Abstract
Background
The prevalence of diet-induced obesity is increasing globally, and posing significant health problems for millions of people worldwide. Diet-induced obesity is a major contributor to the global pandemic of type 2 diabetes mellitus. The reduced ability of muscle tissue to regulate glucose homeostasis plays a major role in the development and prognosis of type 2 diabetes. In this study, an animal model of diet-induced obesity was used to elucidate changes in skeletal muscle insulin signaling in obesity-induced diabetes.
Methods
Adult male Wistar rats were randomized and assigned to either a control group or to a test group. Controls were fed a standard laboratory pellet diet (chow-fed), while the test group had free access to a highly palatable diet (diet-fed). After 8 weeks, the diet-fed animals were subdivided into three subgroups and their diets were altered as follows: diet-to-chow, diet-fed with addition of fenofibrate given by oral gavage for a further 7 weeks, or diet-fed with vehicle given by oral gavage for a further 7 weeks, respectively.
Results
Untreated diet-fed animals had a significantly higher body weight and metabolic profile than the control chow-fed animals. Intramuscular triacylglyceride levels in the untreated obese animals were significantly higher than those in the control chow-fed group. Expression of protein kinase C beta, phosphatidylinositol 3, Shc, insulin receptor substrate 1, ERK1/2, and endothelial nitric oxide synthase was significantly increased by dietary obesity, while that of insulin receptor beta, insulin receptor substrate 1, and protein kinase B (Akt) were not affected by obesity.
Conclusion
These data suggest that diet-induced obesity affects insulin signaling mechanisms, leading to insulin resistance in muscle.
Introduction
Insulin resistance is a comorbidity of overweight and obesity, and is a risk factor for development of type 2 diabetes and cardiovascular disease. In obese individuals and patients with type 2 diabetes, insulin resistance in skeletal muscle is characterized by markedly blunted insulin-stimulated glucose uptake and metabolism.Citation1 Given that skeletal muscle is the primary site of glucose disposal in the human body, the inability of this tissue to take up glucose in response to insulin would account significantly for reduced in vivo disposal of plasma glucose in insulin-resistant individuals. At the cellular level, metabolic insulin resistance in diet-induced obesity shows disrupted protein expression and activation (mainly in skeletal muscle and liver) of signaling via the insulin receptor substrate 1 (IRS-1) phosphatidylinositol (PI)3 kinase pathway, accompanied by a reduction in glucose uptake and utilization.Citation2,Citation3
Systemic lipid accumulation and an increase in the intramuscular concentrations of lipid intermediates, such as fatty acyl-CoA, ceramides, and diacylglycerides, not only correlate with insulin resistance, but also directly and indirectly alter insulin signaling.Citation4–Citation6 Considerable evidence linking increased skeletal muscle lipid content with insulin resistance has been derived from animal studies using acute and chronic high-fat diets.Citation7 Moreover, it has been documented that protein kinase C (PKC) causes insulin resistance in skeletal muscle.Citation8 Furthermore, among the eight PKC isoforms, PKC-β appears to be the only isoform which is increased in insulin-resistant muscle. Indeed, insulin increases PKC-β in the muscle tissue of obese patients but not of lean individuals, while basal PKC-β is reported to be higher in muscle tissue from obese individuals incubated in vitro.Citation9 Moreover, intramuscular lipid intermediates, such as fatty acyl-CoA, ceramides, and diacylglycerides, inhibit steps in the insulin signaling cascade.Citation6 Ceramide activates a protein phosphatase that dephosphorylates protein kinase B (Akt), resulting in inhibition of GLUT4 translocation and glycogen synthesis.Citation10,Citation11
There is a close relationship between insulin resistance and visceral adiposity. It has become apparent that insulin sensitivity is modulated by adiposity, and this has led to a significant interest in adipocytokines, such as tumor necrosis factor alpha, leptin, and C-reactive protein, which may play a pivotal role in insulin signaling and resistance in skeletal muscle.Citation12 Moreover, insulin sensitivity is restored by treatments that reduce intramuscular lipid accumulation, such as lifestyle modification (ie, low-fat feeding, fasting, and exercise) and pharmacological therapy (eg, fenofibrate, an activator of peroxisome proliferator-activated receptor alpha), which improve the lipid profile and insulin resistance.Citation13–Citation16 To elucidate further the exact molecular mechanism(s) involved in insulin transduction in skeletal muscle, this study investigated PI3 kinase and mitogen-activated protein kinase expression in insulin signaling pathways in the skeletal muscle of rats with diet-induced obesity, and analyzed the relationship between lipid profile, protein expression, and induction of insulin resistance in skeletal muscle.
Materials and methods
Experimental protocol
Adult male Wistar rats (n = 40, weighing 195 ± 4 g) were randomized to a control group (n = 10) or to a test group (n = 30). Animals from each group were housed in pairs. Controls were fed a standard laboratory pellet diet (chowfed), while the test group had free access to a highly palatable diet. The standard chow diet provided 1310 kJ/100 g total energy (carbohydrate 60%, protein 30%, and fat 10%) whereas the highly palatable diet provided 1010 kJ/100 g energy (carbohydrate 65%, protein 19%, and fat 16%). The highly palatable diet consisted of 33% powdered chow, 33% condensed milk (Nestle UK Ltd, Croydon, UK), and 7% sucrose (Tate and Lyle, London, UK) by weight, with the remaining being tap water. Chow-fed controls remained on their prospective diet for 15 weeks; after 8 weeks, animals fed the palatable diet were divided further into three subgroups (each group containing 10 animals). In the first subgroup, the palatable diet was removed and the standard chow diet was reintroduced (diet-to-chow); the second subgroup remained on the palatable diet and were given fenofibrate (fenofibrate-treated, 50 mg/kg/day) by oral gavage for a further 7 weeks; and the third subgroup was diet-fed (untreated) and given vehicle (1% carboxymethylcellulose [Sigma, UK] at 1 mL/kg body weight) by oral gavage daily for a further 7 weeks.
On the day of the experiment (after 15 weeks) the rats were weighed and sacrificed by CO2 inhalation after 2 hours of fasting. Total body fat mass was measured immediately using a bioimpedance method (Tobec®, Biotec Instruments Ltd, Kimpton, UK). The gonadal and perirenal fat pads and gastrocnemius muscle were dissected and weighed. The gastrocnemius muscle was snap-frozen in liquid nitrogen for later molecular and biochemical analysis. Plasma was immediately separated by centrifugation before being frozen for later measurement of glucose, insulin, nonesterified free fatty acids, triacylglycerides, and tumor necrosis factor alpha, using commercially available diagnostic kits (Roche and Sigma Diagnostics). All procedures were carried out according to institutional ethical guidelines.
Experimental procedures
Western immunoblotting
Western immunoblotting was carried out on the four experimental groups (chow-fed, vehicle, diet-to-chow, and fenofibrate-treated) to measure protein expression levels, ie, PKC-β, insulin receptor beta (IR-β), IRS-1, Pl3 kinase, Akt, Shc, ERK1/2, and endothelial nitric oxide synthase (eNOS), all of which are involved in the insulin signaling pathway in skeletal muscle.
Nonspecific binding proteins were discriminated by incubating the blot with a blocking buffer (5% milk powder and 1 × phosphate-buffered saline) at room temperature for one hour, followed by immunoblotting with the appropriate primary antibody (1:500 dilution) made up in blocking buffer, and left overnight at 4°C. The following morning, the blots were washed in 1 × phosphate-buffered saline with 1% Tween and incubated with secondary antibody (1:1000), ie, a horseradish peroxidase-linked antirabbit (or antigoat, depending on the source of the species used for primary antibody) for one hour at room temperature. Proteins were detected using an enhanced chemiluminescence method. Positive controls were incubated for standardization of samples between blots, while molecular weight markers (Bio-Rad Laboratories Inc, Hercules, CA) were used for sizing of the bands. Densitometry was used to quantify the bands obtained.
Triacylglyceride content in gastrocnemius skeletal muscle samples
Triacylglyceride content was assessed in gastrocnemius skeletal muscle samples of all animals in the four experimental groups. A small sample of gastrocnemius muscle (160–200 mg) was homogenized in 500 μL of distilled water using a Polytron homogenizer. The homogenate were centrifuged for 5 minutes at 13,000 × g, and the supernatants were transferred to Eppendorf tubes. A triacylglyceride assay was performed on the supernatant to detect triacylglyceride levels in the muscle samples.
Data interpretation and statistical analysis
For Western blotting, the data from the chow-fed (control) animals were expressed as a 100% response, and the results from the other three groups were normalized and then expressed as the percentage of their respective controls. Data are expressed as the mean ± standard error, and have a normal distribution (Shapiro–Wilk test). Statistical significance was tested using the Student’s t-test, repeated-measures (one-way analysis of variance, Bonferroni t-test), or Mann–Whitney U test, as appropriate. Results were considered statistically significant at P < 0.05.
Results
Body weight and metabolic changes
Animals given the palatable diet progressively gained more weight than their chow-fed counterparts. At the end of the study, the untreated diet-fed animals were significantly heavier than the chow-fed, diet-to-chow, and fenofibrate-treated animals, and the body weight of the diet-to-chow and fenofibrate-treated animals was significantly (P < 0.001) lower than that of the untreated diet-fed group, but was significantly (P < 0.05) higher than that of the control chow-fed group (Table 1). Compared with the chow-fed controls, the untreated diet-fed group had a significantly (P < 0.001) higher total and epididymal perirenal fat mass, but a significantly (P < 0.001) lower total lean mass (Table 1). Similarly, plasma insulin, triacylglyceride, nonesterified free fatty acid, and tumor necrosis factor alpha levels were significantly (P < 0.001) higher in the untreated diet-fed group compared with the chow-fed group (Table 1). Removal of the palatable diet or fenofibrate treatment significantly reversed palatable diet-induced changes in fat mass and metabolic parameters (Table 1). There were no significant differences in plasma glucose levels and gastrocnemius muscle mass between the four experimental groups (Table 1). However, the HOMA (Homeostasis Model Assessment) index was significantly (P < 0.001) elevated in the group with untreated diet-induced obesity compared with the control chow-fed group. There was no significant difference in the HOMA index between the fenofibrate-treated and diet- to-chow groups and the chow-fed controls (Table 1).
Triacylglyceride content in gastrocnemius muscle
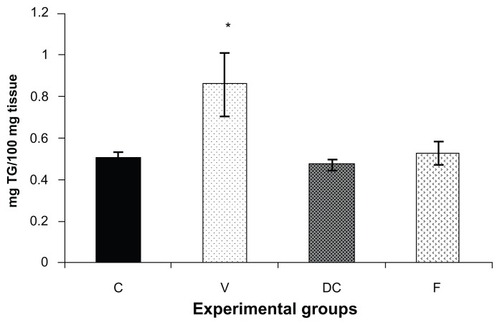
Triacylglyceride content was measured in the skeletal muscle tissue samples () from all four experimental groups, ie, chow-fed control, vehicle, diet-to-chow, and fenofibrate-treated groups. Triacylglyceride levels in muscle samples from the vehicle group (untreated obese animals) were significantly higher (70%, P < 0.05) in comparison with the chow-fed control group, while triacylglyceride levels from the other two groups (diet-to-chow and fenofibrate-treated) were comparable with that in the chow-fed controls, with no significant differences noted ().
Figure 1 Triglyceride content of gastrocnemius muscle in four experimental groups, ie, chow-fed, untreated dietary obese, diet-to-chow, and fenofibrate-treated groups.
Abbreviations: C, chow-fed; V, untreated dietary obese; DC, diet-to-chow; F, fenofibrate-treated; TG, triacylglycerol.
Protein kinase expression in skeletal muscle insulin signaling pathways
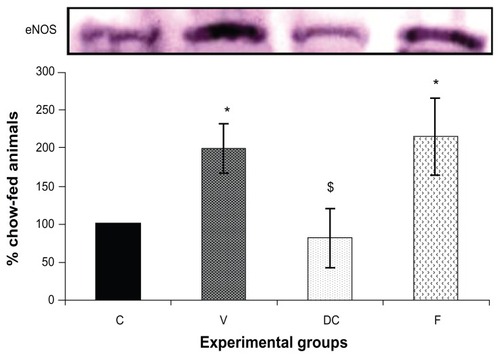
Compared with the chow-fed group, PKC-β expression was significantly increased in the vehicle (113%, P < 0.01) and fenofibrate-treated (137%, P < 0.05) groups, but not in the diet-to-chow group (). IR-β protein expression was similar in all four experimental groups, with no significant difference between them (). Similar to IR-β, IRS-1 expression was comparable in all four groups, again with no significant differences between them (). Interestingly, concentrations of the regulatory subunit p85 of PI3 kinase were significantly increased in the vehicle (77%, P < 0.005) and fenofibrate-treated (68%, P < 0.05) groups, but not in the diet-to-chow group compared with the chow-fed controls (). AKt protein expression in the vehicle group (diet-induced obese animals) did not change significantly in comparison with that in the chow-fed control group (). However, there was a marked reduction in AKt levels in the diet-to-chow (38%, P < 0.001) and fenofibrate-treated (24%, P < 0.05) groups when compared with the chow-fed group (). Expression of two protein kinases important in the mitogen-activated protein kinase pathway, namely Shc and ERK1/2, was measured. Compared with the chow-fed control group, Shc protein levels in the vehicle group were significantly increased (65%, P < 0.01, ), while Shc expression in the other two experimental groups (ie, diet-to-chow and fenofibrate-treated) was not significantly altered (). Expression of ERK1/2 protein in the vehicle group was increased significantly by 95%, P < 0.005) and was markedly decreased in the diet-to- chow group (28%, P < 0.05) when compared with the control group (). Furthermore, ERK1/2 levels in skeletal muscle tissue were not significantly altered by treatment with fenofibrate (). Like PI3 kinase, eNOS protein levels were markedly augmented in the vehicle group (98%, P < 0.001) and in the fenofibrate-treated group (105%, P < 0.001) in comparison with chow-fed controls. However, there were no significant changes in eNOS expression in tissues from the diet-to-chow group compared with that from the chow-fed controls ().
Figure 2 PKC-β protein expression in rat skeletal muscle. Equal amounts (40 mg/well) of protein were separated by SDS-PAGE and immunoblotted with PKC-β antibodies.
Abbreviations: C, chow-fed; V, untreated dietary obese; DC, diet-to-chow; F, fenofibrate-treated; PKC-β, protein kinase C beta; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
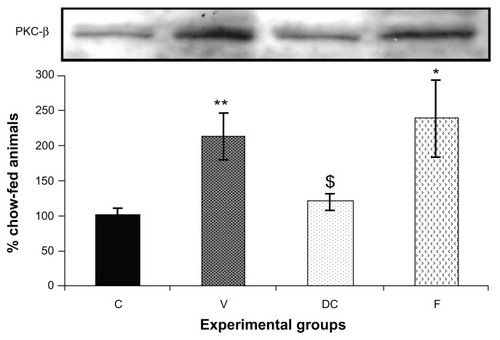
Figure 3 Protein expression of (A) IR-β, and (B) IRS-1 in rat skeletal muscle. Equal amounts (40 mg/well) of protein were separated by SDS-PAGE and immunoblotted with IR-β or IRS-1 antibodies.
Abbreviations: C, chow-fed; V, untreated dietary obese; DC, diet-to-chow; F, fenofibrate-treated; IR-β, insulin receptor beta; IRS-1, insulin receptor substrate 1; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
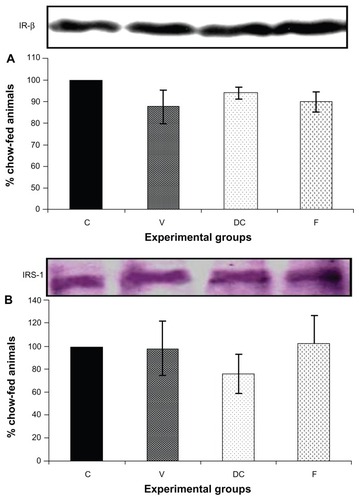
Figure 4 Protein expression of (A) PI3 kinase, and (B) AKt in rat skeletal muscle. Equal amounts (40 mg/well) of protein were separated by SDS-PAGE and immunoblotted with PI3 kinase or AKt antibodies.
Abbreviations: Akt, protein kinase B; C, chow-fed; V, untreated dietary obese; DC, diet-to-chow; F, fenofibrate-treated; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; PI3, phosphatidylinositol 3.
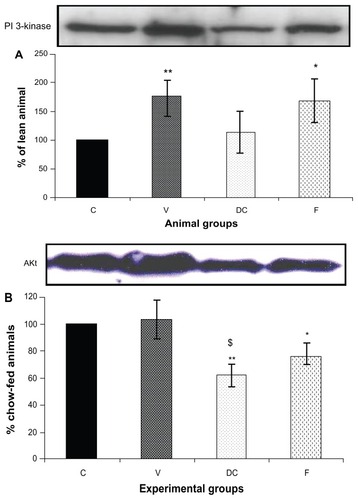
Figure 5 Protein expression of (A) Shc, and (B) ERK1/2, in rat skeletal muscle.
Abbreviations: C, chow-fed; V, untreated dietary obese; DC, diet-to-chow; F, fenofibrate-treated; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
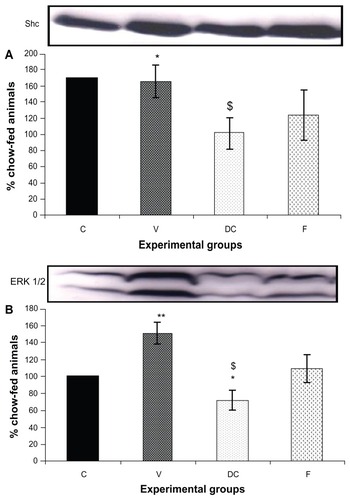
Figure 6 Protein expression of eNOS in rat skeletal muscle.
Abbreviations: C, chow-fed; V, untreated dietary obese; DC, diet-to-chow; F, fenofibrate-treated; insulin receptor substrate 1; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; eNOS, endothelial nitric oxide synthase.
Discussion
Normal muscular insulin signaling is an important factor in the regulation of glucose metabolism. In fact, muscle insulin resistance is one of the major contributing factors in the development of diabetes mellitus. Insulin action in muscle involves a complex signaling pathway composed of a cascade of protein kinases. Their expression and activation/ deactivation play a crucial role in maintaining normal glucose homeostasis. Therefore, any reduced strength in this insulin transduction may lead to skeletal muscle insulin resistance, and ultimately to systemic insulin resistance.
In diet-induced obesity, intramuscular triacylglycerides (and their intermediates, ie, ceramides, fatty acyl-CoA, and diacylglycerides), hyperinsulinemia, and elevated levels of some adipocytokines induce muscular insulin resistance in a cooperative manner. This insulin resistance is thought to be secondary to disruption of protein expression and/or phosphorylation of PI3 and mitogen-activated protein kinase signaling pathways in skeletal muscle.
The present study demonstrates that diet-induced obesity significantly increased the triacylglyceride content in muscle compared with normal chow-fed control animals. Although the mechanism of triacylglyceride accumulation is not fully explained, reports have suggested that this may be in part be due to increased lipid uptake by muscle rather than a decrease in fatty acid oxidation.Citation17 Furthermore, a number of studies have reported a lack of impact of reduction in fatty acid oxidation on increasing accumulation of fatty acyl-CoA (a triacylglyceride intermediate) in muscle.Citation13,Citation17,Citation18 The increased plasma triacylglyceride levels seen in this study and in previous reportsCitation3,Citation19 suggest that it is reasonable to hypothesize a role of increased lipid uptake into muscle in subjects with diet-induced obesity and ultimately development of diet-induced diabetes. Interestingly, both methods of weight reduction (long-term removal of a highly palatable diet and administration of fenofibrate) caused marked reduction of elevated levels of intramuscular and plasma triacylglycerides. It is also reasonable to hypothesize that these improvements occurred via two different mechanisms, ie, removing the highly palatable diet causes a reduction in total energy (calorie) intake and an improved plasma and muscle lipid profile, and fenofibrate treatment increases beta oxidation of fatty acids in muscle, leading to increased fatty acid utilization and clearance.
In the present study, PKC-β expression was markedly increased in the gastrocnemius muscle of rats with diet-induced obesity, and this was associated with insulin resistance, as indicated by an increase in the HOMA index, suggesting a role for PKC-β protein expression during glucose uptake in skeletal muscle.Citation20,Citation21 The triacylglyceride intermediates, fatty acyl-CoA and diacylglycerol, might activate PKC-β directly,Citation22,Citation23 which, in turn, may have negative effects on the insulin transduction cascade in muscle, ultimately reducing insulin-mediated glucose uptake from circulating plasma. In this study, treating obesity by lifestyle modification, ie, long-term withdrawal of a highly palatable diet, achieved complete normalization of PKC-β protein expression as well as improvement in intramuscular lipid accumulation and a subsequent decrease in systemic insulin resistance, indicated by a HOMA index similar to that of the chow-fed controls. However, pharmacological treatment of obesity in this study (ie, administration of fenofibrate) failed to reduce or normalize expression of PKC-β in muscle, despite a marked improvement in intramuscular and plasma lipid concentrations and insulin resistance. It is possible that the elevated PKC-β levels seen in muscle samples from fenofibrate-treated animals could be due to excess production of reactive oxygen species as a result of increased beta oxidation of fatty acids. The principal mechanism of lipid reduction by fenofibrate is stimulation of beta oxidation of fatty acids in tissues such as skeletal muscle.Citation24 Reactive oxygen species are some of the end products of beta oxidation of fatty acids which may have a major role in increasing expression and phosphorylation of PKC-β.Citation25,Citation26 Furthermore, various reports have indicated that fenofibrate is associated with increased production and availability of reactive oxygen species,Citation27–Citation29 further underlined by the increased PKC-β levels seen in the fenofibrate-treated group in our study. Hence, our results indicate that diet-induced obesity can cause increased expression of PKC-β in skeletal muscle, which augments systemic insulin resistance. Interestingly, in the present study, the increased PKC-β levels were reversed completely by long-term withdrawal of the highly palatable diet, but not by pharmacological intervention using fenofibrate, indicating a key role for diet and/or its content in elevation of PKC-β.
Protein expression in the early steps of the PI3 kinase insulin signaling pathway, such as IR-β and IRS-1, was not affected by diet-induced obesity in our study. However, others have reported amelioration of tyrosine phosphorylation of IRS-1 and IR-β in skeletal muscle from obese and diabetic patients.Citation30,Citation31 The deficiencies in phosphorylation could be mediated by increased protein expression and activation of PKC-β, which in turn phosphorylates IR-β and IRS-1 in serine residues, thereby reducing their ability to phosphorylate tyrosine and leading to insulin resistance.Citation20,Citation21,Citation32 However, further studies are required to test this hypothesis.
Although, we did not measure IRS-2 protein expression in skeletal muscle in the current study, several studies have indicated that IRS-1 and IRS-2 are not functionally interchangeable in tissues that are responsible for glucose production and glucose uptake, ie, skeletal muscle.Citation33 Indeed, IRS-1 appears to have its major role in skeletal muscle, whereas IRS-2 appears to regulate hepatic insulin action.Citation34,Citation35
Interestingly, in this study, diet-induced obesity resulted in a significant increase in protein expression of the regulatory subunit p85 of PI3 kinase in skeletal muscle. The increase in protein expression was synchronized with systemic insulin resistance. Therefore, it could be postulated that an increased level of the p85 regulatory subunit might inhibit PI3 kinase activity by competing with phosphotyrosine targets. Hence, disruption of the balance between the amounts of PI3 kinase subunits and excess levels of the p85 regulatory subunit could contribute to insulin resistance. This hypothesis is supported by studies of insulin-resistant states induced by human placental growth hormone, obesity, and type 2 diabetes.Citation36,Citation37 Therefore, if p85 production can be enhanced nutritionally, the resulting changes in the ratio of p85 to p110 may be the earliest manifestation of the ensuing insulin resistance. This could also explain how overfeeding and weight gain trigger insulin resistance.
AKt protein expression in skeletal muscle did not change in response to diet-induced obesity. Similar findings have been reported whereby phosphorylation of AKt in skeletal muscle from insulin-resistant obese diabetic subjects was not changed significantly.Citation38,Citation39 Taken together, it can be argued that AKt in skeletal muscle has no role in inducing muscular insulin resistance. However, chronic withdrawal of the highly palatable diet in our study caused a significant reduction in AKt protein levels compared with the control group, despite complete normalization of systemic insulin resistance as indicated by normalization of the HOMA index, further indicating the lack of a significant role for AKt in insulin resistance in skeletal muscle. Similarly, fenofibrate-treated animals had lower AKt levels in skeletal muscle, despite improved insulin signaling and lipid concentrations in muscle. This is in agreement with previous studies showing marked attenuation of AKt phosphorylation and AKt levels in human cells in vitro in response to fenofibrate.Citation40,Citation41
In the present study, diet-induced obesity significantly increased the expression of ERK1/2 and Shc, whereas removal of the highly palatable diet or treatment with fenofibrate abolished the increase in protein expression of both ERK1/2 and Shc. This is in agreement with previous reports whereby diet-induced obesity increased ERK1/2 expression while hyperinsulinemia resulted in a marked increase in Shc expression.Citation42,Citation43 These findings suggest important roles for ERK1/2 and Shc in insulin resistance in skeletal muscle. Furthermore, eNOS expression was significantly increased in the group fed the highly palatable diet, which may indicate a role of high caloric intake in eNOS expression. We have previously reported a significant increase in eNOS expression in the vascular tissue of obese rats but not in liver samples.Citation3,Citation44 It is suggested that increased eNOS expression may be secondary to increases in adipokines, such as tumor necrosis factor alpha; however, this is in contrast with a previous report of tumor necrosis factor alpha in fact downregulating eNOS expression in muscle tissue.Citation45 Therefore, this hypothesis merits further investigation.
In summary, it could be postulated that dietary-induced muscular insulin resistance is a consequence of a series of defects in the insulin signaling pathway resulting from impaired expression of PKC-β, PI3 kinase, ERK1/2, Shc, and eNOS. The exact mechanism is not fully understood, but it is likely that excessive triacylglyceride levels in muscle tissue may have detrimental effects on protein expression in insulin signaling. This in turn may reduce insulin’s primary action on skeletal muscle as a glucose homeostasis agent in obesity induced diabetes.
Acknowledgment
The authors would like to express their gratitude to Miss Mahdieh Naderali for her editorial assistance in preparing this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
- DohmGTapscottEPoruiesWAn in vitro human muscle preparation suitable for metabolic studies. Decreased insulin stimulation of glucose transport in muscle from morbidly obese and diabetic subjectsJ Clin Invest1988824864943403714
- FataniSItuaIClarkPWongCNaderaliEKThe effects of diet-induced obesity on hepatocyte insulin signaling pathways and induction of non-alcoholic liver damageInt J Gen Med2011421121921475632
- ShulmanGICellular mechanisms of insulin resistance in humansAm J Cardiol1999843J10J
- McGarryJDDysregulation of fatty acid metabolism in the etiology of type 2 diabetesDiabetes20025171811756317
- DespresJPLam LamarcheBMauriègePHyperinsulinemia as an independent risk factor for ischemic heart diseaseN Engl J Med19963349529588596596
- Schmitz-PeifferCProtein kinase C and lipid-induced insulin resistance in skeletal muscleAnn N Y Acad Sci200296714615712079844
- CooneyGJThompsonALFurlerSMYeJKraegenEWMuscle long chain acyl CoA esters and insulin resistanceAnn N Y Acad Sci200296719620712079848
- BossenmaierBMosthafLMischakHUllrichAHaringHProtein kinase C isoforms beta 1 and beta 2 inhibit the tyrosine kinase activity of the insulin receptorDiabetologia1997408638669243110
- ItaniSIZhouQPoriesWJMacdonaldKGDohmGLInvolvement of protein kinase C in human skeletal muscle insulin resistance and obesityDiabetes2000491353135810923637
- LongSPekalaPLipid mediators of insulin resistance: ceramide signal-ling down-regulates GLUT4 gene transcription in 3T3-L1 adipocytesBiochem J19963191791848870666
- ChavezJAKnottsTAWangLPA role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acidsJ Biol Chem2003278102971030312525490
- BastardJMaachiMLagathuCRecent advances in the relationship between obesity, inflammation, and insulin resistanceEur Cytokine Netw20061741216613757
- OakesNBellKFurlerSDiet-induced muscle insulin resistance in rats is ameliorated by acute dietary lipid withdrawal or a single bout of exercise: parallel relationship between insulin stimulation of glucose uptake and suppression of long-chain fatty acyl-CoADiabetes199746202220289392490
- MonzilloLHamdyOHortonEEffect of lifestyle modification on adipokine levels in obese subjects with insulin resistanceObes Res2003111048105412972674
- YeJMDoylePJIglesiasMAWatsonDGCooneyGJKraegenEWPeroxisome proliferator-activated receptor (PPAR) alpha activation lowers muscle lipids and improves insulin sensitivity in high fate-fed rats: comparison with PPAR-gamma activationDiabetes20015041141711272155
- LeeHJChoiSSParkMKFenofibrate lowers abdominal and skeletal adiposity and improves insulin sensitivity in OLETF ratsBiochem Biophys Res Commun200229629329912163016
- HulverMWBerggrenJRCortrightRNSkeletal muscle lipid metabolism with obesityAm J Physiol Endocrinol Metab2003284E741E74712626325
- HegartyBDFurlerSMYeJCooneyGJKraegenEWThe role of intramuscular lipid in insulin resistanceActa Physiol Scand200317837338312864742
- NaderaliEKBrownMJPickavanceLCWildingJPDoylePJWilliamsGDietary obesity in the rat induces endothelial dysfunction without causing insulin resistance: a possible role for triacylglycerolsClin Sci (Lond)200110149950611672455
- SampsonSRCooperDRSpecific protein kinase C isoforms as transducers and modulators of insulin signalingMol Genet Metab200689324716798038
- HoumardJPoriesWDohmGIs there a metabolic program in the skeletal muscle of obese individuals?J Obes2011201125049621603262
- SestiGPathophysiology of insulin resistanceBest Pract Res Clin Endocrinol Metab20062066567917161338
- ZhangLKeungWSamokhvalovVWangWLopaschukGDRole of fatty acid uptake and fatty acid [beta]-oxidation in mediating insulin resistance in heart and skeletal muscleBiochim Biophys Acta2010180112219782765
- BaiXLiHYangWEffects of fenofibrate on gene expression of carnitine palmitoyltransferase 1 in liver and skeletal muscle and its influence on insulin sensitivityZhonghua Yi Xue Za Zhi200888268270 Chinese18361841
- SelivanovVAVotyakovaTVPivtoraikoVNReactive oxygen species production by forward and reverse electron fluxes in the mitochondrial respiratory chainPLoS Comput Biol20117e100111521483483
- Del CarloMLoeserRFChondrocyte cell death mediated by reactive oxygen species-dependent activation of PKC-betaIAm J Physiol Cell Physiol2006290C802C81116236825
- Lores-ArnaizSTravacioMMonserratAJCutrinJCLlesuySBoverisAChemiluminescence and antioxidant levels during peroxisome proliferation by fenofibrateBiochim Biophys Acta199713602222289197464
- JiaoHLZhaoBLCytotoxic effect of peroxisome proliferator fenofibrate on human HepG2 hepatoma cell line and relevant mechanismsToxicol Appl Pharm2002185172179
- JiaoHLYePZhaoBLProtective effects of green tea polyphenols on human HepG2 cells against oxidative damage of fenofibrateFree Radic Biol Med2003351121112814572614
- KimYBKotaniKCiaraldiTPHenryRRKahnBBInsulin-stimulated protein kinase C activity is reduced in skeletal muscle of humans with obesity and type 2 diabetesDiabetes2003521935194212882908
- FareseRVSajanMPStandaertMLInsulin-sensitive protein kinases (atypical protein kinase C and protein kinase B/Akt): actions and defects in obesity and type II diabetesExp Biol Med2005230593605
- ChowLFromASeaquistESkeletal muscle insulin resistance: the interplay of local lipid excess and mitochondrial dysfunctionMetabolism201059708519766267
- VazquezMSilvestreJProusJExperimental approaches to study PPAR gamma agonists as antidiabetic drugsMethods Find Exp Clin Pharmacol20022451552312500431
- BhattacharyaSDeyDRoySMolecular mechanism of insulin resistanceJ Biosci20073240541317435330
- SainiVMolecular mechanisms of insulin resistance in type 2 diabetes mellitusWorld J Diabetes20101687521537430
- BarbourLAShaoJQiaoLHuman placental growth hormone increases expression of the P85 regulatory unit of phosphatidylinositol 3-kinase and triggers severe insulin resistance in skeletal muscleEndocrinology20041451144115014633976
- BandyopadhyayGKYuJGOfrecioJOlefskyJMIncreased p85/55/50 expression and decreased phosphotidylinositol 3-kinase activity in insulin-resistant human skeletal muscleDiabetes2005542351235916046301
- KimYBNikoulinaSECiaraldiTPHenryRRKahnBBNormal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetesJ Clin Invest199910473374110491408
- ChunKHChoiKDLeeDHIn vivo activation of ROCK1 by insulin is impaired in skeletal muscle of humans with type 2 diabetesAm J Physiol Endocrinol Metab2010300E536E54221189360
- KubotaTYanoTFujisakiKItohYOishiRFenofibrate induces apoptotic injury in cultured human hepatocytes by inhibiting phosphorylation of AktApoptosis20051034935815843896
- GrabackaMPlonkaPMUrbanskaKReissKPeroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of AktClin Cancer Res2006123028303616707598
- MoonJHLeeJYKangSBDietary monounsaturated fatty acids but not saturated fatty acids preserve the insulin signaling pathway via IRS-1/PI3K in rat skeletal muscleLipids2010451109111620960069
- Páez-EspinosaEVRochaEMVellosoLABoscheroACSaadMJInsulin-induced tyrosine phosphorylation of Shc in liver, muscle and adipose tissue of insulin resistant ratsMol Cell Endocrinol199915612112910612430
- FataniSNaderaliEKPanchianiSShahFWongCThe effects of dietary obesity on protein expressions of insulin signaling pathway in rat aortaDrug Discov Ther2008225426122504637
- ValerioACardileACozziVTNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodentsJ Clin Invest20061162791279816981010