
Abstract
Introduction
Chondrocyte degeneration and senescence are characteristics of osteoarthritis (OA) and other joint degenerative diseases, and ferroptosis has been observed to regulate the development of OA. However, the role of the N6-methyladenosine (m6A) modification in OA ferroptosis remains unclear.
Methods
This study performed series of assays to investigate the function of the m6A reader IGF2BP1 in OA ferroptosis, including m6A quantitative analysis, Iron (Fe2+) release analysis, Malondialdehyde (MDA) measurement, lipid peroxidation (ROS) detection and Glutathione (GSH) measurement. The molecular interaction and mechanism analysis was performed by Luciferase reporter assay, mRNA stability analysis and RNA immunoprecipitation (RIP) assay.
Results
These results indicate that IGF2BP1 is upregulated in IL-1β-induced chondrocytes. Functionally, IGF2BP1 silencing represses ferroptosis, including iron (Fe2+) accumulation, malondialdehyde, and reactive oxygen species (ROS). Mechanistically, among the potential downstream targets, matrix metalloproteinase-3 (MMP3) was observed to harbor a significant m6A modified site in the 3’-UTR. IGF2BP1 combines with MMP3 through the binding of m6A sites, thereby enhancing MMP3 mRNA stability.
Discussion
In conclusion, our findings revealed the functions and mechanisms of m6A regulator IGF2BP1 in OA chondrocyte’s ferroptosis, providing a novel target for OA treatment.
Introduction
Osteoarthritis (OA) is the most common musculoskeletal disease and is associated with an extremely high disability rate and a decreased quality of life in the elderly population.Citation1 The clinical symptoms of OA include joint swelling, joint stiffness, joint pain, and loss of joint function, which induce difficulties in daily activities.Citation2 Currently, there are few effective treatments for OA other than total knee replacement in patients with advanced OA.Citation3 For patients with early OA, the treatment is to reduce joint wear and relieve pain.Citation4 Furthermore, nonsteroidal anti-inflammatory drugs and surgical interventions are also applied to OA treatment, which have some side effects.Citation5 Therefore, it is necessary to identify novel molecular targets for OA pathophysiology and prevention.
N6-methyladenosine (m6A) is an important ubiquitous internal modification of eukaryotic RNA that plays a critical role in physiological functions.Citation6 The m6A modification has been shown to modify numerous aspects of pathophysiological processes, including RNA metabolism, export, translation, and decay. Three key enzymes regulate the m6A modification, including “writer” proteins (METTL3, METTL14, KIAA1429, WTAP, etc.), “eraser” proteins (ALKBH5, FTO) and “reader” proteins (YTHDF1/2/3 YTHDC1, IGF2BP1/2/3, etc.). In OA pathophysiology, m6A levels in clinical OA samples are higher than those in healthy samples.Citation7 Currently, key m6A modification enzymes are known to act as risk factors for OA, or might provide potential therapeutic targets for OA treatment. For example, METTL3 is highly expressed in clinical samples from patients with OA, and low METTL3 expression inhibits chondrocyte pyroptosis.Citation7
Ferroptosis is an iron-dependent subtype of programmed cell death (iron-dependent type) involved in oxidative stress-induced cell death.Citation8–10 Several studies have demonstrated that redox imbalance plays an important role in OA development. Previous studies have indicated that iron can trigger bovine articular cartilage degeneration by enhancing the release of sulfated glycosaminoglycan. For example, chondrocytes undergo inflammation and iron overload that can trigger ferroptosis. Recent findings have indicated that ferroptosis plays an important role in the occurrence and development of OA. For instance, the ferroptosis-related genes LPCAT3 and PGD, which have both been identified as effective diagnostic biomarkers for OA patients, are decreased in OA chondrocytes with an IL-1β-induced condition.Citation11 There was remarkable relevance of m6A and ferroptosis in OA pathophysiology. Recently, more and more researches reveal the pathophysiological processes of OA. For instance, Zhang (2023) reported that maintaining hypoxia environment by HIF-1α in subchondral bone could alleviate the OA progression.Citation12 Besides, the enhanced ferroptosis is also correlated to immune response.Citation13
The present study aimed to explore the expression patterns of the m6A reader IGF2BP1 in OA chondrocytes and identify the mechanism that mediates ferroptosis phenotypes in chondrocytes. IGF2BP1 silencing represses ferroptosis, including iron accumulation, malondialdehyde (MDA), and reactive oxygen species (ROS). IGF2BP1 combines with matrix metalloproteinase-3 (MMP3) through the binding of m6A sites, thereby enhancing MMP3 mRNA stability. These findings reveal the function and mechanism of IGF2BP1 in OA chondrocyte ferroptosis, and may provide a novel target for OA treatment.
Materials
Cartilage Samples Collection
Cartilage tissues were acquired from patients with OA who underwent total knee replacement. Normal control tissues were obtained from patients with femoral neck fractures who underwent total hip replacement surgery without a history of OA or rheumatoid arthritis. All the patients involved in the study had signed informed consent forms. This study was designed in accordance with the Declaration of Helsinki and approved by the ethics committee of Tianjin Hospital.
Chondrocytes Isolation and Culture
Chondrocytes were isolated from acquired OA cartilage and healthy control cartilage and cultured according to previously reported methods.Citation14,Citation15 Briefly, chondrocytes were isolated from digested tissue samples after grinding, digestion, and centrifugation. The isolated chondrocytes were cultured in DMEM-F12 supplemented with 10% fetal bovine serum (FBS; Gibco) and antibiotics. When chondrocytes reached 70–80% density, the cells were digested and transferred to T75 for further experiments. For the OA cellular model, primary chondrocytes were treated with IL-1β (10 ng/mL) for 24 h.
m6A Quantitative Analysis
The global m6A level in chondrocytes was measured using an m6A RNA methylation assay kit (Colorimetric, Abcam, Cat No. ab185912) according to the manufacturer’s protocol.
Real-Time PCR
Total RNA was extracted from chondrocytes using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Next, the total RNA sample (1 μg) was reverse-transcribed to first-strand cDNA using a reverse transcription system kit (Promega, Madison, WI, USA). cDNAs were amplified using SYBR Premix Taq (Applied Biosystems, Foster City, CA, USA) on an Applied Biosystems 7500 step-one plus system (Applied Biosystems). Primer sequences used in this study are listed in Table S1. The relative mRNAs expression levels were calculated using the 2−ΔΔct method.
Iron (Fe2+) Release Analysis
The levels of iron (Fe2+) released from chondrocytes were tested using an iron colorimetric assay kit (Cat No. E1042, Applygen, Beijing, China) referring to the manufacturer’s instructions. The surgical procedure was based on previous literature.Citation16
Malondialdehyde (MDA) Measurement
Cellular MDA concentrations were determined using a lipid peroxidation MDA assay kit (cat. S0131S, Beyotime), according to the manufacturer’s instructions. After IL-1β treatment, cells were harvested and lysed using a radioimmunoprecipitation assay (RIPA) lysis solution. The lysate was centrifuged and the supernatant was collected for analysis.
Lipid Peroxidation (ROS) Detection
Intracellular lipid peroxidation was tested using C11-BODIPY581/591 (Cat No. D3861, Thermo Fisher Scientific). After 24 h, C11-BODIPY were incubated in medium for 30 min at 37 °C. The data were analyzed using FlowJo V10.0 software.
Glutathione (GSH) Measurement
Cellular GSH levels were measured using a GSH ratio detection assay kit (cat. S0053; Beyotime), according to the manufacturer’s instructions. The amount of GSH in the supernatant was determined by using a standard curve.
Cell Viability Assay
Chondrocyte viability was determined using cell counting kit-8 (Cat No. MA0218, Meilunbio). Chondrocytes were seeded in 96-well plates at a density of 5000 cells/well. After removing the culture medium, 10% CCK-8 solution (100 μL) was added to each well and incubated at 37 °C in the dark for 2 h. Absorbance was measured at 450 nm using a microplate reader.
Western Blot
The chondrocytes were homogenized in RIPA lysis buffer containing protease inhibitors (Diagnostics, Mannheim, Germany). The protein concentration of each sample was measured using a bicinchoninic acid (BCA) kit and separated by SDS-PAGE. After transfer to polyvinylidene difluoride (PVDF) membranes, protein samples were blocked with nonfat milk (5%) for 1 h and then incubated with antibodies against IGF2BP1 (Cat No. 22803-1-AP, Proteintech, 1:1000) and β-actin (Cat No. 81115-1-RR, Proteintech, 1:5000). After overnight incubation with the primary antibodies, the membranes were incubated with secondary antibodies. Finally, bands were visualized using enhanced chemiluminescence reagents.
Cell Death Analysis
Cell death was analyzed using flow cytometry and detected as previously described.Citation17 Cell death was measured using Annexin V-FITC/propidium iodide (PI) apoptosis detection kit (Vazyme) double staining. Briefly, primary chondrocytes were harvested, resuspended in 100 mL of binding buffer, and stained with FITC-Annexin V/PI. Death was analyzed using a BD FACSCalibur instrument (BD Biosciences, Franklin Lakes, NJ, USA).
mRNA Stability Analysis
Chondrocytes were seeded into 6-well plates and transfected with control (NC) and IGF2BP1 knockdown (shIGF2BP1) cells treated with actinomycin D (Act D, 8 μg/mL) for 0, 4, and 12 h. RNA was extracted from chondrocytes and reverse-transcribed for qRT-PCR. Relative expression levels were calculated using the 2−ΔΔCt method and normalized to β-actin levels. The half-life (t1/2) of MMP3 mRNA was calculated using the SPSS software.
Luciferase Reporter Assays
The dual-luciferase reporter gene vectors of target MMP3, wild type, and mutants with mutations in the IGF2BP1-binding sites, were formulated by GenePharma (Shanghai, China) respectively, including Luc-MMP3-3’UTR-WT and Luc-MMP3-3’UTR-MUT. Two other plasmids (sh-NC and sh-IGF2BP1) were co-transfected into 293T cells, and the cells were lysed for 36 h after transfection. The supernatant was collected and analyzed using a dual-luciferase reporter assay system (Promega). The firefly luminescence and Renilla luciferase working solutions were normalized to those of Renilla.
RNA Immunoprecipitation (RIP) and MeRIP-PCR Assays
The binding between IGF2BP1 and MMP3 was identified using the RNA Binding Protein Immunoprecipitation Kit (Magna). Briefly, at 4 °C overnight, IGF2BP1 antibody (5 μg) was incubated with A/G magnetic beads and then subjected to immunoprecipitation. The extracted and purified RNA was precipitated using the magnetic beads. Later, the relative precipitated MMP3 mRNA was determined by qPCR. For the m6A-modified level on MMP3 mRNA, the MeRIP-PCR assay was performed with the ferroptosis inhibitor Fer-1 administration.
Statistical Analysis
All data are expressed as mean ± standard deviation (SD) and were analyzed using GraphPad software (GraphPad Inc., La Jolla, CA, USA). Differences within groups were analyzed using Student’s t-test and one-way analysis of variance (ANOVA). Statistical significance was set at p < 0.05.
Results
IGF2BP1 Up-Regulated in IL-1β-Stimulate Primary Chondrocytes with Ferroptosis Characteristic
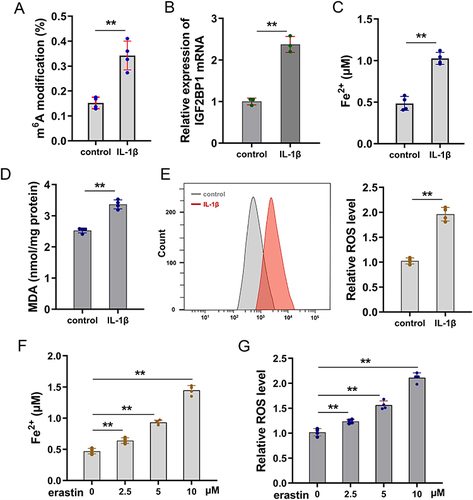
For the OA cellular model, primary chondrocytes were treated with IL-1β (10 ng/mL) for 24 h. First, m6A modification quantitative analysis showed that the m6A modification level increased compared to that in the control group (). In this OA cellular model, the level of m6A reader IGF2BP1 was also increased compared to that in the control group (). To identify the characteristics of ferroptosis in OA chondrocytes, the ferroptosis indicators were assessed. The iron ion concentration analysis illustrated that Fe2+ concentration was upregulated in the OA cellular model (). Malondialdehyde (MDA) and reactive oxygen species (ROS) analyses revealed that MDA and lipid ROS levels were upregulated in the OA cellular model ( and ). Given that ferroptosis characteristics increased in IL-1β-stimulate primary chondrocytes and erastin-activated ferroptosis, more assays were performed to verify ferroptosis compared to erastin-treated cells. In erastin-treated primary chondrocytes, both Fe2+ concentration and lipid ROS levels were upregulated in a dose-dependent manner ( and ). Overall, these findings illustrate that IGF2BP1 up-regulated in IL-1β-stimulate primary chondrocytes, showing significant ferroptosis characteristics.
Figure 1 IGF2BP1 up-regulated in IL-1β-stimulate primary chondrocytes with ferroptosis characteristic. (A) Primary chondrocytes were treated by IL-1β (10 ng/mL) for 24 h for the OA cellular model. The m6A modification quantitative analysis was performed. (B) RT-PCR analysis detected the IGF2BP1 level in OA model group and comparing control group. (C) The iron ion (Fe2+) concentration analysis illustrated the Fe2+ concentration in OA cellular model. (D) Malondialdehyde (MDA) and (E) reactive oxygen species (ROS) analysis were performed to detect the MDA and ROS levels. (F and G) Erastin-treated primary chondrocytes constructed to mimic ferroptosis characteristics. (F) Fe2+ concentration and (G) lipid ROS levels were tested in erastin-treated primary chondrocytes with dose-dependent manner (0, 2.5, 5, 10 μM). **p < 0.01.
Silencing of IGF2BP1 Repressed the IL-1β-Stimulated Ferroptosis in Primary Chondrocytes
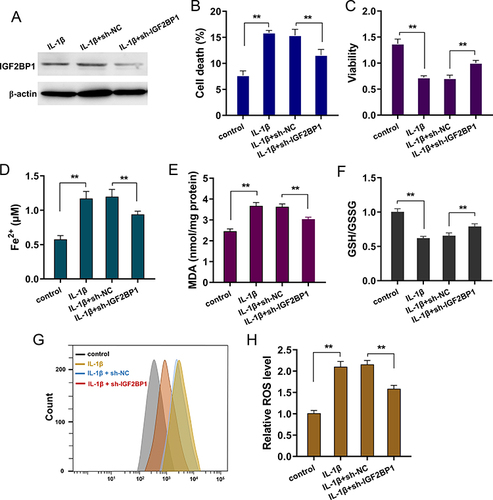
Given the high expression of IGF2BP1 in IL-1β-treated primary chondrocytes, IGF2BP1 was silenced for further in vitro analysis (). Cell death analysis revealed that IL-1β-stimulation induced the death rate, whereas silencing of IGF2BP1 inhibited the cell death rate of primary chondrocytes (). Viability analysis by CCK-8 indicated that IL-1β-stimulation reduced the proliferative ability, and silencing of IGF2BP1 recovered the proliferative ability (). For the ferroptosis related characteristic, the iron (Fe2+) accumulation, malondialdehyde, cellular glutathione (GSH) and reactive oxygen species (ROS) were tested in IL-1β-stimulated primary chondrocytes. The results indicated that IL-1β-stimulation increased Fe2+ accumulation (), MDA () and lipid ROS levels ( and ) and reduced GSH levels (). Silencing of IGF2BP1 ameliorated Fe2+ accumulation (), MDA () and lipid ROS levels ( and ) and exacerbated GSH levels (). Overall, the above findings illustrate that silencing IGF2BP1 represses IL-1β-stimulated ferroptosis in primary chondrocytes.
Figure 2 Silencing of IGF2BP1 repressed the IL-1β-stimulated ferroptosis in primary chondrocytes. (A) Western blot analysis revealed the silencing transfection of IGF2BP1 (sh-IGF2BP1, sh-NC) in vitro assay of IL-1β-stimulated primary chondrocytes. (B) Cell death analysis by flow cytometry revealed the death rate of IL-1β-stimulated primary chondrocytes with IGF2BP1 silencing transfection and control transfection. (C) Viability analysis by CCK-8 revealed the proliferative ability of IL-1β-stimulated primary chondrocytes. (D) Iron (Fe2+) accumulation, (E) malondialdehyde (MDA), (F) cellular glutathione (GSH) and (G and H) reactive oxygen species (ROS) were tested in IL-1β-stimulated primary chondrocytes. **p < 0.01.
MMP3 was Verified as Downstream Target of IGF2BP1
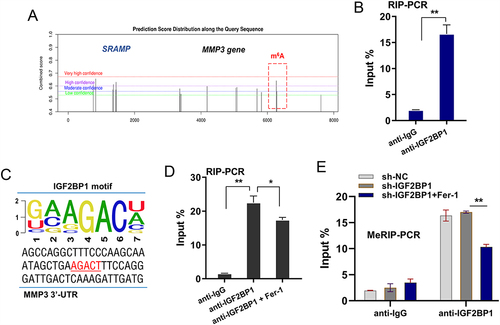
In the pre-assay investigation, we found potential m6A modified sites in the 3’-UTR of MMP3 (). In the molecular interaction analysis, RIP-PCR was performed using an anti-IGF2BP1 antibody, and the data showed that IGF2BP1 was significantly associated with MMP3 mRNA in chondrocytes (). In the 3’-UTR of MMP3, the possible m6A binding motif was AGACT (). To test whether the ferroptosis microenvironment affects the molecular interaction between IGF2BP1 and MMP3 mRNA, RIP-PCR was performed using ferrostatin-1. Ferrostatin 1(Fer-1) is a selective inhibitor of erastin-induced ferroptosis. The results indicated that the inhibition of ferroptosis by Fer-1 mitigated MMP3 mRNA immunoprecipitation with the anti-IGF2BP1 antibody (). To test whether m6A modification was altered in the ferroptosis microenvironment, MeRIP-PCR was performed to detect the m6A modification level in MMP3 mRNA. The results indicated that inhibition of ferroptosis by Fer-1 mitigated the m6A modification level of MMP3 mRNA (). Overall, these findings illustrate that MMP3 is a downstream target of IGF2BP1 in an m6A-dependent manner in the ferroptosis microenvironment.
Figure 3 MMP3 was verified as downstream target of IGF2BP1. (A) There were potential m6A modified sites on the 3’-UTR of MMP3 gene, which was predicted by online SRAMP (http://www.cuilab.cn/sramp). (B) RIP-PCR was performed using anti-IGF2BP1 antibody to test the molecular interaction within IGF2BP1 and MMP3 mRNA in chondrocytes. (C) The possible m6A binding motif was AGACT in the 3’-UTR sequences of MMP3 gene. (D) RIP-PCR was performed with ferrostatin-1 (Fer-1) addition in chondrocytes. (E) MeRIP-PCR was performed using anti-m6A antibody to detect the m6A modification level of MMP3 mRNA. *p < 0.05, **p < 0.01.
IGF2BP1 Enhanced MMP3 mRNA Stability in Ferroptosis Microenvironment
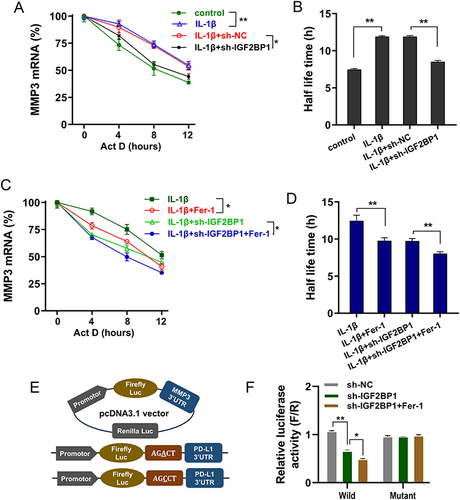
Previous findings had revealed that IGF2BP1 accelerated the ferroptosis in IL-1β-stimulated chondrocytes, and MMP3 acted as the downstream of IGF2BP1. Additional data were obtained in subsequent assays. Studies have shown that IGF2BP1, as an m6A reader, can enhance its target mRNA stability in an m6A-dependent manner; therefore, this study also tested whether IGF2BP1 enhances MMP3 mRNA stability in a ferroptosis microenvironment in an m6A-dependent manner. RNA decay analysis revealed that IL-1β-stimulation up-regulated MMP3 mRNA stability and half-life (t1/2), whereas IGF2BP1 silences the repressed half-life (t1/2) ( and ). Moreover, to test whether the ferroptosis microenvironment affected RNA stability, MeRIP-PCR was performed to detect the m6A modification level of MMP3 mRNA following administration of the ferroptosis inhibitor Fer-1. Results indicated that Fer-1 administration repressed the half-life (t1/2) of MMP3 mRNA ( and ). The luciferase assay also indicated that the luciferase activity of the wild-type plasmid transfection tended to decrease when IGF2BP1 was silenced. Moreover, Fer-1 administration reduced the luciferase activity ( and ). Overall, these findings illustrate that IGF2BP1 enhances MMP3 mRNA stability in the ferroptosis microenvironment.
Figure 4 IGF2BP1 enhanced MMP3 mRNA stability in ferroptosis microenvironment. (A) RNA decay analysis was performed on chondrocytes with Act D administration (8 μg/mL, actinomycin (D) by IL-1β treatment or sh-IGF2BP1 transfection. The MMP3 mRNA level was detected at the indicated time point by RT-PCR. (B) Half-life time (t1/2) of MMP3 mRNA was calculated using ln2/slope and GAPDH was used for normalization. (C) RNA decay analysis was performed by chondrocytes with Act D administration by IL-1β treatment or sh-IGF2BP1 transfection or ferroptosis inhibitor Fer-1 administration. (D) Half-life time (t1/2) of MMP3 mRNA was calculated. (E) The construction of luciferase wild type plasmids and mutant plasmids. (F) The luciferase activity of wild type/mutant plasmids transfection. *p < 0.05, **p < 0.01.
IGF2BP1/MMP3 Axis Deteriorated the Ferroptosis of Primary Chondrocytes via m6A-Dependent Manner
Rescue assays were performed to identify the regulatory pathways of the IGF2BP1/MMP3 axis. MMP3 overexpression upregulated Fe2+ (), MDA (), cell death () and lipid ROS levels ( and ). In addition, cotransfection with IGF2BP1 silencing reduced these effects. Moreover, MMP3 overexpression repressed proliferative viability () and GSH levels (), and co-transfection with IGF2BP1 silencing promoted these effects. Overall, the above findings illustrate that the IGF2BP1/MMP3 axis deteriorates the ferroptosis of primary chondrocytes in an m6A-dependent manner ().
Figure 5 IGF2BP1/MMP3 axis deteriorated the ferroptosis of primary chondrocytes via m6A-dependent manner. (A) Iron (Fe2+) accumulation, (B) malondialdehyde (MDA), (C) cell death analysis by flow cytometry, (D) CCK-8 assay, (E) cellular glutathione (GSH), (F and G) reactive oxygen species (ROS) assays were performed in erastin-treated primary chondrocytes. Primary chondrocytes were transfected with MMP3 overexpression plasmids and co-transfection of IGF2BP1 silencing (sh-IGF2BP1). *p < 0.05, **p < 0.01.
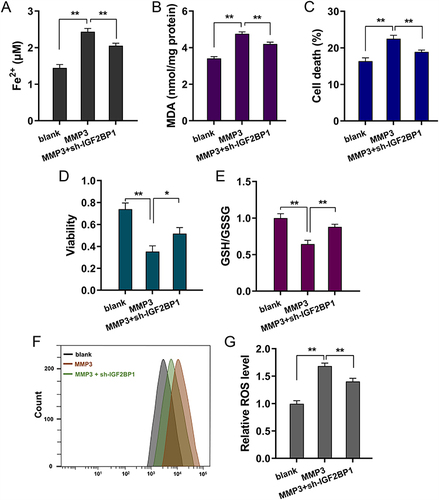
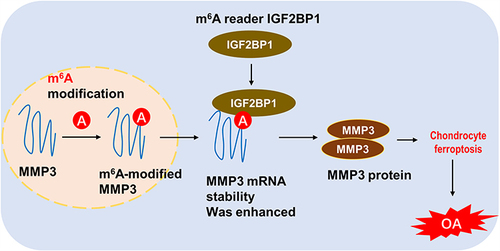
Figure 6 IGF2BP1 accelerates chondrocyte ferroptosis in osteoarthritis through targets m6A/MMP3 axis.
Discussion
Osteoarthritis (OA) acts as the most common disabling joint disease, which seriously compromises the quality of osteoarthritic cartilages.Citation18,Citation19 OA is characterized by the loss of cartilage in the primary lesion.Citation20 Primary OA treatment approaches involve nonpharmacological interventions combined with pharmacotherapy or surgical intervention.Citation21 As we all know, with the increase of aging, the prevention of OA is more relevant and meaningful.
Previous findings from numerous studies have indicated that dysregulation of m6A modifications and several m6A correlated enzymes participate in OA development and progression. For example, the m6A methyltransferase WTAP is differentially upregulated in chondrocytes stimulated by lipopolysaccharide, and WTAP knockdown suppresses apoptosis and extracellular matrix degradation and enhances proliferation in the LPS-induced chondrocyte injury model.Citation22 METTL3 is highly expressed in OA patient samples, and silencing METTL3 expression inhibits chondrocyte pyroptosis in combination with NEK7, thereby inhibiting OA progression by promoting chondrocytes pyroptosis.Citation7 Therefore, m6A modification and m6A modified enzymes may regulate OA progression and pathophysiological processes.
Here, we found that the m6A reader IGF2BP1 is highly expressed during IL-1β-stimulated ferroptosis in primary chondrocytes, which simulates the cellular OA chondrocyte injury model. Previous literature has indicated that m6A reader IGF2BP1 participates in the mesenchymal stem cell (MSC) senescence in articular cartilage. For instance, IGF2BP1 is identified as the m6A reader restraining the CYP1B1 mRNA degradation via m6A-modified manner to aggravate spontaneous OA in mice.Citation23 To further elucidate the mechanism by which IGF2BP1 modulates OA progression, this study focused on ferroptosis of chondrocytes. Previous studies have shown that OA and ferroptosis share certain features, including abnormal accumulation of iron metabolism, mitochondrial dysfunction, and lipid peroxidation generation.Citation24–26 The genes or regulators involved in the pathophysiological process of ferroptosis is still scarce. IL-1β-stimulation increased Fe2+ accumulation, MDA, and lipid ROS levels, and reduced GSH levels. Silencing IGF2BP1 ameliorated Fe2+ accumulation, MDA, and lipid ROS levels and exacerbated GSH levels. Erastin promotes ferroptosis in chondrocytes and upregulates the level of matrix metalloproteinases MMPs. Ferrostatin 1 (Fer-1) is a selective inhibitor of erastin-induced ferroptosis These data indicate that Fer-1 administration represses ferroptosis. These findings indicate that IGF2BP1 modulates ferroptosis during OA development.
Boy et al (2023) identified MMP3 as a potential hub biomarker in an OA SD rat model with significant associations with m6A regulators and ferroptosis.Citation27 Because the role of matrix metalloproteinase-3 (MMP3), an important enzyme mediating cartilage degradation, has been sufficiently reported in chondrocytes. The present research may shift to molecular targets with the potential to regulate ferroptosis in OA. Our findings revealed that MMP3 was a downstream target of IGF2BP1. In the pre-assay investigation, we found potential m6A modified sites in the 3’-UTR of MMP3. RIP-PCR molecular interaction analysis showed that IGF2BP1 is significantly associated with MMP3 mRNA expression in chondrocytes. Induction of ferroptosis may increase MMPs protein expression and decrease collagen II expression in chondrocytes.Citation28,Citation29
Overall, the above findings illustrate that silencing IGF2BP1 represses IL-1β-stimulated ferroptosis in primary chondrocytes by enhancing MMP3 mRNA stability. However, the shortcomings of the study are also need our attention and further study to solve the problem. For example, due to the lack or immaturity of in vivo delivery methods of IGF2BP1, rigorous in vivo experiments in mice were not carried out. This difficulty requires us to continue to explore, or rely on the application of more advanced technology. In conclusion, this study demonstrates the function and mechanism of the m6A regulator IGF2BP1 in OA chondrocyte ferroptosis, providing a novel target for OA treatment.
Ethics Approval and Consent to Participate
Written informed consent was obtained from each patient, and the study was approved by the Ethical Committee of the Tianjin Hospital of Tianjin University.
Disclosure
All authors declare no conflicts of interest in this work.
Data Sharing Statement
Data and materials are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Ghanta RB, Tsay EL, Feeley B. Augmented baseplates in reverse shoulder arthroplasty: a systematic review of outcomes and complications. JSES Rev Rep Tech. 2023;3(1):37–43. doi:10.1016/j.xrrt.2022.08.008
- Kim HA. Osteoarthritis - insights from recent research. J Rheum Dis. 2022;29(3):132–139. doi:10.4078/jrd.2022.29.3.132
- Tamaddon M, Gilja H, Wang L, et al. Osteochondral scaffolds for early treatment of cartilage defects in osteoarthritic joints: from bench to clinic. Biomater Transl. 2020;1(1):3–17. doi:10.3877/cma.j.issn.2096-112X.2020.01.002
- Park JS, Lee HJ, Jo YH, Lee MK, Lee BG. Surgical trends of shoulder arthroplasty: nationwide epidemiologic study in South Korea. Clin Orthopedic Surg. 2023;15(2):290–299. doi:10.4055/cios22163
- Sudah SY, Menendez ME, Moverman MA, et al. The role of the anterior shoulder joint capsule in primary glenohumeral osteoarthritis. JSES Rev Rep Tech. 2023;3(1):21–27. doi:10.1016/j.xrrt.2022.09.005
- Shen H, Xie K, Tian Y, Wang X. N6-methyladenosine writer METTL3 accelerates the sepsis-induced myocardial injury by regulating m6A-dependent ferroptosis. Apoptosis. 2023;28(3–4):514–524. doi:10.1007/s10495-022-01808-y
- Xiong X, Xiong H, Peng J, Liu Y, Zong Y. METTL3 regulates the m(6)A modification of NEK7 to inhibit the formation of osteoarthritis. In: Cartilage. SAGE Publications; 2023:19476035231200336.
- Wang H, Fu L, Li Y, et al. m6A methyltransferase WTAP regulates myocardial ischemia reperfusion injury through YTHDF1/FOXO3a signaling. Apoptosis. 2023;28(5–6):830–839. doi:10.1007/s10495-023-01818-4
- Wang K, Wang G, Li G, et al. m6A writer WTAP targets NRF2 to accelerate bladder cancer malignancy via m6A-dependent ferroptosis regulation. Apoptosis. 2023;28(3–4):627–638. doi:10.1007/s10495-023-01817-5
- Liu L, Jin H, Dong M, et al. Identification of ferroptosis-related signature with potential implications in prognosis and immunotherapy of renal cell carcinoma. Apoptosis. 2022;27(11–12):946–960. doi:10.1007/s10495-022-01766-5
- Wang L, Ye S, Qin J, Tang M, Dong M-Y, Fang J. Ferroptosis-related genes LPCAT3 and PGD are potential diagnostic biomarkers for osteoarthritis. J Orthopaedic Surg Res. 2023;18(1):699. doi:10.1186/s13018-023-04128-2
- Zhang H, Wang L, Cui J. Maintaining hypoxia environment of subchondral bone alleviates osteoarthritis progression. Sci Adv. 2023;9(14):eabo7868. doi:10.1126/sciadv.abo7868
- Wang S, Zhu L, Li T, et al. Disruption of MerTK increases the efficacy of checkpoint inhibitor by enhancing ferroptosis and immune response in hepatocellular carcinoma, Cell reports. Medicine. 2024;5(2):101415. doi:10.1016/j.xcrm.2024.101415
- Shen S, Wu Y, Chen J, et al. CircSERPINE2 protects against osteoarthritis by targeting miR-1271 and ETS-related gene. Ann Rheumatic Dis. 2019;78(6):826–836. doi:10.1136/annrheumdis-2018-214786
- Zhao C, Sun G, Li Y, et al. Forkhead box O3 attenuates osteoarthritis by suppressing ferroptosis through inactivation of NF-κB/MAPK signaling. J Orthop Transl. 2023;39:147–162. doi:10.1016/j.jot.2023.02.005
- Ji FH, Fu XH, Li GQ, He Q, Qiu XG. FTO prevents thyroid cancer progression by SLC7A11 m6A methylation in a ferroptosis-dependent manner. Front Endocrinol. 2022;13:857765. doi:10.3389/fendo.2022.857765
- Tang Y, Hong F, Ding S, et al. METTL3-mediated m(6)A modification of IGFBP7-OT promotes osteoarthritis progression by regulating the DNMT1/DNMT3a-IGFBP7 axis. Cell Rep. 2023;42(6):112589. doi:10.1016/j.celrep.2023.112589
- Wang H, Liu X, Yang H, et al. Activation of the Nrf-2 pathway by pinocembrin safeguards vertebral endplate chondrocytes against apoptosis and degeneration caused by oxidative stress. Life Sci. 2023;333:122162. doi:10.1016/j.lfs.2023.122162
- Sun H, Peng G, Chen K, et al. Identification of EGFR as an essential regulator in chondrocytes ferroptosis of osteoarthritis using bioinformatics, in vivo, and in vitro study. Heliyon. 2023;9(9):e19975. doi:10.1016/j.heliyon.2023.e19975
- Wang Y, Chen Y, Wei Y. Osteoarthritis animal models for biomaterial-assisted osteochondral regeneration. Biomater Transl. 2022;3(4):264–279. doi:10.12336/biomatertransl.2022.04.006
- Cheng B, Zhang J, Shen Q, et al. Liproxstatin-1 alleviates cartilage degradation by inhibiting chondrocyte ferroptosis in the temporomandibular joint. Biol Cell. 2023;116(1):e202300042.
- Lin Z, Jiang T, Zheng W, et al. N6-methyladenosine (m6A) methyltransferase WTAP-mediated miR-92b-5p accelerates osteoarthritis progression. Cell Commun Signaling. 2023;21(1):199. doi:10.1186/s12964-023-01228-8
- Ye G, Li J, Yu W, et al. ALKBH5 facilitates CYP1B1 mRNA degradation via m6A demethylation to alleviate MSC senescence and osteoarthritis progression. Exp Mol Med. 2023;55(8):1743–1756. doi:10.1038/s12276-023-01059-0
- Maslov LN, Popov SV, Naryzhnaya NV, et al. The regulation of necroptosis and perspectives for the development of new drugs preventing ischemic/reperfusion of cardiac injury. Apoptosis. 2022;27(9–10):697–719. doi:10.1007/s10495-022-01760-x
- Han X, Zhang J, Liu J, et al. Targeting ferroptosis: a novel insight against myocardial infarction and ischemia-reperfusion injuries. Apoptosis. 2023;28(1–2):108–123. doi:10.1007/s10495-022-01785-2
- Luo M, Yan J, Hu X, et al. Targeting lipid metabolism for ferroptotic cancer therapy. Apoptosis. 2023;28(1–2):81–107. doi:10.1007/s10495-022-01795-0
- Wang Y, Zhang X, Chen Y, Zhu B, Xing Q. Identification of hub biomarkers and exploring the roles of immunity, M6A, ferroptosis, or cuproptosis in rats with diabetic erectile dysfunction. Andrology. 2023;11(2):316–331. doi:10.1111/andr.13265
- Chen K, Zhu P, Chu M, et al. What do osteoporosis and osteoarthritis have in common? An integrated study of overlapping differentially expressed genes in bone mesenchymal stem cells of osteoporosis and osteoarthritis. Gene. 2023;893:147914. doi:10.1016/j.gene.2023.147914
- Ma T, Ruan H, Lv L, et al. Oleanolic acid, a small-molecule natural product, inhibits ECM degeneration in osteoarthritis by regulating the Hippo/YAP and Wnt/β-catenin pathways. Food Funct. 2023;14(22):9999–10013. doi:10.1039/D3FO01902K