
Abstract
Background
Carrier screening is the most effective means of controlling the prevalence of alpha-thalassemia. However, due to the differences in ethnic populations and genotypes, the distribution of mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH) and hemoglobin A2 (HbA2) varies in different regions. This study aimed to examine screening efficiency of these indicators in different genotypes of alpha-thalassemia in Fujian Province, China.
Methods
The data of 13,294 subjects collected from May 2016 to December 2019 were reviewed. The participants were categorized as alpha-thalassemia group and negative-for-alpha-thalassemia group based on the results of the genetic analysis. The distribution of MCV, MCH, and HbA2 in different groups was analysed statistically. And the screening efficiency of different indicators and schemes was compared in different genotypes. The positive criteria of MCV < 80fL, MCH < 27pg, and Hb A2< 2.5% were applied.
Results
Among the 13,294 subjects, 2658 were alpha-thalassemia carriers. The genotypes of –SEA/αα and -α3.7/αα are the most prevalent with 63.9% and 21.9% in Fujian Province, China. There were significant differences in the distribution of the three indicators in different groups. The detection rate of the three indicators combined screening was 92.6%.
Conclusion
The distribution of the three indicators overlapped partly between alpha-thalassemia group and negative-for-alpha-thalassemia group. They showed significant differences in the median comparison of seven common genotypes. Combined screening with MCV, MCH and HbA2 improved the detection rate of alpha-thalassemia. The results of this study provide a data basis for clinical laboratories and a reliable reference for clinical consultation.
Introduction
Thalassemia is a group of genetic disorders with varied phenotypes caused by human globin gene synthesis disorders.Citation1 It is one of the top five most common birth defects.Citation2 Thalassemia is widespread globally, particularly in Southeast Asian countries.Citation3–Citation5 According to the type of defective globin, it can be divided into two common categories, alpha- and beta-thalassemia.Citation6,Citation7 The prevalence of alpha-thalassemia is 3.17% in Fujian Province, China, which is higher than that of beta-thalassemia.Citation8 Furthermore, alpha-thalassemia can lead to anemia, hemoglobin H (HbH) disease, and hydrops foetalis syndrome.Citation9 The disease severity is determined by the amount of alpha-chain produced. Alpha-thalassemia major has imposed an enormous burden on society and adversely affects the quality of life of the population.Citation10 However, to date, no ideal treatment is available for patients with thalassemia major, except bone marrow transplantationCitation11 and gene therapy.Citation12–Citation15 The detection of carriers using screening programs is considered the most effective way to control symptomatic alpha-thalassemia.
In general, the clinical criteria for suspected alpha-thalassemia were determined by hypochromic microcytic anaemia, including decreased mean corpuscular volume (MCV) and/or mean corpuscular hemoglobin (MCH) levels and/or decreased hemoglobin A2 (HbA2) levels measured using a robust method such as high-performance liquid chromatography (HPLC) or capillary electrophoresis (CE).Citation16 However, the distribution and screening efficiency of these indicators varies in different regions due to the differences in ethnic populations and genotypes which is difficult to estimate in practice. In the present study, we performed a large-scale investigation to analyse the distribution of these indicators in different groups. The data were used to evaluate the effectiveness of different screening schemes and establish reliable associations between clinical phenotypes and genotypes for the clinical diagnosis of alpha-thalassemia.
Methods
Subjects and Hematological Indicator Detection
The present study was approved by the Protection of Human Ethics Committee of Fujian Maternity and Child Health Hospital, Affiliated Hospital of Fujian Medical University (No. 2016-101). All participants were informed and signed a written informed consent. All experiments were performed in accordance with the Declaration of Helsinki and National Regulations for Ethics of Biological Medical Sciences on Human Studies released by Ministry of Health, China. In total, 13,294 individuals were recruited and tested from May 2016 to December 2019. Among them, 10,636 participants who tested negative for alpha- and beta-thalassemia genes were categorized as healthy subjects (2978 men and 7658 women; median age: 29.3 years). The other 2658 participants tested positive for alpha-thalassemia and negative for beta-thalassemia genes and were designated alpha-thalassemia carriers (850 men and 1808 women; median age: 28.7 years). Those who did not undergo further genetic testing, with iron-deficiency anemia, beta-thalassemia and other hemoglobinopathies, or blood transfusion within one year were excluded. The MCV and MCH were measured using an automated analyser (XN3000; Sysmex, Japan). The positive criteria of MCV <80fL and MCH <27pg were applied.
Capillary Electrophoresis
Hemoglobin analysis was performed using an automated capillary electrophoresis analyser (Capillarys2TM; Sebia, France). It can measure the percentages of HbA2, as well as any variants, including hemoglobin constant spring (HbCS), hemoglobin H (HbH), and hemoglobin Barts (Hb Barts). The positive criteria of Hb A2<2.5% was applied.
Common Genotype Test
DNA was extracted using the DNA Blood Extraction Kit (Yaneng Biosciences, Shenzhen, China). The deletions (–SEA/, -α4.2/, and -α3.7/) and the mutations (αcsα/, αQsα/, and αwsα/) of alpha-thalassemia, and seventeen mutations of beta-thalassemia commonly found in Chinese populations were analyzed by PCR reverse dot blot assay using commercial kits (Yaneng Biosciences, Shenzhen, China) as described.Citation17
–THAI and HKαα Genotype Test
–THAI genotype was tested using gap polymerase chain reaction (gap-PCR), and HKαα genotype was tested by single PCR and nested PCR, as described previously.Citation18,Citation19
Statistical Analysis
The data were analysed using the Statistical Package for the Social Sciences (SPSS) version 24 (IBM Inc., Chicago, USA). Histograms were created demonstrating the distributions of MCV, MCH and HbA2 in the subjects. Continuous data were checked for normality using the Kolmogorov–Smirnov test. Mann–Whitney U-test was used to compare these non-parametric variables between alpha-thalassemia group and negative-for-alpha-thalassemia group. The Kruskal–Wallis test was used to compare the three indicators among the seven common genetic mutation groups. The results with P < 0.05 were considered statistically significant. The screening efficiency of different indicators and schemes was compared in different genotypes and the decision criteria of MCV/MCH primary screening was positive for one item or both of MCV and MCH, and MCV/MCH/HbA2 primary screening was positive for one item or more of MCV, MCH and HbA2.
Results
The participants investigated in this study comprised 13,294 individuals (3828 men and 9466 women; median age: 29.1 years). They were grouped according to the genetic test results: 10,636 participants were categorized as negative-for-alpha-thalassemia group, whereas 2658 participants were designated alpha-thalassemia group. The different genotypes were identified and are listed in . The genotypes of –SEA/αα and -α3.7/αα were the most frequent with 63.9% and 21.9% prevalence.
Table 1 Genotypes Identified Among Alpha-Thalassemia Carriers
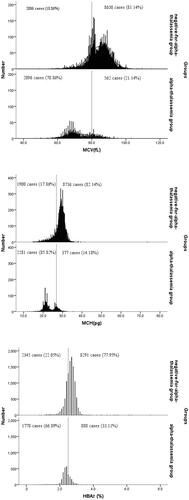
In the alpha-thalassemia group, 2096 of 2658 (78.86%) subjects had MCV < 80 fL, 2281 of 2658 (85.82%) subjects had MCH < 27 pg, and 1778 of 2658 (66.89%) subjects had HbA2< 2.5%. But 21.14% of subjects had MCV, 14.18% had MCH and 33.11% had HbA2 higher than these cut-off values. At the same time, in the negative-for-alpha-thalassemia group, 2006 of 10,636 (18.86%) subjects in MCV, 1900 of 10,636 (17.86%) subjects in MCH and 2345 of 10,636 (22.05%) subjects in HbA2 were abnormal. A partial overlap was observed between the groups. The distributions of MCV, MCH and HbA2 values in the alpha-thalassemia and negative-for-alpha-thalassemia groups are depicted in . The distributions of the three indicators in the two groups were not normal. The three indicators were significantly lower in the alpha-thalassemia group than in the negative-for-alpha-thalassemia group (P < 0.05).
Figure 1 Histogram showing number of alpha-thalassemia carriers and negative-for-alpha-thalassemia participants with MCV, MCH and HbA2. Dash lines indicate the cutoff MCV of 80fL, MCH of 27pg and HbA2 of 2.5% used for screening of alpha-thalassemia. The proportions of cases found at below and above these cutoff levels are provided.
The distribution of MCV, MCH and HbA2 in the seven most common genotypes (including the genotypes of –SEA/αα, -α3.7/αα, -α4.2/αα, αQSα/αα, αcsα/αα, –THAI/αα, α wsα/αα) was not normal (P < 0.05). There was a significant difference in the three indicators among these genotypes (p < 0.05). The different genotypes shown in are represented by the letters a~g. In the comparison of MCV, there were significant differences between group a and b/c/d/e/g, group d and b/c, group f and b/c/d/e/g. In MCH, there were significant differences between group a and b/c/d/e/g, group d and b/c/g, group f and b/c/d/e/g. In HbA2, there were significant differences between group a and b/c/d/e, group e and b/c/d, group f and b/d/e, group g and a/b/c/e/f. The median of MCV and MCH in the genotype of –THAI/αα and –SEA/αα were the lowest and HbA2 in the genotype of αcsα/αα was the lowest.
Table 2 Hematological Values for Each Genotype and the Difference Between Groups
The detection rate of different screening schemes in different genotypes varied with the alpha-thalassemia genotype (). The total detection rates of MCV/MCH, HbA2 and MCV/MCH/HbA2 were 88.0%, 66.9% and 92.6%, respectively.
Table 3 Detection Rates of the Different Screening Schemes
Discussion
Thalassemia is mainly distributed on the Mediterranean coast, the Middle East and Southeast Asian countries.Citation20–Citation22 In China, the most frequent provinces are Guangxi, Guangdong, Yunnan and Hainan.Citation23,Citation24 Since 2015, Fujian has become a pilot province of the National Health and Family Planning Commission to prevent and control thalassemia. An epidemiological survey showed that the carrying rate was 4.41% in Fujian Province.Citation8
In this study, –SEA/αα was the most common genotype of alpha-thalassemia, followed by -α3.7/αα. It illustrated that the two genotypes, accounting for 85.8% of alpha-thalassemia, had the highest carrying rate in Fujian Province, China. This result was consistent with that for the genotype of the Yulin area in ChinaCitation6 and Lao Loum Group in the Lao People’s Democratic Republic.Citation25
Carrier screening is crucial because the confirmatory genotype test is not available for all subjects in many regions. Presently, large-scale screening of alpha-thalassemia still relies on blood cell parameters and hemoglobin component analysis. In clinical practice, the criteria of MCV < 80 fL, MCH < 27 pg, and HbA2 < 2.5% was used basing on the requirements of technical service specifications for thalassaemia prevention and control pilot projects issued by the General Office of the National Health and Family Planning Commission of China. In the study, we identified 2658 alpha-thalassemia carriers and 10,636 negative-for-alpha-thalassemia participants, and the sensitivity and specificity of MCV, MCH and HbA2 were 78.86%/81.14%, 85.82%/82.14% and 66.89%/77.95%, respectively. The screening efficiency of HbA2 in alpha-thalassemia was relatively low. Furthermore, some other diseases, such as malaria, lead poisoning, iron-deficiency anemia and bone marrow proliferative diseases, affected to the accuracy of HbA2.Citation26 On the contrary, the Youden index (85.82%+82.14% −1) of MCH was the highest among the three indicators. And MCH is considerably more stable than MCV during the storage of blood specimensCitation27 and less influenced by age.Citation16 Thus, it is the best parameter of the three indicators for screening alpha-thalassemia. However, partial overlap in the distribution of the three indicators existed between the alpha-thalassemia group and negative-for-alpha-thalassemia group (). Screening with a single indicator can lead to a false diagnosis.
In this study, we compared the three hematological indicators of the seven most common genotypes listed in . The highest and lowest MCV mean values were analysed among -α4.2/αα and –THAI/αα carriers, respectively (ranging from 81.30 to 67.10 fL). The highest and lowest MCH mean values were analysed among αwsα/αα and –SEA/αα carriers, respectively (ranging from 27.30 to 21.40 pg/cell). The highest and lowest HbA2 mean values were analysed among αwsα/αα and αcsα/αα (ranging from 2.70 to 2.20% of the total Hb). These data were helpful for clinical laboratories and contribute to the revision of clinical testing guides in the region. Additionally, valuable clues are provided for physicians in clinical diagnosis and consultations. The significant reductions of MCV and MCH associated with the genotypes of –SEA/αα and –THAI/αα suggested that the effect was related to the size of the deletion fragment. But it needed to be confirmed by further research. Among three-point mutations of alpha-thalassemia, the αQSα/αα genotype was associated with lower MCV and MCH, but the αCSα/αα genotype caused lower HbA2.
In clinical practice, MCV and MCH are tested simultaneously. The detection rate in our study reached 88.0% when the two indicators were combined for screening. But the detection rate of all three indicators combined screening was 100% in 14 of the 19 genotypes and the total positive rate reached 92.6% (). Capillary electrophoresis also analyse abnormal hemoglobin in addition to detect HbA2. In our study, two cases of αcsα/αα with normal HbA2 were diagnosed because of their zone(c) peaks in the electrophoretogram indicating alpha-thalassemia. Therefore, the simultaneous screening of alpha-thalassemia using hematological parameters and hemoglobin analysis in the prevalent region is recommended. However, it is worth noting that the possibility of alpha-thalassemia could not be ruled out completely and genetic analysis and sequencing should also be performed when necessary, even if the three indicators were normal.
Although genotype tests can diagnose most common alpha-thalassaemia, rare deletions or point mutations may still be missed. However, patients with large-fragment deletions generally have clinical manifestations, and combined screening of several indicators tends to be positive. We identified one novel mutation of alpha-thalassaemia in this study. The patient presented decreased MCV, MCH and HbA2 levels, and second-generation sequencing was performed because of the negative results of the conventional genotype test. It revealed a mutation in codons 90–93 (−8 bp) (-AGCTTCGG) which was first found in Fujian Province, China. Furthermore, 67 cases of hemoglobinopathy, such as Hb Q-Thailand, Hb New York, Hb J-Bangkok, Hb G-Coushatta, Hb G-Honolulu, Hb Dunn and Hb J-Wenchang-Wuming, were confirmed by DNA sequencing and/or second-generation sequencing after capillary electrophoresis indicating abnormal hemoglobin but the negative results of the genotype test in our study. Several pathological variants may cause moderate or severe anemia if associated with thalassemia. Therefore, combined screening including hematological indices and capillary electrophoresis should not be ignored.
The main limitation of this study was the bias caused by incomplete data from retrospective analysis. Some participants with normal result of initial screening test did not undergo further molecular diagnosis and were excluded from the study. This limitation indicates that the prevalence found in our study may not represent the true situation of our region. However, almost all participants suspected of having alpha-thalassemia were subjected to genetic analysis, so the proportion of each genotype could basically represent the distribution in the local population. Furthermore, our results could reflect the reality of the three indicators in practice since the data were derived from routine standard practice.
Conclusion
In summary, our results indicated that the different genotypes causing alpha-thalassemia affected the MCV, MCH and HbA2 to varying degrees. These findings may help clinicians relate phenotypes to genotypes for better genetic counselling.
Disclosure
The authors declare no conflicts of interest.
Additional information
Funding
References
- Muncie HL Jr, JS Campbell. Alpha and Beta Thalassemia . Am Fam Physician. 2009;80(4):339–344.
- Dong BQ, Chen BY, Liang QY, et al. Study on the characteristics of major birth defects in 1.69 million cases of fetus in Guangxi Zhuang Autonomous Region. Zhonghua Liu Xing Bing Xue Za Zhi. 2019;40(12):1554–1559.
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79(8):704–712.
- Baysal E. Alpha-thalassemia syndromes in the United Arab Emirates. Hemoglobin. 2011;35(5–6):574–580. doi:10.3109/03630269.2011.634698
- Alkindi S, Al Zadjali S, Al Madhani A, et al. Forecasting hemoglobinopathy burden through neonatal screening in Omani neonates. Hemoglobin. 2010;34(2):135–144. doi:10.3109/03630261003677213
- He S, Li J, Li DM, et al. Molecular characterization of alpha- and beta-thalassemia in the Yulin region of Southern China. Gene. 2018;655:61–64. doi:10.1016/j.gene.2018.02.058
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155–167. doi:10.1016/S0140-6736(17)31822-6
- Xu LP, Huang HL, Wang Y, et al. Molecular epidemiological analysis of alpha- and beta-thalassemia in Fujian province. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2013;30(4):403–406.
- Waye CD, Waye JS. Hydrops fetalis caused by alpha-thalassemia: an emerging health care problem. Blood. 1998;91(7):2213–2222. doi:10.1182/blood.V91.7.2213
- Choudhry VP. Quality of life in thalassemia major. Indian J Pediatr. 2018;85(11):957–958. doi:10.1007/s12098-018-2792-z
- Yao XY, Yu J, Chen SP, et al. Prevalence and genetic analysis of alpha-thalassemia and beta-thalassemia in Chongqing area of China. Gene. 2013;532(1):120–124. doi:10.1016/j.gene.2013.09.031
- Angelucci E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99(5):811–820. doi:10.3324/haematol.2013.099747
- Algiraigri AH, Wright NAM, Paolucci EO, A Kassam Hydroxyurea for nontransfusion-dependent beta-thalassemia: a systematic review and meta-analysis. Hematol Oncol Stem Cell Ther. 2017;10(3):116–125. doi:10.1016/j.hemonc.2017.02.002
- Srivastava A, Shaji RV. Cure for thalassemia major - from allogeneic hematopoietic stem cell transplantation to gene therapy. Haematologica. 2017;102(2):214–223. doi:10.3324/haematol.2015.141200
- Ferrari G, CavazzanaF Mavilio M. Gene therapy approaches to hemoglobinopathies. Hematol Oncol Clin North Am. 2017;31(5):835–852. doi:10.1016/j.hoc.2017.06.010
- Ryan K, Bain BJ, Worthington D, et al. Significant haemoglobinopathies: guidelines for screening and diagnosis. Br J Haematol. 2010;149(1):35–49. doi:10.1111/j.1365-2141.2009.08054.x
- Xu C, Liao B, Qi Y, et al. Analysis of gene mutation types of alpha- and beta-thalassemia in Fuzhou, Fujian Province in China. Hemoglobin. 2018;42(3):143–147. doi:10.1080/03630269.2018.1496096
- Chong SS, Boehm CD, Cutting GR, Higgs DR. Simplified multiplex-PCR diagnosis of common Southeast Asian deletional determinants of alpha-thalassemia. Clin Chem. 2000;46(10):1692–1695. doi:10.1093/clinchem/46.10.1692
- Wang W, Chan AY, Chan LC, MaS ES, Chong S. Unusual rearrangement of the alpha-globin gene cluster containing both the -alpha3.7 and alphaalphaalphaanti-4.2 crossover junctions: clinical diagnostic implications and possible mechanisms. Clin Chem. 2005;51(11):2167–2170. doi:10.1373/clinchem.2005.054189
- Zeng YT, Huang SZ. Alpha-globin gene organisation and prenatal diagnosis of alpha-thalassaemia in Chinese. Lancet. 1985;1(8424):304–307.
- Lal A, Goldrich ML, Haines DA, et al. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med. 2011;364(8):710–718. doi:10.1056/NEJMoa1010174
- Vichinsky E. Advances in the treatment of alpha-thalassemia. Blood Rev. 2012;26(Suppl 1):S31–34. doi:10.1016/S0268-960X(12)70010-3
- Lin M, Wang Q, Zheng L, et al. Prevalence and molecular characterization of abnormal hemoglobin in eastern Guangdong of southern China. Clin Genet. 2012;81(2):165–171. doi:10.1111/j.1399-0004.2011.01627.x
- Zheng CG, Liu M, Du J, et al. Molecular spectrum of alpha- and beta-globin gene mutations detected in the population of Guangxi Zhuang Autonomous Region, People’s Republic of China. Hemoglobin. 2011;35(1):28–39. doi:10.3109/03630269.2010.547429
- Phanmany S, Chanprasert S, Munkongdee T, Svasti S, K Leecharoenkiat. Molecular prevalence of thalassemia and hemoglobinopathies among the Lao Loum Group in the Lao People’s Democratic Republic. Int J Lab Hematol. 2019;41(5):650–656. doi:10.1111/ijlh.13080
- Bain BJ. Haemoglobinopathy diagnosis: algorithms, lessons and pitfalls. Blood Rev. 2011;25(5):205–213. doi:10.1016/j.blre.2011.04.001
- Karnpean R, Pansuwan A, Fucharoen G, Fucharoen S. Evaluation of the URIT-2900 automated hematology analyzer for screening of thalassemia and hemoglobinopathies in Southeast Asian populations. Clin Biochem. 2011;44(10–11):889–893. doi:10.1016/j.clinbiochem.2011.04.009