Abstract
Background:
Sanjad–Sakati syndrome (SSS), also known as hypoparathyroidism–mental retardation–dysmorphism syndrome, or HRD, is a rare disorder characterized by growth and developmental delay, and by mental retardation and dysmorphic features.
Objective:
The objective of this study was to clarify the clinical and neurological features of SSS.
Patients:
Twenty-four patients were included in the study. They were seen at two hospitals in Kuwait.
Methods:
This was a retrospective study of patients with SSS who attended the pediatric endocrinology, genetic, and neurology clinics in the Aladan and Alfarawanya hospitals in Kuwait from September 2007 to September 2012. Clinical and radiological data were obtained from each patient’s medical records.
Results:
All 24 patients had the characteristic dysmorphic features and laboratory findings of SSS. Consanguinity was reported in 75% of parents. Neurological manifestations in the form of microcephaly, developmental delay, mental retardation, and seizures were reported in all patients. Computerized tomography scans and/or magnetic resonance imaging showed evidence of intracranial calcifications in 29.2% of patients. Two patients showed a thin corpus callosum, and one patient showed intraventricular hemorrhaging.
Conclusion:
Patients with SSS display a variety of dysmorphic features and neurological manifestations, including microcephaly, mental retardation, intracranial calcification, and epilepsy.
Introduction
Sanjad–Sakati syndrome (SSS) is a newly described syndrome originating mainly from the Middle East and Arabian Gulf countries.Citation1–Citation3 It is an autosomal recessive disorder first reported in 1988Citation1 and confirmed by a definitive report in 1991.Citation2 Children affected by this condition are born with intrauterine growth retardation and present with hypocalcemic tetany and seizures due to hypoparathyroidism at an early stage in their lives. They have consistent physical features, such as long, narrow faces; small, deep-set eyes; beaked noses; large, floppy ears and micrognathia; a failure to thrive; and mild to moderate mental retardation.Citation1–Citation5
The molecular pathology of this syndrome was shown to be due to mutations in the tubulin-folding cofactor E (TBCE) gene in chromosomal area 1q42-q43.Citation5–Citation6 The incidence of SSS is one case out of every 100,000 live births reported in Saudi Arabia.Citation7 Some authors consider SSS to be a variant of Kenny–Caffey syndrome (KCS) type 1.Citation8–Citation11 The neurological manifestations of SSS range from microcephaly and seizures to mental retardation and developmental delay. The objective of the present study is to clarify the clinical and neurological features of this rare genetic disorder.
Subjects and methods
Our work is a retrospective study of patients diagnosed with SSS who attended the pediatric endocrinology, genetic, and neurology clinics in the Aladan and Alfarawanya hospitals in Kuwait from September 2005 to September 2012. Twenty-four patients were diagnosed as having SSS. The diagnosis of SSS was based on history (intrauterine growth retardation, neonatal apnea or convulsions, and family history of similar conditions), physical examination (dysmorphic features), laboratory investigations (hypocalcemia, hypoparathyroidism), and genetic study.
Clinical, radiological, and laboratory data were obtained from the patients’ medical records. The following data were extracted and reviewed: perinatal history; age at symptom onset; presenting complaint (apnea, seizures, and dysmorphic features); clinical examination of the dysmorphism; ophthalmological manifestations; laboratory investigations, including calcium serum levels, phosphorus, alkaline phosphatase enzyme counts, magnesium, parathyroid hormones, and urine for calcium/creatinine ratios; intelligence quotient assessment; skeletal surveys; genetic studies; renal ultrasound; brain computerized tomography (CT) scans; brain magnetic resonance imaging (MRI) scans; and electroencephalography (EEG). Arrangements were made to recall the patients if any of the biochemical or radiological studies were inadequate.
Statistical analysis
SPSS software (v18; IBM Corporation, Armonk, NY, USA) was used to analyze the demographic data; neurological and physical examinations; laboratory and genetic investigation results; EEG, brain CT, and MRI findings; and renal ultrasound results.
Results
The results are summarized in and . Twenty-four children (11 males and 13 females) were identified as having SSS as their final diagnosis. Six patients died, all from pneumonia and respiratory failure (at the ages of 2–10 years). Six out of 16 families (37.5%) had more than one affected child (two families with three affected siblings and four families with two affected siblings; – show three siblings from the same family). Parental consanguinity was observed in 12 families (75%), but all families belong to the same tribe (blood relations). The mean parental age when the children were born was 21.4 years for mothers and 25.3 years for fathers. The patients’ ages ranged from 9 months up to 12 years (mean 4 years and 9 months) and nine of our patients (37.5%) were older than 5. All patients were born with intrauterine growth retardation, and three were preterm. Two children were delivered with meconium-stained amniotic fluid.
Figure 1 Facial features of a 12-year-old girl with Sanjad–Sakati syndrome.
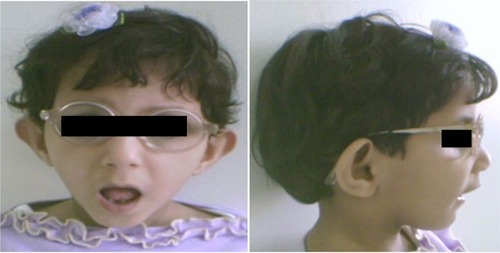
Figure 2 Facial features of an 8-year-old girl with Sanjad–Sakati syndrome.
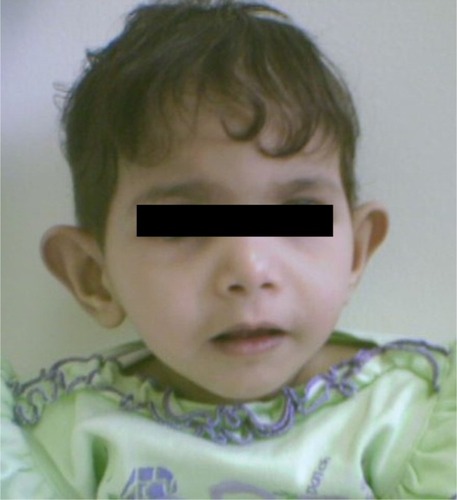
Figure 3 Facial features of a 1-year-old girl with Sanjad–Sakati syndrome.
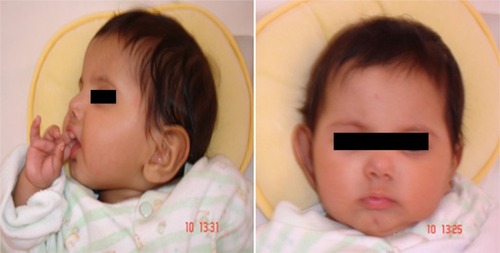
Figure 4 The small hands of a 1-year-old girl with Sanjad–Sakati syndrome.
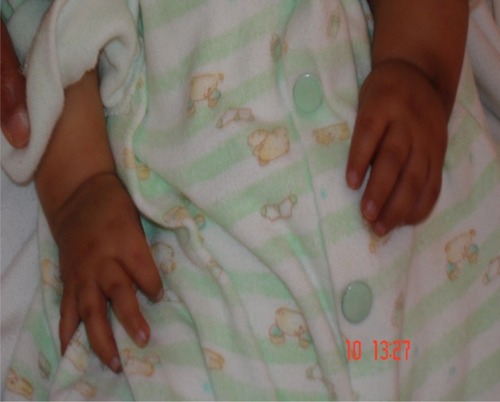
Table 1 Epidemiological, clinical, and radiological data for the studied patients
Table 2 Laboratory, radiological, and EEG data for the studied population
The presenting symptoms were usually in the form of apnea (six patients, 25%), convulsions (11 patients, 45.8%), and dysmorphic features (seven patients, 29.2%). Symptom onset was usually between 10 and 30 days (mean 17 days). Short statures, small hands and feet, and dysmorphic facial features (deep-set eyes, peaked noses, blue sclera, and large floppy ear lobules) were present in all patients (–).
Neurologically, microcephaly was observed in all 24 patients. All patients demonstrated some degree of developmental delay, although this was mild to moderate but not severe. All patients had walked unaided after the age of 2 years. All patients had mild to moderate mental retardation. Speech was not a problem, but five patients were attending or had attended speech therapy for better articulation; all developed intelligible speech. None of the children older than 4 years showed severe neurological disabilities, as all of them had successfully toilet trained and were capable of self-feeding. All patients had developed seizures at some stage of their life – 20 of them in the first year. EEG was done on 18 patients and was abnormal in five, but two of these had a family history of epilepsy.
Serum calcium and parathyroid hormones were decreased in all patients. Sixteen patients (66.6%) had ultrasound evidence of renal calcifications. The age of discovery of renal calcifications was between 18–36 months (mean 24.3 months). Fortunately, however, all patients had normal kidney functions. Elevated urine calcium/creatinine ratios were reported in 22 patients (91.2%) at any stage of their life at least two to three times after the start of 1 alpha -hydroxycholecalciferol and calcium therapy.
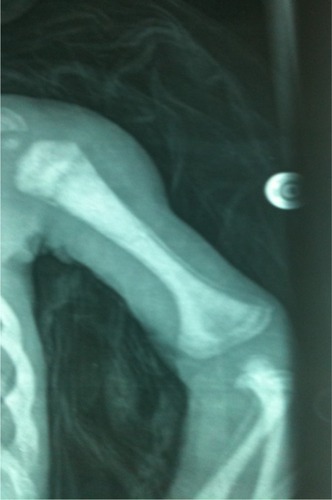
Skeletal surveys showed medullary stenosis in only two patients (8.3%). Brain MRI and CT were performed on and reviewed for only 18 patients. Intracranial calcification was observed in seven patients (38.9%). Two patients showed a thin corpus callosum, and one patient had evidence of intraventricular hemorrhage (an ex-preterm baby).
Chromosomal and fluorescence in situ hybridization studies revealed normal findings in all cases. Molecular study using polymerase chain reaction/single-strand conformation polymorphism analysis of exon 3 of the TBCE gene revealed 12 bp deletions (155–166del) in all affected children. The affected individuals were homozygous with respect to this mutation. Screening for this deletion in the parents revealed that both parents of all patients were heterozygous carriers for this mutation.
Discussion
Although SSS is a rare genetic disorder, it is not uncommon in the Gulf region, especially Saudi Arabia and Kuwait, as it is distributed in certain Bedouin tribes. The syndrome can be confused with autosomal recessive KCS, caused by a mutation in the TBCE gene, which shares similar phenotypic and genotypic features, but also osteosclerosis, medullary stenosis of the long bones, and normal intelligence. Previous reports that reviewed most of the KCS-diagnosed cases in Kuwait concluded that all patients fulfilled the criteria of SSS.Citation3,Citation7–Citation9,Citation10,Citation12 shows the differences between the two syndromes.Citation12
Table 3 Differentiation between Sanjad–Sakati syndrome and Kenny–Caffey syndrome, types 1 and 2
To date, fewer than 20 reports have been published worldwide in English discussing SSS. According to a previous report by Naguib et al,Citation3 the 2009 estimated incidence of the syndrome in Kuwait was 7–18 per 100,000 live births – higher than the estimated incidence in Saudi Arabia, which was 1 in 40,000 to 1 in 100,000 live births.Citation7 The difference could be attributed to the recent awareness of SSS and/or to a high frequency of heterozygous carriers among the Kuwaiti population.
In the present study, affected siblings were reported in only four non-consanguineous families (25%). Overall, consanguinity was found in 75% of our patients. Naguib et alCitation3 reported fewer incidences of consanguineous marriage among patients with SSS, a difference that could be explained by the wider distribution of the affected tribe in the population around the studied hospitals. In another study of seven families in Jordan, the incidence of consanguinity was 100%.Citation13
Patients with SSS typically present in the neonatal period with tetany, seizures, or apnea due to hypocalcemia and recurrent infections, probably due to immune defects.Citation1–Citation3 In the present study, most of the cases had been diagnosed during the neonatal period due to hypocalcemia/seizures or apnea, phenotypic pictures, or raised awareness of the syndrome among affected families. The syndrome has a wide variety of clinical features, including deep-set eyes, micrognathia, thin lips, small maxilla, severely decayed teeth, beaked noses, depressed nasal bridges, external ear anomalies, small hands and feet, short stature, and learning difficulties.Citation14,Citation15 In addition, hypoparathyroidism and hypocalcemia are constant findings.Citation1–Citation3,Citation15 All of our patients showed the cardinal features described previously (–).
Neurological manifestations of SSS have not been discussed in detail before. Microcephaly, developmental delay, delayed speech, and some degree of mental retardation were observed in all our patients. Seizures were reported in all patients, but abnormal EEGs were found in only five patients (20.8%). The seizures were mostly due to hypocalcemia, but could also be explained in some of our patients by the abnormal EEG. Twenty-two percent of our patients showed intracranial calcifications upon brain CT and/or MRI due to excessive use of vitamin D and calcium therapy, evidenced by the renal calcification reported in 66.6% of the studied population and the elevated urine calcium/creatinine ratio in 91.2% of our patients. A thin corpus callosum was reported in two patients (8.3%); this factor was reported in a previous study with a higher incidence rate.Citation13 In a skeletal survey, only 8.3% of patients showed medullary stenosis (), which is more common in KCS.Citation1,Citation11
Figure 5 Medullary stenosis with osteoscelosis in a patient with Sanjad–Sakati syndrome.
The common characteristic laboratory findings of SSS are hypocalcemia, low parathyroid hormone levels, and hyperphosphatasia. These cardinal laboratory findings were also reported in all of our patients. Most of our patients also showed hypomagnesemia, which was previously reported.Citation1–Citation3 Detailed studies of T-cell function were not routinely done on our patients, but repeated infection – especially pneumonia – was observed in those patients and was the cause of death for all of our deceased patients.
The syndrome is related to a locus situated at 1q42-q43.Citation5,Citation14,Citation15 Recently, Parvari et alCitation6 demonstrated mutations in the TBCE gene in SSS. All Middle Eastern patients showed a deletion in exon 3 of the TBCE gene.Citation3 All patients in this study were homozygous for the deletion of 12 bp (155–166del) in the TBCE gene, and their parents were ––heterozygous carriers for this mutation.
Therapy for SSS is focused on correcting hypocalcemia and treating infections.Citation15,Citation16 Marsden et alCitation16 reported the use of growth hormones for a child with SSS with good outcome. Two of our patients had started growth hormone injections, but there was no benefit to their heights and both developed kyphosis. Prevention could potentially be achieved through pre-implantation genetic diagnosis and carrier detection.
Conclusion
SSS is a rare autosomal recessive disorder that is nonetheless not uncommon in the Gulf area. Clinically, it resembles KCS; however, microcephaly, less cortical thickening, medullary stenosis, and subnormal mentality are features characteristic of SSS. Patients with SSS have some neurological manifestations in the form of microcephaly, submentality, early seizures, and intracranial calcifications. Prevention of this syndrome may be achieved in the future through pre-implantation genetic diagnosis and carrier detection.
Disclosure
The authors report no conflicts of interest in this work.
References
- SanjadSSakatiNAbu-OsbaYCongenital hypoparathyroidism with dysmorphic features: a new syndromePediatr Res198823271A
- SanjadSASakatiNAAbu-OsbaYKKaddoraRMilnerRDGA new syndrome of congenital hypoparathyroidism, seizure, growth failure and dysmorphic featuresArch Dis Child1991661931962001103
- NaguibKKGoudaSAElshafeyASanjad-Sakati syndrome/Kenny-Caffey syndrome type 1: a study of 21 cases in KuwaitEast Mediterr Health J200915234535219554981
- Al-MalikMIThe dentofacial features of Sanjad-Sakati syndrome: a case reportInt J Paediatr Dent20041413614015005702
- KellyTEBlantonSSaifRSanjadSSakatiNConfirmation of the assignment of the Sanjad-Sakati (congenital hypoparathyroidism syndrome (OMIM 241410) locus to chromosome 1q42-43J Med Genet2000371636410691411
- ParvariRHershkovitzEGrossmanNMutation of TBCE causes hypoparathyroidism-retardation-dysmorphism and autosomal recessive Kenny-Caffey syndromeNat Genet20023244845212389028
- FranceschiniPTestaABogettiGKenny-Caffey syndrome in two sibs born to consanguineous parents: evidence for an autosomal recessive variantAm J Med Gen199242112116
- SabryMAZakiMAbul HassanSJKenny-Caffey syndrome is part of the CATCH 22 haploinsufficiency clusterJ Med Genet19983531369475091
- SabryMAZakiMShaltoutAGenotypic/phenotypic heterogeneity of Kenny-Caffey syndromeJ Med Genet199835105410559863613
- DiazGAKhanKTSGelbBDThe autosomal recessive Kenny-Caffey syndrome locus maps to chromosome 1q42-q43Genomics19985413189806825
- OMIM – Online Mendelian Inheritance in Man [database on the Internet]Baltimore, MDJohns Hopkins University Available from: http://www.ncbi.nlm.nih.gov/omim/. Accessed April 13, 2009.
- TahseenKKhanTUmaRKenny-Caffey syndrome in six Bedouin sibships: autosomal recessive inheritance is confirmedAm J Med Genet19976921261329056548
- AlbaramkiJAklKAl-MuhtasebASanjad Sakati syndrome: a case series from JordanEast Mediterr Health J201218552753122764442
- DiazGAGelbBDAliFSanjad-Sakati and autosomal recessive Kenny-Caffey syndromes are allelic: evidence for an ancestral founder mutation and locus refinementAm J Med Genet199985485210377012
- ParvariRHershkovitzEKanisAHomozygosity and linkage-disequilibrium mapping of the syndrome of congenital hypoparathyroidism, growth and mental retardation, and dysmorphism to a 1-cM interval on chromosome 1q42-43Am J Human Genet1998631631699634513
- MarsdenDNyhanWLSakatiNOSyndrome of hypoparathyroidism, growth hormone deficiency, and multiple minor anomaliesAm J Med Genet1994523343387810565