegravir or novel IN inhibitors with or without more potent inducers of HIV-1 expression. Other areas of raltegravir use being investigated include prevention of perinatal infection and postexposure prophylaxis.
Conclusions
The pattern of development of resistance to INSTIs has been extensively investigated. Three primary mutations occurring independently at residues Q148, N155, and Y143 result in high-level resistance to first-generation INSTIs. These mutations are often accompanied by other mutations in IN which increase resistance and/or improve replication capacity of the virus. Multiple cell-based and biochemical assays have been used to understand the effects of the mutations associated with resistance to INSTIs. The recent availability of the structural model of an HIV-1 intasome that has been obtained following the elucidation of the crystal structure of the PFV intasome has significantly augmented the understanding of the mechanisms of resistance to INSTIs. The availability of the HIV-1 intasome structural model will undoubtedly bolster the strategic design of new inhibitors and will help predict how HIV-1 might evolve resistance to the drugs. The structural model could also allow improvements to be made to existing IN inhibitors with the goal of reducing the chances for the development of resistance.
The increase in the classes and numbers of ARVs, which now includes IN inhibitors, has significantly contributed to the success of HAART. This has allowed the complete suppression of viral replication in infected individuals, resulting in undetectable HIV-1 RNA levels of <50 copies/mL. This can be achieved not only in ART-naïve individuals, but also in individuals who have experienced treatment failure on previous regimens. An understanding of the evolution of drug resistance in the new classes of ARVs is crucial to maintaining or improving on this success as well as in the design of next generation drugs in these new classes.
Disclosure
The authors report no conflicts of interest in this work.
References
- BroderSThe development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemicAntiviral Res201085111820018391
- PalellaFJJrDelaneyKMMoormanACDeclining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study InvestigatorsN Engl J Med1998338138538609516219
- WalenskyRPPaltielADLosinaEThe survival benefits of AIDS treatment in the United StatesJ Infect Dis20061941111916741877
- CihlarTRayASNucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudineAntiviral Res2010851395819887088
- De BethuneMPNon-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989–2009)Antiviral Res2010851759019781578
- WensingAMvan MaarseveenNMNijhuisMFifteen years of HIV protease inhibitors: raising the barrier to resistanceAntiviral Res2010851597419853627
- CraigieRHIV integrase, a brief overview from chemistry to therapeuticsJ Biol Chem200127626232132321611346660
- EngelmanAReverse transcription and integrationKurthABannertNRetroviruses: Molecular Biology, Genomics and PathogenesisNorfolk (UK)Caister Academic Press2010129159
- HazudaDJFelockPWitmerMInhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cellsScience2000287545364665010649997
- PommierYJohnsonAAMarchandCIntegrase inhibitors to treat HIV/AIDSNat Rev Drug Discov20054323624815729361
- MarkowitzMNguyenBYGotuzzoEProtocol 004 Part II Study TeamRapid and durable antiretroviral effect of the HIV-1 integrase inhibitor raltegravir as part of combination therapy in treatment-naïve patients with HIV-1 infection: results of a 48-week controlled studyJ Acquir Immune Defic Syndr200746212513317721395
- CooperDASteigbigelRTGatellJMBENCHMRK Study TeamsSubgroup and resistance analyses of raltegravir for resistant HIV-1 infectionN Engl J Med2008359435536518650513
- SteigbigelRTCooperDAKumarPNBENCHMRK Study TeamsRaltegravir with optimized background therapy for resistant HIV-1 infectionN Engl J Med2008359433935418650512
- BukrinskyMISharovaNMcDonaldTLPushkarskayaTTarpleyWGStevensonMAssociation of integrase, matrix, and reverse transcriptase antigens of human immunodeficiency virus type 1 with viral nucleic acids following acute infectionProc Natl Acad Sci U S A19939013612561297687060
- MillerMDFarnetCMBushmanFDHuman immunodeficiency virus type 1 preintegration complexes: studies of organization and compositionJ Virol1997717538253909188609
- BushmanFDHost proteins in retroviral cDNA integrationAdv Virus Res19995230131710384239
- LinCWEngelmanAThe barrier-to-autointegration factor is a component of functional human immunodeficiency virus type 1 preintegration complexesJ Virol20037785030503612663813
- RaghavendraNKShkriabaiNGrahamRLJHessSKvaratskheliaMWuLIdentification of host proteins associated with HIV-1 preintegration complexes isolated from infected CD4+ cellsRetrovirology201076620698996
- WoodwardCLPrakobwanakitSMosessianSChowSAIntegrase interacts with nucleoporin NUP153 to mediate the nuclear import of human immunodeficiency virus type 1J Virol200983136522653319369352
- YoderKEBushmanFDRepair of gaps in retroviral DNA integration intermediatesJ Virol20007423111911120011070016
- SchroderARShinnPChenHBerryCEckerJRBushmanFHIV-1 integration in the human genome favors active genes and local hotspotsCell2002110452152912202041
- MitchellRSBeitzelBFSchroderARRetroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferencesPLoS Biol200428E23415314653
- WuXBurgessSMIntegration target site selection for retroviruses and transposable elementsCell Mol Life Sci20046119–202588259615526164
- MaxfieldLFFraizeCDCoffinJMRelationship between retroviral DNA-integration-site selection and host cell transcriptionProc Natl Acad Sci U S A200510251436144115659548
- DydaFHickmanABJenkinsTMEngelmanACraigieRDaviesDRCrystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferasesScience19942665193198119867801124
- ChenJCKrucinskiJMierckeLJCrystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA bindingProc Natl Acad Sci U S A200097158233823810890912
- WangJYLingHYangWCraigieRStructure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact proteinEMBO J200120247333734311743009
- McCollDJChenXStrand transfer inhibitors of HIV-1 integrase: bringing IN a new era of antiretroviral therapyAntiviral Res201085110111819925830
- EngelmanAHickmanABCraigieRThe core and carboxyl-terminal domains of the integrase protein of human immunodeficiency virus type 1 each contribute to nonspecific DNA bindingJ Virol1994689591159178057470
- KrishnanLLiXNaraharisettyHLHareSCherepanovPEngelmanAStructure-based modeling of the functional HIV-1 intasome and its inhibitionProc Natl Acad Sci U S A201010736159101591520733078
- HareSGuptaSSValkovEEngelmanACherepanovPRetroviral intasome assembly and inhibition of DNA strand transferNature2010464728623223620118915
- MarchandCMaddaliKMetifiotMPommierYHIV-1 IN inhibitors: 2010 update and perspectivesCurr Top Med Chem20099111016103719747122
- KobayashiMNakaharaKSekiTSelection of diverse and clinically relevant integrase inhibitor-resistant human immunodeficiency virus type 1 mutantsAntiviral Res200880221322218625269
- PacePRowleyMIntegrase inhibitors for the treatment of HIV infectionCurr Opin Drug Discov Devel2008114471479
- ShimuraKKodamaESakagamiYBroad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137)J Virol200882276477417977962
- GoodmanDHluhanichRWatersJIntegrase inhibitor resistance involves complex interactions among primary and secondary resistance mutations: a novel mutation L68V/I associates with E92Q and increases resistance [abstract]Antivir Ther200813Suppl 3A15
- FunAvan BaelenKvan LelyveldSFMutation Q95K enhances N155H-mediated integrase inhibitor resistance and improves viral replication capacityJ Antimicrob Chemother201065112300230420736234
- FernsRBKirkSBennettJWilliamsIEdwardsSPillayDThe dynamics of appearance and disappearance of HIV-1 integrase mutations during and after withdrawal of raltegravir therapyAIDS200923162159216419571721
- Ceccherini-SilbersteinFArmeniaDD’ArrigoRVirological response and resistance in multi-experienced patients treated with raltegravir [abstract]Antivir Ther200813Suppl 3A20
- CanducciFMarinozziMCSampaoloMGenotypic/phenotypic patterns of HIV-1 integrase resistance to raltegravirJ Antimicrob Chemother201065342543320056687
- MyersREPillayDAnalysis of natural sequence variation and covariation in human immunodeficiency virus type 1 integraseJ Virol200882189228923518596095
- FransenSGuptaSDanovichRLoss of raltegravir susceptibility by human immunodeficiency virus type 1 is conferred via multiple non-overlapping genetic pathwaysJ Virol20098322114401144619759152
- RheeSYLiuTFKiuchiMNatural variation of HIV-1 group M integrase: implications for a new class of antiretroviral inhibitorsRetrovirology200857418687142
- DelelisOMaletINaLThe G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutationNucleic Acids Res20093741193120119129221
- MaletIDelelisOSoulieCQuasispecies variant dynamics during emergence of resistance to raltegravir in HIV-1-infected patientsJ Antimicrob Chemother200963479580419221102
- NakaharaKWakasa-MorimotoCKobayashiMSecondary mutations in viruses resistant to HIV-1 integrase inhibitors that restore viral infectivity and replication kineticsAntiviral Res200981214114619027039
- MetifiotMMaddaliKNaumovaAZhangXMarchandCPommierYBiochemical and pharmacological analyses of HIV-1 integrase flexible loop mutants resistant to raltegravirBiochemistry201049173715372220334344
- ReigadasSAniesGMasquelierBThe HIV-1 integrase mutations Y143C/R are an alternative pathway for resistance to raltegravir and impact the enzyme functionsPLoS One201054e1031120436677
- MaletIDelelisOValantinMAMutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitroAntimicrob Agents Chemother20085241351135818227187
- GroblerJAStillmockKAMillerMDHazudaDJMechanism by which the HIV integrase active-site mutation N155H confers resistance to raltegravir [abstract]Antivir Ther200813Suppl 3A41
- DelelisOThierrySSubraFImpact of Y143 HIV-1 integrase mutations on resistance to raltegravir in vitro and in vivoAntimicrob Agents Chemother201054149150119901095
- CharpentierCKarmochkineMLaureillardDDrug resistance profiles for the HIV integrase gene in patients failing raltegravir salvage therapyHIV Med20089976577018651855
- SichtigNSierraSKaiserREvolution of raltegravir resistance during therapyJ Antimicrob Chemother2009641253219447792
- PradaNMarkowitzMNovel integrase inhibitors for HIVExpert Opin Investig Drugs201019910871098
- UnderwoodMJohnsBSatoAFujiwaraTSpreenWS/GSK1349572: a next generation integrase inhibitor with activity against integrase inhibitor-resistant clinical isolates from patients experiencing virologic failure while on raltegravir therapy [abstract]5th IAS Conference on HIV Pathogenesis, Treatment and Prevention2009 Jul 19–22Cape Town, South Africa
- SatoAKobayashiMYoshinagaTS/GSK1349572 is a potent next generation HIV integrase inhibitor [abstract]5th IAS Conference on HIV Pathogenesis, Treatment and Prevention2009 Jul 19–22Cape Town, South Africa
- SekiTKobayashiMWakasa-MorimotoCS/GSK1349572 is a potent next generation HIV integrase inhibitor and demonstrates a superior resistance profile substantiated with 60 integrase mutant molecular clones [abstract]17th Conference on Retroviruses and Opportunistic Infections2010 Feb 16–19San Francisco, CA
- World Health Organization (WHO)Antiretroviral therapy for HIV infection in adults and adolescents: recommendations for a public health approach2010 Available from: http://whqlibdoc.who.int/publications/2010/9789241599764_eng.pdf. Accessed January 13, 2011.
- SteigbigelRTCooperDATepplerHBENCHMRK Study TeamsLong-term efficacy and safety of raltegravir combined with optimized background therapy in treatment-experienced patients with drug-resistant HIV infection: week 96 results of the BENCHMRK 1 and 2 Phase III trialsClin Infect Dis201050460561220085491
- LennoxJLDeJesusELazzarinASTARTMRK InvestigatorsSafety and efficacy of raltegravir-based versus efavirenz-based combination therapy in treatment-naïve patients with HIV-1 infection: a multicentre, double-blind randomised controlled trialLancet2009374969279680619647866
- ReynesJLawalAPulidoFLopinavir/ritonavir combined with raltegravir demonstrated similar antiviral efficacy and safety as lopinavir/ritonavir combined with tenofovir disoproxil fumarate/emtricitabine in treatment-naïve HIV-1 infected subjects [abstract]XVIII International AIDS Conference2010 Jul 18–23Vienna, Austria
- KozalMLupoSDeJesusESPARTAN Study TeamThe SPARTAN study: a pilot study to assess the safety and efficacy of an investigational NRTI- and RTV-sparing regimen of atazanavir (ATV) experimental dose of 300 mg BID plus raltegravir (RAL) 400 mg BID (ATV+RAL) in treatment-naïve HIV-infected subjects [abstract]XVIII International AIDS Conference2010 Jul 18–23Vienna, Austria
- PowderlyWGIntegrase inhibitors in the treatment of HIV-1 infectionJ Antimicrob Chemother201065122485248820852268
- CohenCShamblawDRuanePSinglet-tablet, fixed-dose regimen of elvitegravir/emtricitabine/tenofovir disoproxil fumarate/GS-9350 achieves a high rate of virologic suppression and GS-9350 is an effective booster [abstract]17th Conference on Retroviruses and Opportunistic Infections2010 Feb 16–19San Francisco, CA
- EronJJYoungBCooperDASWITCHMRK 1 and 2 InvestigatorsSwitch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicentre, double-blind, randomised controlled trialsLancet2010375971239640720074791
- MartinezELarrousseMLlibreJMSPIRAL Study GroupSubstitution of raltegravir for ritonavir-boosted protease inhibitors in HIV-infected patients: the SPIRAL studyAIDS201024111697170720467288
- FinziDBlanksonJSilicianoJDLatent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapyNat Med19995551251710229227
- PalmerSMaldarelliFWiegandALow-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapyProc Natl Acad Sci U S A2008105103879388418332425
- CroteauDLetendreSBestBMCHARTER GroupTotal raltegravir concentrations in cerebrospinal fluid exceed the 50-percent inhibitory concentration for wild-type HIV-1Antimicrob Agents Chemother201054125156516020876368
- ArchinNMCheemaMParkerDAntiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infectionPLoS One201052e939020186346
- GandhiRZhengSBoschRRaltegravir (RAL) intensification does not reduce low-level residual viremia in HIV-1-infected patients on antiretroviral therapy (ART): results from ACTG A5244 [abstract]5th IAS Conference on HIV Pathogenesis, Treatment and Prevention2009 Jul 19–22Cape Town, South Africa
- GoethalsOVosAvan GinderenMPrimary mutations selected in vitro with raltegravir confer large fold changes in susceptibility to first-generation integrase inhibitors, but minor fold changes to inhibitors with second-generation resistance profilesVirology2010402233834620421122
- AniesGDa SilvaDRecordon-PinsonPClonal analysis of raltegravir-resistant patterns including mutations at positions 143 and 155 [abstract]16th Conference on Retroviruses and Opportunistic Infections2009 Feb 8–11Montreal, Canada
- GoethalsOClaytonRvan GinderenMResistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitorsJ Virol20088221103661037418715920
- MillerMDDanovichRMKeYon behalf of the P005 Study TeamLongitudinal analysis of resistance to the HIV-1 integrase inhibitor raltegravir: results from P005, a phase II study in treatment-experienced patients [abstract]Antivir Ther200813Suppl 3A8
- HuZKuritzkesDREffect of raltegravir resistance mutations in HIV-1 integrase on viral fitnessJ Acquir Immune Defic Syndr201055214815520634701
- GerettiAMFearnhillECeccherini-SilbersteinFPrevalence and patterns of raltegravir resistance in treated patients in Europe [abstract]Antivir Ther201015Suppl 2A62
- BlancoJLBuzonMJCalvoMEvolution of viral load and integrase resistance mutations after interrupting raltegravir (RAL) in HIV-infected subjects with virological failure on a RAL-containing regimen: the Raldyn study [abstract]Antivir Ther201015Suppl 2A133
- BaldantiFPaolucciSGulminettiRBrandoliniMBarbariniGMaseratiREarly emergence of raltegravir resistance mutations in patients receiving HAART salvage regimensJ Med Virol201082111612219950236
- McCollDJFransenSGuptaSResistance and cross-resistance to first generation integrase inihibitors: insights from a Phase II study of elvitegravir (GS-9137) [abstract]Antivir Ther200712Suppl 1S11
- GallienSDelaugerreCHuZIntegrase inhibitor resistance mutations in treatment-experienced HIV-1-infected patients with low-level viraemia receiving raltegravir-containing antiretroviral therapy: an ANRS 138-EASIER Trial substudy [abstract]Antivir Ther201015Suppl 2A60