
Abstract
Purpose
This study aimed to evaluate the functions of critical N6-methyladenosine (m6A)-related long non-coding RNAs (lncRNAs) and their correlations with immunotherapeutic targets in clear cell renal cell carcinoma (ccRCC).
Methods
m6A-related lncRNAs were analyzed using the dataset from The Cancer Genome Atlas database via Pearson correlation analysis. Then, their prognostic functions in patients with ccRCC were determined via univariate Cox analysis. A prognostic m6A-related lncRNA signature (MRLS) in ccRCC was established using the least absolute shrinkage and selection operator (LASSO) Cox regression model. In addition, the correlations between these prognostic m6A-related lncRNAs with immune checkpoints were further evaluated in clinical samples.
Results
MRLS was established by the LASSO Cox regression model on the basis of seven prognostic m6A-related lncRNAs. The risk score for each patient was calculated using the MRLS model, and the patients were further stratified into high- and low-risk subgroups. The MRLS model was validated with a robust prognostic ability by the stratification analysis. On the basis of age, grade, stage, and risk score, a nomogram was developed with a strong reliability in forecasting the overall survival percentages of the patients with ccRCC. Moreover, seven prognostic m6A-related lncRNAs enrolled in the MRLS model were found to be correlated with various immunotherapeutic targets, namely, PD-1, PD-L1, CTLA4, and LAG3, and the expression levels of which in the high-risk subgroup were significantly higher than those in the low-risk subgroup. The significant correlations between LINC00342 and the aforementioned immunotherapeutic targets were also confirmed in clinical samples.
Conclusion
In this study, seven m6A-related lncRNAs were identified as potential biomarkers for forecasting the prognosis of patients with ccRCC and evaluating the efficacy of immunotherapy for these patients. Furthermore, a prognostic and predictive MRLS model with a high reliability was constructed to predict the overall survival probability of patients with ccRCC.
Introduction
Renal cell carcinoma (RCC), one of the most frequent cancer types in the urinary system,Citation1 accounts for approximately 3% of all kinds of cancer diagnoses and cancer-specific mortalities worldwide.Citation2,Citation3 The incidence rate of RCC has increased annually by 2% worldwide during the last two decades,Citation2,Citation3 translating into an average increase of 295,000 newly diagnosed RCC cases and 134,000 cancer-related deaths every year.Citation1,Citation4 Clear cell RCC (ccRCC), the most common pathological type of RCC, accounts for approximately 75% of RCCs.Citation5 Notably, the five-year survival rate of patients with RCC in clinical stage I is about 80–95%, but this rate dramatically decreases to less than 10% in patients with stage IV RCC,Citation6 whose average overall survival length is only 10–15 months, indicating the great clinical significance of early diagnosis and accurate prognosis of patients with RCC to formulate the optimal therapeutic regimens and improve the patients’ quality of life. RCC is widely perceived as an immunogenic disease, but it is known to mediate immune dysfunction to a large extent by stimulating the infiltration of immune-inhibitory cells into tumor microenvironment.Citation7–Citation9 Several underlying mechanisms have been developed to interpret the invalidity of the antitumor immune response caused by these multiple tumor-infiltrating cells, including inactivation of effector T cells and antigen-presenting cells via upregulation of suppressive factors, such as checkpoint molecules.Citation8
Targeting immune suppression by inhibiting checkpoints has achieved positive therapeutic effects on some patients harboring ccRCC. To date, combinatorial methods containing checkpoint blockades are the standard choice for patients with advanced ccRCC.Citation8 However, a substantial proportion of patients still do not benefit from checkpoint blockades. Under the circumstances, the identification of reliable biomarkers of response to checkpoint inhibition is of great clinical significance to facilitate improvements in the clinical efficacy of these therapies.
Many studies demonstrated that the immune checkpoints, including CTLA4, PD-1, PD-L1, and LAG3, could be effective targets for stimulating T cell-mediated antitumor immunity.Citation8 However, up to date, no clinical biomarkers can accurately predict the prognosis of patients with ccRCC and evaluate the efficacy of immune checkpoint inhibitors. Hence, researchers were striving to break ground in intricate biological processes and molecular mechanisms of ccRCC to identify the biomarkers as potential prognosis factors in ccRCC and therapeutic targets.
Many biological processes, including the regulation of RNA post-transcriptional stability,Citation10 localization,Citation11 translocation, slicing,Citation12 and translation,Citation13 are modulated by various RNA modifications, among which RNA methylation accounts for over 60%.Citation13 N6-methyladenosine (m6A) modification, an invertible and dynamical RNA epigenetic process regulated by m6A regulators containing “writers” (methyltransferases), “readers” (signal transducers), and “erasers” (demethylases),Citation14 is the most prevalent epigenetic methylation of messenger RNAs (mRNAs) and noncoding RNAs (ncRNAs).Citation15,Citation16 Recent studies reported that m6A modification participates in the regulation of oncogenesis and tumor progression in various kinds of malignancies,Citation17–Citation19 including RCC.Citation20,Citation21 For example, lncRNA DMDRMR cooperating with IGF2BP3 exhibits essential oncogenic roles in ccRCC in an m6A-dependent manner.Citation22 Zhang et alCitation23 found that METTL14-mediated m6A modification negatively regulates the mRNA stability of bromodomain PHD finger transcription factor to drive lung metastasis in patients with RCC. In addition, both Zhou et alCitation21 and Zhang et alCitation20 suggested that the genetic alterations of m6A regulators in RCC are strongly related to the potential prognosis of this disease.
The pullulation of transcriptome sequencing over the past decade has indicated that more than 70% of the genome is transcribed into RNAs, most of which are ncRNAs.Citation24 As a major type of ncRNAs equipped with more than 200 nucleotides in length, long ncRNAs (lncRNAs) are also considered to play a vital role in various biological processes with the prosperity of a wide range of researches in lncRNAs.Citation15,Citation24,Citation25 lncRNAs exhibit diverse biological functions through the regulation of gene expression levels and functions at the transcriptional, translational, and post-translational levels.Citation26 Dysregulation of lncRNAs is widely involved in the pathogenesis of many malignancies,Citation24,Citation26,Citation27 including ccRCC.Citation28 Accumulating evidences indicate that lncRNAs may serve as both prognostic biomarkers and therapeutic targets in many cancer types, especially in cancers in the urinary tract.Citation24 However, the connection between m6A regulators with the dysregulation of lncRNAs in cancers remains unclear. Thus far, no study has explored the underlying mechanisms by which m6A modifications contribute to lncRNA-dependent ccRCC genesis and evolution.
To close this gap of cogent evidence on the utility of m6A-related lncRNAs in forecasting the prognosis of patients with ccRCC and their potential as therapeutic targets for this disease, we determined the prognostic value of m6A-related lncRNAs via bioinformatics and statistical analyses of the data of patients with ccRCC from The Cancer Genome Atlas (TCGA) database. Univariate Cox regression analysis revealed 39 m6A-related lncRNAs with prognostic value in patients with ccRCC. Afterward, a prognostic m6A-related lncRNA signature (MRLS) was developed in accordance with the ability of seven m6A-related lncRNAs to predict the overall survival of patients with ccRCC. Then, an accurate nomogram was formulated with a high reliability to evaluate the overall survival of patients bearing ccRCC. The correlations of the seven critical m6A-related lncRNAs with immune checkpoints and the expression levels of immune checkpoint molecules in the high- and low-risk subgroups were evaluated to assess the efficacy of immune checkpoint inhibitors for the treatment of ccRCC. Furthermore, in order to evaluate the clinical values of the MRLS model, the focus next turned toward the expressions of some critical m6A-related lncRNAs immersed in the model and the connections between critical m6A-related lncRNAs and immune checkpoints in the TCGA samples and 20 ccRCC tissues and paired adjacent normal tissues of our own cohort.
Materials and Methods
Datasets and m6A-Related Genes
The Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/) and the cBioPortal website (https://www.cbioportal.org/) were employed to acquire the mRNA expression files (fragments per kilobase of transcript per million mapped reads [FPKM] normalized) and the corresponding clinicopathological data, respectively. Only ccRCC patients with data on overall survivals were enrolled in the final analysis to attenuate the impact of statistical bias on the results. The inclusion criteria were as follows: (1) patients were definitely diagnosed with ccRCC; (2) patients were followed up for overall survival. The exclusion criteria were as follows: (1) existences of other primary cancers; (2) patients without any follow-up data. Finally, 530 patients harboring ccRCC were extracted from the TCGA datasets. Thirty-five m6A RNA methylation regulators were selected on the ground of the results from previously published studies.Citation13,Citation19,Citation29 The expression matrices of 35 m6A-related genes were also collected from the TCGA datasets.
Annotation of lncRNAs
The lncRNAs extracted from the TCGA dataset were annotated in compliance with the lncRNA annotation file of the Genome Reference Consortium Human Build 38, which was acquired from the GENCODE website (https://www.gencodegenes.org/human/). A total of 10,876 lncRNAs were recognized in the TCGA dataset after the ensemble IDs of the genes were identified. Eight transcript forms covering incRNA, antisense, processed transcript, sense intronic, 3ʹ overlapping ncRNA, sense overlapping, and macro lncRNA of lncRNAs were included in the analysis.
Bioinformatics Analysis
The m6A-related lncRNAs were screened from the dataset via Pearson correlation analysis (Pearson R > 0.7 and p value < 0.001). Prognostic m6A-related lncRNAs were identified via univariate Cox regression analysis.
On the basis of the expression levels of the identified prognostic m6A-related lncRNAs, the patients bearing ccRCC were stratified into subgroups via consensus clustering by using the “ConsensusClusterPlus” package.Citation13 Survival probability and KEGG pathway analyses were also carried out between the two subgroups by using R software 3.6.1. The PD-L1 expression and immunescore calculated for each patient were compared between different clusters. Furthermore, the relationship of the prognostic m6A-related lncRNAs identified herein with PD-L1 was evaluated via Pearson correlation analysis. The compositions of various immune cells within tumor microenvironment were also compared between different clusters.
Least absolute shrinkage and selection operator (LASSO) Cox regression was performed with the package “glmnet”Citation15 to establish the optimal prognostic MRLS for patients afflicted with ccRCC that involved the seven m6A-related lncRNAs, which were perceived as indispensable biomarkers. MRLS risk score was calculated using the formula , where Expri represents the relative expression of m6A-related lncRNA i in the signature, and coefi indicates the coefficient of lncRNA I. The entire TCGA cohort was randomly grouped into training and test sets at a ratio of 1:1. Afterward, the patients were divided into high- and low-risk arms at the median cut-off on the basis of MRLS risk scores. The MRLS risk scores were further validated in the test set via survival analysis, risk plot, and ROC curve. In addition, the correlations between risk scores and clinicopathological variables were investigated using R software 3.6.1.
Independent prognostic factors were confirmed by both univariate and multivariate Cox regression analyses with the combination of MRLS and clinical variables in the training and test sets. After that, a predictive nomogram was constructed on the basis of the selected independent prognostic variables by using R software 3.6.1. Three packages containing “rms”, “nomogramEx”, and “regplot” were employed to delineate the nomogram. Furthermore, the prognostic and predictive accuracy of the nomogram was evaluated with calibration curves.
The correlations of the seven critical m6A-related lncRNAs with immune checkpoints, including CTLA4, PD-1, PD-L1, and LAG3, were assessed via Pearson correlation analysis. Furthermore, the difference in the expression of each of these immune checkpoint molecules was analyzed between the high- and low-risk subgroups. In addition, the CIBERSORT algorithm, which is a tool for the deconvolution of the expression matrix of human immune cell subtypes according to the principle of linear support vector regression, was employed to evaluate the immune cell infiltration of the high- and low-risk subgroups.
The relative expression levels of LINC00342 in ccRCC and normal tissues, as well as in different subgroups, were examined and analyzed on the basis of different clinical characteristics by using the RNA sequencing data of TCGA samples in the UALCAN database. In addition, the connections between LINC00342 and the immune checkpoints were assessed by analyzing the RNA sequencing data from GEPIA. Furthermore, the expression levels of LINC00342, PD-1, PD-L1, LAG3, and CTLA4, were detected in the 20 ccRCC tissues and paired adjacent tissues of our own cohort to evaluate the correlations between LINC00342 and the immune checkpoints via RT-qPCR.
RNA Extraction and RT-qPCR
The use of clinical samples was approved by the Ethics Committee of the Second Affiliated Hospital of Nanchang University. The total RNA from the ccRCC tissues and adjacent normal tissues was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) in compliance with the manufacturer’s instructions. The reverse transcriptions of the RNA samples were completed with PrimeScript RT reagent kit (TaKaRa, Dalian, China). RNA expression was detected using SYBR Premix Ex Taq II Kit (TaKaRa, Dalian, China) on a StepOne qPCR platform (Applied Biosystems, Foster City, CA, USA). β-actin was used as an internal reference gene. The 2−ΔΔCt method was employed to quantify the relative expression levels. Each analysis was performed in triplicate. The primers used were as follows: LINC00342, 5ʹ-CCCAAAGCAGTCCTTCACTACA-3ʹ (forward) and 5ʹ-CTGCAGTTCACTCTGCTGCTT-3ʹ (reverse); β-actin, 5ʹ-CATGTACGTTGCTATCCAGGC-3ʹ (forward) and 5ʹ-CTCCTTAATGTCACGCACGAT-3ʹ (reverse); PD-1 (PDCD1), 5ʹ-CAGTTCCAAACCCTGGTGGT-3ʹ (forward) and 5ʹ-GGCTCCTATTGTCCCTCGTG-3ʹ (reverse); PD-L1 (CD274), 5ʹ-TACTGGCATTTGCTGAACGC-3ʹ (forward) and 5ʹ-TCCAGATGACTTCGGCCTTG-3ʹ (reverse); CTLA4, 5ʹ-AGCCACAGCTGGTGGTATCT-3ʹ (forward) and 5ʹ-GAATTGGGCCCATCGAACTG-3ʹ (reverse); LAG3, 5ʹ-CGACTAGAGGATGTGAGCCAG-3ʹ (forward) and 5ʹ-GATCCAGGTGACCCAAAGGAT-3ʹ (reverse).
Statistical Analyses
The statistical correlation between two variables was estimated via Spearman correlation analysis. Hazard ratios (HRs) with 95% confidence intervals (CIs) were calculated for each variable and independent prognostic factors were identified via univariate and multivariate Cox regression analyses. Statistical significance for the parameters between two groups was assessed by independent t-test. Corrections between the MRLS risk scores and clinicopathological variables were calculated via χ2 test. The survival probabilities of patients with ccRCC in the two subgroups were evaluated via the Kaplan–Meier method with Log rank test by utilizing the “survminer” package in R. A nomogram and calibration curves were developed in line with the suggestions proposed by Iasonos et al.Citation24,Citation30 The correlations of the seven critical m6A-related lncRNAs with the immune checkpoint molecules were validated via Pearson correlation analysis. All statistical analyses were conducted using R software 3.6.1. Statistical significance was defined a two-sided p value of < 0.05.
Results
A total of 14,086 lncRNAs and the expression matrices of 35 m6A-related genes were gathered from the TCGA dataset with the file downloaded from the GENCODE website (). Only when the expression value of a lncRNA was significantly associated with one or more of the 35 m6A-related genes (Pearson R > 0.7 and p value < 0.001) would the lncRNA be regarded as a m6A-related lncRNA. Identified from the TCGA dataset after applying Pearson correlation analysis were 356 m6A-related lncRNAs (), of who 39 were defined as prognostic m6A-related lncRNAs via univariate Cox regression analysis (). The expression levels of these m6A-related lncRNAs in normal and tumor tissues are displayed in .
Figure 1 (A) The components of m6A RNA methylation regulators in writer-, reader-, and eraser-complex; (B) network of m6A-related lncRNAs and m6A-related genes; (C) forest plot of 39 m6A-related prognostic lncRNAs; (D) the expression levels of 39 prognostic m6A-related lncRNAs in renal cell carcinoma tissue and normal tissue.
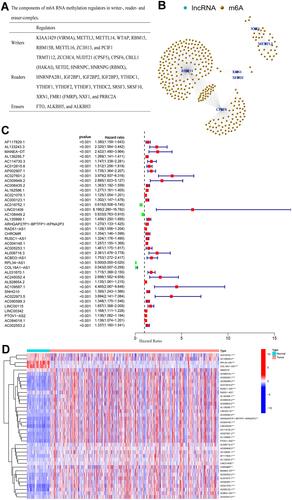
The impacts of the m6A-related lncRNAs identified herein on ccRCC progression were evaluated by dividing the tumorous tissues into different subgroups via consensus clustering according to the expression of the 39 prognostic m6A-related lncRNAs. The cumulative distribution function (CDF) of the consensus clustering with k = 2–9 and the relative change in area under the curve (AUC) increment are delineated in . Given the maximum AUC increment and the divergence in the expression corrections of the prognostic m6A-related lncRNAs between within groups and between groups, k = 2 was determined as the number of clusters (). The tracking plot of the subgroups for 2 to 9 clusters is presented in . The consensus matrix for optimal k = 2 is delineated in . The difference between these two clusters was discriminated by evaluating the survival probabilities of patients with ccRCC via survival analysis. Results showed that the patients in cluster 2 had better overall survivals than those in cluster 1 (p < 0.001) (HR: 1.860; 95% CIs: 1.375–2.516) (). The median PD-L1 expression in cluster 2 was significantly higher than that in cluster 1 (p < 0.01) (), but no significant difference was found between the two subgroups in terms of the median immunescore (p = 0.31) (). Intriguingly, CD8 T cells were filled in cluster 2 (). Moreover, 16 of the 39 prognostic m6A-related lncRNAs were found to be correlated with PD-L1 expression (). Gene set enrichment analysis (GSEA) revealed several enriched tumor signaling pathways in cluster 2, including the MTOR, Notch, phosphatidylinositol, and VEGF signaling pathways (–). These results demonstrated that the stratification of the prognostic m6A-related lncRNAs identified herein was a clinically meaningful method, and their expression was strongly associated with the prognosis of patients with ccRCC.
Figure 2 (A) Consensus CDF for k = 2 to 9; (B) area under CDF curve increment for k = 2 to 9; (C) tracking plot for k = 2 to 9; (D) consensus matrix for optimal k = 2; (E) Kaplan–Meier overall survival curves for patients in distinct clusters (p < 0.001); (F) PD-L1 expression levels in distinct clusters (** p < 0.01); (G) immunescore for each patient in distinct clusters (p = 0.31); (H) various types of immune cells in distinct clusters; (I) Correlation matrix of PD-L1 and 39 prognostic m6A-related lncRNAs; (J–M) gene set enrichment analysis indicating that tumor signal pathways were enriched in the cluster 2 subgroup.
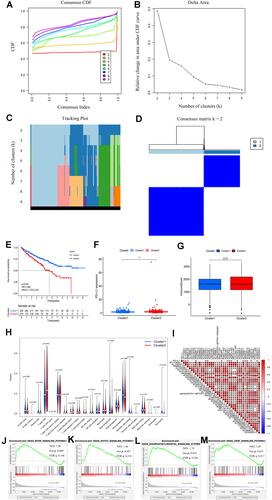
An MRLS model for evaluating the survival probabilities of patients with ccRCC was developed via LASSO Cox analysis on the basis of the 39 prognostic m6A-related lncRNAs identified herein. Seven lncRNAs coupled with their coefficients stuck out (–). Each patient in the TCGA dataset was valued with a risk score on the basis of the coefficients for the seven lncRNAs. The patients in the low-risk subgroup had better overall survivals than those in the high-risk subgroup in both the training (p < 0.001; HR: 1.644; CIs: 1.430–1.889) and test (p < 0.001; HR: 1.512; CIs: 1.345–1.699) sets ( and , respectively). The risk plots of the training and test sets corroborated the prognostic reliability of the risk scores ( and ). The AUCs of time-dependent receiver operating characteristic (ROC) curves revealed that the accuracy of prediction was 0.724 (95% CIs: 0.688–0.760) () and 0.672 (95% CIs: 0.638–0.706) () in the training and test sets, respectively. The relative expressions of seven critical m6A-related lncRNAs in the high- and low-risk subgroups of the training and test sets are shown in and , respectively. All the immune checkpoint molecules, including PD-L1 (), PD-1 (), CTLA4 (), and LAG3 (), were related to more than three of the seven critical m6A-related lncRNAs, suggesting the strong connections between the immune checkpoints with the critical m6A-related lncRNAs. The Kaplan–Meier analyses revealed that the expression levels of all the seven critical m6A-related lncRNAs were significantly correlated with the prognosis of patients with ccRCC (all p values < 0.01) (–).
Figure 3 (A–B) Least absolute shrinkage and selection operator (LASSO) regression, calculating the minimum criteria; (C) coefficients of selected lncRNAs; (D) Kaplan–Meier curves showed that the high-risk subgroup had worse overall survival than the low-risk subgroup in the training set; (E) distributions of risk scores and survival status of renal cell carcinoma patients in the training set; (F) receiver operating characteristic (ROC) curves of m6A-related lncRNAs significance for predicting the overall survival in the training set; (G) Kaplan–Meier curves showed that the high-risk subgroup had worse overall survival than the low-risk subgroup in the test set; (H) distributions of risk scores and survival status of renal cell carcinoma patients in the test set; (I) receiver operating characteristic (ROC) curves of m6A-related lncRNAs significance for predicting the overall survival in the test set; (J) heatmap of the expression of m6A-related lncRNAs in the high- and low-risk subgroups of the training set; (K) heatmap of the expression of m6A-related lncRNAs in the high- and low-risk subgroups of the test set; (L) correlation matrix of PD-L1 and 7 critical prognostic m6A-related lncRNAs; (M) correlation matrix of PD-1 and 7 critical prognostic m6A-related lncRNAs; (N) correlation matrix of CTLA4 and 7 critical prognostic m6A-related lncRNAs; (O) correlation matrix of LAG3 and 7 critical prognostic m6A-related lncRNAs; (P–V) the expression levels of AC108449.2, AF117829.1, AL008718.3, AL031670.1, CHROMR, COL18A1−AS1, and LINC00342 were significantly correlated with the prognosis of patients with ccRCC (all p values <0.01).
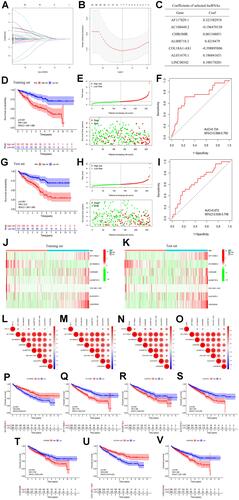
The connections of risk scores, clinicopathological variables, and expression levels of constituent m6A-related lncRNAs are presented in , which suggests that the patients in the high-risk subgroup were significantly related to a higher mean age, high mean immunescore, and a higher rate of T3-4 or G4 disease than those in the low-risk subgroup. Furthermore, the risk score was correlated with older age, G3-4 disease, and cluster 2 (–). Patients in the high-risk subgroup had a worse overall survival than those in the low-risk subgroup in different stratifications (–). These results demonstrated that the prognostic signature combining the seven critical m6A-related lncRNAs could independently forecast the prognosis of patients with ccRCC. In addition, the number of memory B cells, CD8 T cells, follicular helper T cells, and T regulatory cells (Tregs) in the high-risk subgroup was significantly higher than that in the low-risk subgroup (p = 0.008, p = 0.003, p < 0.001, and p < 0.001, respectively) ().
Figure 4 (A) Heatmap of the associations between the expression levels of the 7 critical m6A-related lncRNAs and clinicopathological features in the entire cohort; (B–I) risk scores of renal cell carcinoma patients with different clinicopathological features (including age, gender, grade, cluster, T stage, N stage, M stage, clinical stage); (J–U) Kaplan–Meier survival stratification analyses in multiple subgroups of renal cell carcinoma patients (including patients aged ≤65 or >65 years, male or female patients, patients with G1-2 or G3-4 tumors, patients with T1-2 or T3-4 tumors, patients with N0 or N1 diseases, patients with M0 or M1 diseases); (V) various types of immune cells in the low- and high-risk subgroups.
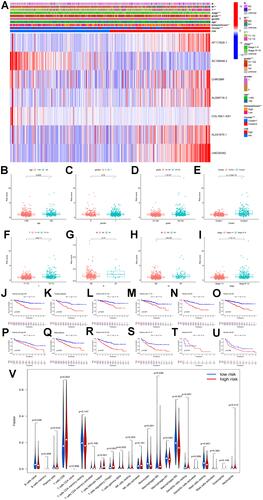
After all the clinicopathological features were integrated with MRLS, univariate COX regression analysis revealed that the variables, including patient age, tumor grade and stage, and risk score, were significantly predictive for patients with ccRCC in the training () and test sets (). However, multivariate COX regression analysis indicated that tumor grade was no longer an independent factor for forecasting the prognosis of patients with ccRCC ( and ). The survival probability of each patient was evaluated with a total point from each variable by using the nomogram established on the basis of these three independent parameters (ie, patient age, tumor stage, and risk score) (). The higher the total point, the lower the survival possibility of patient bearing ccRCC. The calibration curves regarding the 1-year (), 2-year (), and 3-year () survival rates revealed that the predictive ability of the nomogram was similar to that of the ideal model, disclosing the high reliability of the nomogram for clinical utility.
Figure 5 (A and B) Univariate and multivariate Cox analyses of risk score along with clinicopathological features (including age, gender, grade, and stage) in the training set; (C and D) univariate and multivariate Cox analyses of risk score along with clinicopathological features (including age, gender, grade, and stage) in the test set; (E) nomogram based on risk score, age, grade, and stage in the entire cohort; (F–H) calibration plots of the nomogram for predicting the probability of overall survival at 1, 2, and 3 years in the TCGA dataset; (I–L) the expression levels of PD-L1, PD-1, CTLA4, and LAG3 in the high- and low-risk subgroup.
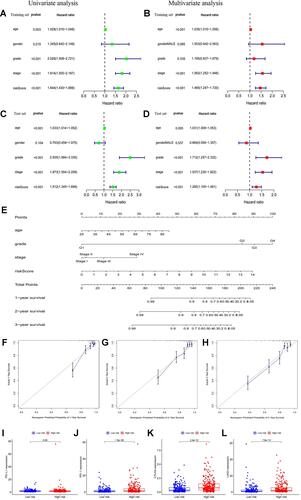
In addition, the expression levels of all immune checkpoint molecules, including PD-1 (), PD-L1 (), CTLA4 (), and LAG3 (), in the high-risk subgroup were significantly higher than those in the low-risk subgroup, suggesting the potential high efficacy of immune checkpoint inhibitors for the treatment of patients with a high risk score.
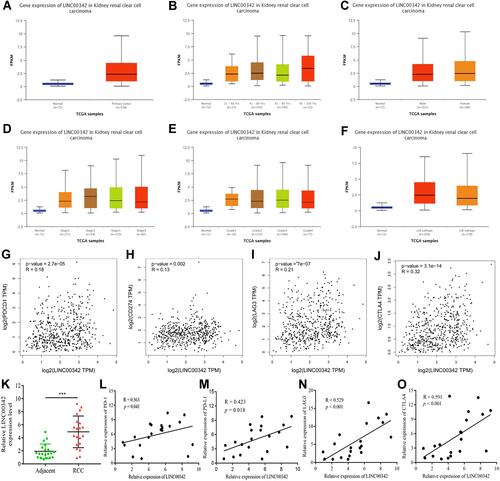
Consequently, the expression levels of LINC00342 in the ccRCC tissues were significantly increased than those in normal kidney tissues from the TCGA cohort (UALCAN) (). Furthermore, the relative expression levels of LINC00342 in the normal populations were significantly lower with that in ccRCC patients of any age at diagnosis, gender, and clinical stages (–). In the meanwhile, significant correlations were also detected between the expression levels of LINC00342 and pathological grades and subtypes ( and ). In addition, LINC00342 expression was positively correlated with immune checkpoints (PDCD1: R = 0.18, p = 2.7e-05; CD274: R = 0.13, p = 0.002; LAG3: R = 0.21, p = 7e-07; CTLA4: R = 0.32, p = 3.1e-14) (–). In our own cohort, RT-qPCR showed that the expression levels of LINC00342 in the ccRCC tissues were significantly higher than those in the adjacent normal tissues (p < 0.001) (). The positive correlations between LINC00342 and the immune checkpoints were also confirmed in our own cohort (PDCD1: R = 0.363, p = 0.041; CD274: R = 0.423, p = 0.018; LAG3: R = 0.529, p < 0.001; CTLA4: R = 0.591, p < 0.001) (–).
Figure 6 (A) The relative expressions of LINC00342 in normal individuals and renal cell carcinoma (RCC) patients (UALCAN); (B) the relative expressions of LINC00342 in normal individuals or in RCC patients with different ages (UALCAN); (C) the relative expressions of LINC00342 in normal individuals of either gender and in male or female RCC patients (UALCAN); (D) the relative expressions of LINC00342 in normal individuals or in RCC patients in stage 1, 2, 3, or 4 (UALCAN); (E) the relative expressions of LINC00342 in normal individuals or in RCC patients with Grade 1, Grade 2, Grade 3, and Grade 4 diseases (UALCAN); (F) the relative expressions of LINC00342 in normal individuals or in RCC patients with different tumor subtypes (UALCAN); (G–J) PD-1, PD-L1, LAG3, and CTLA4 genes positively correlated with LINC00342 in RCC patients (GEPIA); (K) the relative expressions of LINC00342 in 20 RCC tissues and paired adjacent normal tissues of our own cohort using qRT-PCR; (L–O) PD-1, PD-L1, LAG3, and CTLA4 genes positively correlated with LINC00342 in 20 RCC tissues and paired adjacent normal tissues of our own cohort using qRT-PCR.
Discussion
Increasing transcriptomic data stored in public databases, such as TCGA and Gene Expression Omnibus, have become available with the technical advancement in sequencing. By analyzing the profiles acquired from these publicly accessible databases or own prospectively maintained institutes, many studies found that m6A modification may serve as a leading factor contributing to cancer pathogenesis,Citation15 and the expression of ncRNAs, including microRNAs,Citation31 lncRNAs,Citation26 and circular RNAs,Citation24 could greatly impact tumorigenesis and cancer progression. However, the mechanism by which m6A modification promotes ccRCC progression in an lncRNA-dependent manner and the connections between m6A-related lncRNAs and immune checkpoints of the immunotherapy for ccRCC remain unclear. In this context, the TCGA dataset was reviewed to extract information regarding 530 ccRCC patients to identify prognostic m6A-related lncRNAs. Confirmed with prognostic significance in the TCGA dataset were 39 m6A-related lncRNAs, 7 of which were utilized to develop an MRLS model for evaluating the overall survival probabilities of patients with ccRCC. The stratification of these patients into high- and low-risk subgroups was done according to the risk score calculated based on the MRLS model. Patients in the low-risk subgroup exhibited an overall survival benefit compared with those in the high-risk subgroup, and GSEA analysis revealed that certain malignant pathways were enriched in the high-risk subgroup. Furthermore, multivariate Cox regression analysis confirmed MRLS as an independent biomarker for predicting the overall survival percentage of patients with ccRCC. A nomogram was constructed to predict the prognosis of these patients on the basis of three independent parameters, namely, patient age, tumor stage, and risk score. All the immune checkpoint molecules, including PD-1, PD-L1, CTLA4, and LAG3, were found to be strongly correlated with the seven critical prognostic lncRNAs and the risk score calculated based on MRLS, indicating the potential efficacy of immune checkpoint inhibitors for the treatment of high-risk patients.
In view of the potential function of lncRNAs in cancer pathogenesis, lncRNAs have been recently become a hot point in cancer research. Several lncRNAs, such as PCAT1,Citation32 LINC00467,Citation33 and CCAT1,Citation34 play a vital role in prostate cancer progression and invasion. The significantly upregulated lncRNAs CASC11,Citation35 RMRP,Citation36 and UCA1Citation24 enhance cell proliferation and migration in bladder cancer. The overexpression of the lncRNAs UCA1Citation28 and EGFR-AS1Citation37 also promotes cell growth and metastasis in renal cancer.
m6A RNA methylation, the most common modification in RNAs, especially in miRNAs and lncRNAs, is closely related to the pathogenesis of various kinds of cancers.Citation17–Citation19 An important underlying mechanism accounting for the effects of m6A regulators on cancer development is the modification of specific lncRNAs.Citation15 The lncRNA GAS5, which is negatively regulated by the m6A reader YTHDF3, can inhibit colorectal cancer cell proliferation by interacting with and triggering YAP phosphorylation and degradation.Citation38 Lan et alCitation39 found that KIAA1429 can facilitate liver cancer progression via m6A-dependent modification of the lncRNA GATA3. The onset and development of cancer disease could be affected by the m6A-dependent modification of lncRNAs.Citation15 Taking these evidences into accounts, it was reasonable to conclude that m6A modification could be targeted at specific lncRNAs to exert their functions. More attention should be devoted to investigating the interactions and functions of m6A modification and lncRNAs to identify prognostic biomarkers or therapeutic targets of cancers.
Notably, the immune escape mediated by the complex tumor microenvironment could result in the inefficiency of immunotherapy.Citation40 Many lncRNAs are reportedly upregulated during the development, differentiation, and activation of immune cells, including macrophages, dendritic cells, neutrophils, T cells, B cells, and bone marrow mesenchymal stem cells.Citation41 lncRNAs are involved in various processes of the immune response in the tumor microenvironment, as well as in the activation of tumor immunosuppression and immunotherapeutic response in various cancers, such as RCC,Citation42,Citation43 endometrial cancer,Citation44 and liver cancer.Citation45 Memory B cells, T follicular helper cells, and Tregs are important factors contributing to worse outcomes in patients suffering from ccRCC.Citation46 In this study, the relative expression levels of PD-1, PD-L1, CTLA-4, and LAG3 were significantly upregulated in the high-risk patients, who were more likely to achieve clinically positive responses to immunotherapy. The risk score was in a positive connection with the infiltration of memory B cells, CD8 T cells, T follicular helper cells, and Tregs, indicating that m6A-related lncRNAs participate in regulating the immune microenvironment, a presumption that may help in interpreting the worse outcomes of ccRCC patients in the high-risk subgroup.
The MRLS model was constructed on the basis of seven prognostic m6A-related lncRNAs, namely, AF117829.1, AC108449.2, CHROMR, AL008718.3, COL18A1−AS1, AL031670.1, and LINC00342, some of which had been reported to be in close connection with cancer pathogenesis. LINC00342 is overexpressed in nonsmall cell lung cancerCitation47,Citation48 and colorectal cancer,Citation49,Citation50 and it promotes cancer proliferation and progression as a competing endogenous RNA and functions as a potential biomarker of the prognosis of patients bearing these diseases. Wang et alCitation51 found that COL18A1−AS1 is strongly associated with the overall survival of patients with ccRCC. The lncRNA CHROME is a central component of the noncoding RNA regulatory network maintaining cholesterol homeostasis,Citation52,Citation53 whose imbalance is the underlying factor of several diseases, such as cancers.Citation54 However, few studies have investigated the impacts of these lncRNAs on ccRCC initiation and progression. Furthermore, studies on the interactions of lncRNAs and m6A-related genes are even scarcer. In this study, prognostic m6A-related lncRNAs were identified, and their impacts on the survival probability of patients with ccRCC and cancer development were evaluated.
Of note, several limitations cannot be ignored in this study. Our conclusions were drawn on the basis of the dataset extracted from one public database, namely TCGA, and the prognostic m6A-related lncRNAs selected herein should be further confirmed with other independent ccRCC cohorts. In addition, the interactions and functions of these prognostic lncRNAs and m6A-related genes were evaluated via bioinformatics analysis and thus should be validated by in vitro and in vivo experiments.
Conclusions
This study comprehensively evaluated m6A-related lncRNAs in ccRCC. Seven m6A-related lncRNAs were confirmed as potential biomarkers for forecasting the prognosis of patients with ccRCC and evaluating the efficacy of immunotherapy for these patients. Furthermore, a prognostic and predictive MRLS with a high reliability was constructed to predict the overall survival probability of patients with ccRCC, thus providing insights into the potential roles of m6A-related RNAs in ccRCC tumorigenesis and development.
Abbreviations
m6A, N6-methyladenosine; lncRNA, long non-coding RNAs; RCC, renal cell carcinoma; LASSO, least absolute shrinkage and selection operator; ROC, receiver operating characteristics; AUC, area under the curve; MRLS, m6A-related lncRNAs significance; HR, Hazard ratios; CI, confidence interval; TCGA, The Cancer Genome Atlas; RT-qPCR, Real-Time Quantitative PCR; GEPIA, Gene Expression Profiling Interactive Analysis.
Data Sharing Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study was conducted in accordance with the Ethical Standards of the Institutional Ethics Committee of Second Affiliated Hospital of Nanchang University and with the 1964 Helsinki declaration and its later amendments or comparable Ethical Standards. All patients provided informed consent.
Disclosure
The authors declare that they have no competing interests.
Additional information
Funding
References
- Yang H, Xiong X, Li H. Development and interpretation of a genomic instability derived lncRNAs based risk signature as a predictor of prognosis for clear cell renal cell carcinoma patients. Front Oncol. 2021;11:678253. doi:10.3389/fonc.2021.678253
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
- Ljungberg B, Albiges L, Abu-Ghanem Y, et al. European Association of Urology guidelines on renal cell carcinoma: the 2019 update. Eur Urol. 2019;75(5):799–810. doi:10.1016/j.eururo.2019.02.011
- Hsieh JJ, Purdue MP, Signoretti S, et al. Renal cell carcinoma. Nat Rev. Dis Prim. 2017;3(1):17009. doi:10.1038/nrdp.2017.9
- Wang X, Kong W, Wang Y, et al. Analysis of CT, MRI imaging features of renal cell carcinoma with different histopathological types. J BUON. 2021;26(5):2053–2058.
- Zeng Q, Zhang W, Li X, Lai J, Li Z. Bioinformatic identification of renal cell carcinoma microenvironment-associated biomarkers with therapeutic and prognostic value. Life Sci. 2020;243:117273. doi:10.1016/j.lfs.2020.117273
- Hah YS, Koo KC. Immunology and immunotherapeutic approaches for advanced renal cell carcinoma: a comprehensive review. Int J Mol Sci. 2021;22(9):4452. doi:10.3390/ijms22094452
- Díaz-Montero CM, Rini BI, Finke JH. The immunology of renal cell carcinoma. Nat Rev Nephrol. 2020;16(12):721–735. doi:10.1038/s41581-020-0316-3
- Zambrana F, Carril-Ajuria L, Gómez de Liaño A, et al. Complete response and renal cell carcinoma in the immunotherapy era: the paradox of good news. Cancer Treat Rev. 2021;99:102239. doi:10.1016/j.ctrv.2021.102239
- Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42. doi:10.1038/nrm.2016.132
- Fustin JM, Doi M, Yamaguchi Y, et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013;155(4):793–806. doi:10.1016/j.cell.2013.10.026
- Molinie B, Wang J, Lim KS, et al. m(6)A-LAIC-seq reveals the census and complexity of the m(6)A epitranscriptome. Nat Methods. 2016;13(8):692–698. doi:10.1038/nmeth.3898
- Xu J, Liu Y, Liu J, et al. The identification of critical m(6)A RNA methylation regulators as malignant prognosis factors in prostate adenocarcinoma. Front Genet. 2020;11:602485. doi:10.3389/fgene.2020.602485
- Chen M, Nie ZY, Wen XH, Gao YH, Cao H, Zhang SF. m6A RNA methylation regulators can contribute to malignant progression and impact the prognosis of bladder cancer. Biosci Rep. 2019;39(12):12. doi:10.1042/BSR20192892
- Tu Z, Wu L, Wang P, et al. N6-methylandenosine-related lncRNAs are potential biomarkers for predicting the overall survival of lower-grade glioma patients. Front Cell Develop Biol. 2020;8:642. doi:10.3389/fcell.2020.00642
- Ma S, Chen C, Ji X, et al. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. 2019;12(1):121. doi:10.1186/s13045-019-0805-7
- Chen XY, Zhang J, Zhu JS. The role of m(6)A RNA methylation in human cancer. Mol Cancer. 2019;18(1):103. doi:10.1186/s12943-019-1033-z
- He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18(1):176. doi:10.1186/s12943-019-1109-9
- Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother. 2019;112:108613. doi:10.1016/j.biopha.2019.108613
- Zhang QJ, Luan JC, Song LB, et al. m6A RNA methylation regulators correlate with malignant progression and have potential predictive values in clear cell renal cell carcinoma. Exp Cell Res. 2020;392(1):112015. doi:10.1016/j.yexcr.2020.112015
- Zhou J, Wang J, Hong B, et al. Gene signatures and prognostic values of m6A regulators in clear cell renal cell carcinoma - a retrospective study using TCGA database. Aging. 2019;11(6):1633–1647. doi:10.18632/aging.101856
- Gu Y, Niu S, Wang Y, et al. DMDRMR-mediated regulation of m(6) A-modified CDK4 by m(6)A reader IGF2BP3 drives ccRCC progression. Cancer Res. 2021;81(4):923–934. doi:10.1158/0008-5472.CAN-20-1619
- Zhang C, Chen L, Liu Y, et al. Downregulated METTL14 accumulates BPTF that reinforces super-enhancers and distal lung metastasis via glycolytic reprogramming in renal cell carcinoma. Theranostics. 2021;11(8):3676–3693. doi:10.7150/thno.55424
- Cao R, Yuan L, Ma B, Wang G, Tian Y. Immune-related long non-coding RNA signature identified prognosis and immunotherapeutic efficiency in bladder cancer (BLCA). Cancer Cell Int. 2020;20(1):276. doi:10.1186/s12935-020-01362-0
- Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62. doi:10.1038/nrg.2015.10
- Chi Y, Wang J, Wang J, Yu W, Yang J. Long non-coding RNA in the pathogenesis of cancers. Cells. 2019;8(9):1015. doi:10.3390/cells8091015
- Sanchez Calle A, Kawamura Y, Yamamoto Y, Takeshita F, Ochiya T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018;109(7):2093–2100. doi:10.1111/cas.13642
- Wang W, Hu W, Wang Y, et al. Long non-coding RNA UCA1 promotes malignant phenotypes of renal cancer cells by modulating the miR-182-5p/DLL4 axis as a ceRNA. Mol Cancer. 2020;19(1):18. doi:10.1186/s12943-020-1132-x
- Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20(10):608–624. doi:10.1038/s41580-019-0168-5
- Iasonos A, Schrag D, Raj GV, Panageas KS. How to build and interpret a nomogram for cancer prognosis. J Clin Oncol. 2008;26(8):1364–1370. doi:10.1200/JCO.2007.12.9791
- Li R, Jiang S, Li W, et al. Exploration of prognosis-related microRNA and transcription factor co-regulatory networks across cancer types. RNA Biol. 2019;16(8):1010–1021. doi:10.1080/15476286.2019.1607714
- Shang Z, Yu J, Sun L, et al. LncRNA PCAT1 activates AKT and NF-κB signaling in castration-resistant prostate cancer by regulating the PHLPP/FKBP51/IKKα complex. Nucleic Acids Res. 2019;47(8):4211–4225. doi:10.1093/nar/gkz108
- Jiang H, Deng W, Zhu K, et al. LINC00467 promotes prostate cancer progression via M2 macrophage polarization and the miR-494-3p/STAT3 axis. Front Oncol. 2021;11:661431. doi:10.3389/fonc.2021.661431
- You Z, Liu C, Wang C, et al. LncRNA CCAT1 promotes prostate cancer cell proliferation by interacting with DDX5 and MIR-28-5P. Mol Cancer Ther. 2019;18(12):2469–2479. doi:10.1158/1535-7163.MCT-19-0095
- Luo H, Xu C, Le W, Ge B, Wang T. lncRNA CASC11 promotes cancer cell proliferation in bladder cancer through miRNA-150. J Cell Biochem. 2019;120(8):13487–13493. doi:10.1002/jcb.28622
- Cao HL, Liu ZJ, Huang PL, Yue YL, Xi JN. lncRNA-RMRP promotes proliferation, migration and invasion of bladder cancer via miR-206. J Cell Biochem. 2019;23(3):1012–1021.
- Wang A, Bao Y, Wu Z, et al. Long noncoding RNA EGFR-AS1 promotes cell growth and metastasis via affecting HuR mediated mRNA stability of EGFR in renal cancer. Cell Death Dis. 2019;10(3):154. doi:10.1038/s41419-019-1331-9
- Ni W, Yao S, Zhou Y, et al. Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m(6)A reader YTHDF3. Mol Cancer. 2019;18(1):143. doi:10.1186/s12943-019-1079-y
- Lan T, Li H, Zhang D, et al. KIAA1429 contributes to liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol Cancer. 2019;18(1):186. doi:10.1186/s12943-019-1106-z
- Osipov A, Saung MT, Zheng L, Murphy AG. Small molecule immunomodulation: the tumor microenvironment and overcoming immune escape. J Immunother Cancer. 2019;7(1):224. doi:10.1186/s40425-019-0667-0
- Safarzadeh E, Asadzadeh Z, Safaei S, et al. MicroRNAs and lncRNAs-A new layer of myeloid-derived suppressor cells regulation. Front Immunol. 2020;11:572323. doi:10.3389/fimmu.2020.572323
- Sun Z, Jing C, Xiao C, Li T. Long non-coding RNA Profile Study identifies an immune-related lncRNA prognostic signature for kidney renal clear cell carcinoma. Front Oncol. 2020;10:1430. doi:10.3389/fonc.2020.01430
- Zhang Z, Tang Y, Liu Y, et al. A novel immune-related lncRNA-based model for survival prediction in clear cell renal cell carcinoma. Journal of Immunol Res. 2021;2021:9921466. doi:10.1155/2021/9921466
- Liu J, Mei J, Wang Y, et al. Development of a novel immune-related lncRNA signature as a prognostic classifier for endometrial carcinoma. Int J Biol Sci. 2021;17(2):448–459. doi:10.7150/ijbs.51207
- Zhang Y, Zhang L, Xu Y, Wu X, Zhou Y, Mo J. Immune-related long noncoding RNA signature for predicting survival and immune checkpoint blockade in hepatocellular carcinoma. J Cell Physiol. 2020;235(12):9304–9316. doi:10.1002/jcp.29730
- Pan Q, Wang L, Chai S, Zhang H, Li B. The immune infiltration in clear cell renal cell carcinoma and their clinical implications: a study based on TCGA and GEO databases. J Cancer. 2020;11(11):3207–3215. doi:10.7150/jca.37285
- Chen QF, Kong JL, Zou SC, et al. LncRNA LINC00342 regulated cell growth and metastasis in non-small cell lung cancer via targeting miR-203a-3p. J Cell Biochem. 2019;23(17):7408–7418.
- Tang H, Zhao L, Li M, Li T, Hao Y. Investigation of LINC00342 as a poor prognostic biomarker for human patients with non-small cell lung cancer. J Cell Biochem. 2019;120(4):5055–5061. doi:10.1002/jcb.27782
- Miao Z, Liu S, Xiao X, Li D. LINC00342 regulates cell proliferation, apoptosis, migration and invasion in colon adenocarcinoma via miR-545-5p/MDM2 axis. Gene. 2020;743:144604. doi:10.1016/j.gene.2020.144604
- Shen P, Qu L, Wang J, et al. LncRNA LINC00342 contributes to the growth and metastasis of colorectal cancer via targeting miR-19a-3p/NPEPL1 axis. Cancer Cell Int. 2021;21(1):105. doi:10.1186/s12935-020-01705-x
- Wang J, Zhang C, He W, Gou X. Construction and comprehensive analysis of dysregulated long non-coding RNA-associated competing endogenous RNA network in clear cell renal cell carcinoma. J Cell Biochem. 2018;120(2):2576–293.
- Hennessy EJ, van Solingen C, Scacalossi KR, et al. The long noncoding RNA CHROME regulates cholesterol homeostasis in primate. Nat Metab. 2019;1(1):98–110. doi:10.1038/s42255-018-0004-9
- Le Bras A. The lncRNA CHROME regulates cholesterol homeostasis. Nat Rev Cardiol. 2019;16(2):71. doi:10.1038/s41569-018-0154-1
- Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. 2020;21(4):225–245. doi:10.1038/s41580-019-0190-7