
Abstract
Myelofibrosis (MF) is a rare chronic BCR-ABL1 (breakpoint cluster region-Abelson murine leukemia viral oncogene homologue 1)-negative myeloproliferative neoplasm characterized by progressive bone marrow fibrosis, inefficient hematopoiesis, and shortened survival. The clinical manifestations of MF include splenomegaly, consequent to extramedullary hematopoiesis, cytopenias, and an array of potentially debilitating abdominal and constitutional symptoms. Dysregulated Janus kinase (JAK)-signal transducer and activator of transcription signaling underlies secondary disease-associated effects in MF, such as myeloproliferation, bone marrow fibrosis, constitutional symptoms, and cachexia. Common fatal complications of MF include transformation to acute leukemia, thrombohemorrhagic events, organ failure, and infections. Potential complications from hepatosplenomegaly include portal hypertension and variceal bleeding, whereas extramedullary hematopoiesis outside the spleen and liver – depending on the affected organ – may result in intracranial hypertension, spinal cord compression, pulmonary hypertension, pleural effusions, lymphadenopathy, skin lesions, and/or exacerbation of abdominal symptoms. Although allogeneic stem cell transplantation is the only potentially curative therapy, it is suitable for few patients. The JAK1/JAK2 inhibitor ruxolitinib is effective in improving splenomegaly, MF-related symptoms, and quality-of-life measures. Emerging evidence that ruxolitinib may be associated with a survival benefit in intermediate- or high-risk MF suggests the possibility of a disease-modifying effect. Consequently, ruxolitinib could provide a treatment backbone to which other (conventional and novel) therapies may be added for the prevention and effective management of specific MF-associated complications.
Introduction
Myelofibrosis (MF) is a chronic BCR-ABL1 (breakpoint cluster region-Abelson murine leukemia viral oncogene homologue 1)-negative stem cell myeloproliferative neoplasm (MPN) characterized by bone marrow fibrosis, ineffective hematopoiesis, extramedullary hematopoiesis (EMH), splenomegaly, shortened survival and progressive abdominal and constitutional symptoms, as well as other general chronic debilitating complaints.Citation1,Citation2 The MF-associated consequences and medical complications often result in premature death from infection, thrombohemorrhagic events, cardiac or pulmonary failure, and leukemic transformation.Citation3,Citation4 MF is an uncommon malignancy. Recent estimates of MF prevalence in the USA range from 3.6 to 5.7 per 100,000 persons, whereas estimates of MF incidence range from 1.7 to 2.4 per 100,000 persons.Citation5
Although the etiology of MF is unknown, environmental factors may well be relevant since MF has been linked in a small number of patients to radiation and exposure to petrochemicals such as benzene and toluene.Citation6–Citation8 MF can be primary (termed “primary myelofibrosis” [PMF]; formerly termed “idiopathic MF,” “agnogenic myeloid metaplasia,” or “myeloid metaplasia with MF”) or secondary, developing from polycythemia vera (PV; currently termed “post-PV MF” [PPV-MF]) or essential thrombocythemia (ET; currently termed “post-ET MF”).Citation9 The past decade has witnessed considerable progress in the understanding of the cellular and molecular biology of MPNs, and this has recently resulted in the addition of the Janus kinase (JAK) 1 and JAK2 inhibitor ruxolitinib to our therapeutic armamentarium.Citation10 Ruxolitinib is highly effective in the clinical management of patients with intermediate- or high-risk MF, particularly in those with disease-related symptoms and splenomegaly.Citation11–Citation13 Importantly, recent updates from two prospective, randomized, Phase III studies showed that patients with MF treated with ruxolitinib had improved survival over placebo and best available therapy, suggesting an overall survival benefit.Citation14,Citation15 However, the overall prognosis for advanced MF remains guarded, owing to a potentially remaining substantive burden of disease-related morbidities. The basis for these morbidities is the emergence of a remarkably broad array of general medical complications associated with this rare – and, until recently, rather therapeutically neglected – malignancy. Some of these complications are directly linked to excessive clonal myeloproliferation (the end result of which is leukemic transformation); however, most MF-associated complications are of more protean nature and deserve a deeper discourse. Here, we discuss some of the important issues related to the diagnosis and management of these complications.
Definition and pathogenetic features of MF
The current diagnostic criteria for PMF were defined by the World Health Organization in 2008 and are depicted in .Citation16 Available evidence indicates that PMF is a bona fide clonal stem cell malignancy.Citation17 MPNs comprise clonal hematologic diseases that are thought to arise from a transformation of a hematopoietic stem cell. The notion of “clonality” gained popularity in 1974 due to the astute seminal observations of Prchal and Axelrad,Citation18 and thereafter was confirmed by Fialkow et al,Citation19,Citation20 as well as various other investigators.Citation21 Currently, in contrast to our detailed understanding of chronic myeloid leukemia pathogenesis, which is defined by a single causative molecular lesion, the BCR-ABL1 fusion gene, we only have some essential clues to the molecular pathogenetic mechanisms for PV, ET, and PMF. A major clue was the recognition of increased signaling through the JAK-signal transducer and activator of transcription (STAT) pathway, comprised of JAKs and STATs, as well as through the phosphatidylinositol 3-kinase (PI3K)-AKT (also known as protein kinase B) pathway in erythroid and myeloid cells.Citation22–Citation24 The most significant clue to date came in 2005 with the identification of the somatic mutation JAK2V617F.Citation25–Citation28 This mutation in JAK2 exon 14, which occurs in at least 95% of patients with PV and about 60% of those with PMF and ET, results in a valine (V) to phenylalanine (F) substitution at codon 617.Citation29 This codon is located in the JH2 pseudokinase domain of JAK2, and the mutation is generally considered to negatively affect the JH2-mediated auto-inhibitory functionality of the enzyme, resulting in constitutive activation of the tyrosine kinase function. This in turn results in dysregulation of JAK-dependent signal transduction and activation of multiple downstream effectors, including STAT3 and STAT5.Citation13,Citation30 Dysregulated JAK-STAT signaling is now recognized as the central mechanism of MF pathobiologyCitation31 beyond aberrant myeloproliferation ( and ). For example, the efficacy of JAK inhibitors appears to play a role, in large part, in the reversal of secondary disease-related phenomena, such as inflammation and cachexia,Citation11,Citation32 that have no obvious relationship to myeloproliferation but are primary drivers of the MF symptom burden.
Figure 1 Pathogenic mechanisms in myelofibrosis involving dysregulated JAK-STAT signaling. Mutations affecting cytokine receptor function (eg, MPL mutations causing ligand-autonomous activation of the thrombopoietin receptor) or JAK2 mutations resulting in constitutive JAK2 activity lead to over-activation of JAK-STAT signaling in hematopoietic stem cells, with consequent myeloproliferation and excess production of proinflammatory cytokines.Citation108
Abbreviations: JAK, Janus kinase; MPL, myeloproliferative leukemia virus oncogene; STAT, signal transducer and activator of transcription.
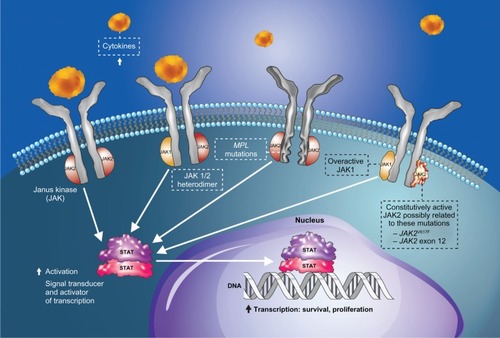
Figure 2 Pathobiology, main clinical manifestations, and common complications of myelofibrosis.
Abbreviations: CD34, cluster of differentiation 34; EMH, extramedullary hematopoiesis; Fb, fibroblast; HSC, hematopoietic stem cell; JAK, Janus kinase; MK, megakaryocyte; MO, monocyte; MPL, myeloproliferative leukemia virus oncogene; Ne, neutrophil; Ob, osteoblast; Oc, osteoclast; STAT, signal transducer and activator of transcription.
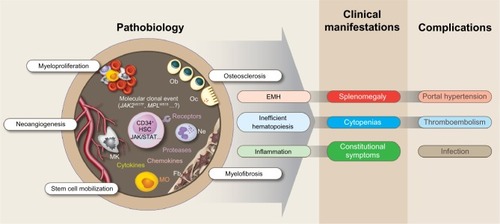
Table 1 World Health Organization (WHO) diagnostic criteria for primary myelofibrosis (PMF)Citation16
Although JAK2V617F is the most prevalent somatic mutation among patients with MF, a large proportion of patients with MF are JAK2V617F negative and, even in those who are JAK2V617F positive, the mutation is unlikely to be the disease-initiating event.Citation33,Citation34 An increasing number of mutations that directly or indirectly affect JAK-STAT signaling are being implicated in the pathobiology of MPNs, including PMF,Citation33–Citation35 revealing an unexpected genetic and epigenetic complexity of these neoplasms.Citation29,Citation36 Notably, findings of multiple mutations in the same patientCitation33 are consistent with the notion that MPNs are multiclonal diseases with as yet poorly understood clonal hierarchies.Citation37–Citation39
In 2006, mutations in exon 10 of the thrombopoietin receptor gene MPL (myeloproliferative leukemia virus oncogene) were noted in some patients with JAK2V617F-negative MPNs,Citation40 and further work confirmed their pathogenicity.Citation33 In 2007, mutations within exon 12 of JAK2 were also described in some patients with JAK2V617F-negative MPNs, but the precise role in the pathogenicity of MPNs remains enigmatic.Citation41,Citation42 To date, mutations in at least 12 genes have been described that can be involved in MPN pathogenesis.Citation43 In addition, recent work has established the important contribution of STAT5 to the molecular pathogenesis of MPNs,Citation44 confirming the central role of the STAT family of phosphorylation-regulatable nuclear transcription factors in the induction and sustenance of excessive proliferation of clonal myeloid progenitor cells in these neoplasms, including MF. Clearly, much has been learned about the molecular biology of MPNs, but the precise causative (“initiator” culprit) lesion remains, for the moment, an enigma.Citation34,Citation45
Natural history of MF
The evolution of bone marrow fibrosis, a cardinal feature of MF, is poorly understood. It is thought to represent a polyclonal reaction to several cytokines, particularly transforming growth factor-β, basic fibroblast growth factor, epidermal growth factor, platelet-derived growth factor, vascular endothelial growth factor, and calmodulin.Citation46 These moieties are mostly locally produced from the malignant clone throughout its various stages of aberrant differentiation (megakaryocytes, monocytes, or both).Citation47 The effects of these growth factors are mainly para- and autocrine; however, in advanced MF, as well as advanced PV and ET, progression of fibrosis appears to involve increasingly altered crosstalk between hematopoietic and stromal cells, resulting in the liberation of fibrogenic cytokines and the escape of malignant stem cells in the circulation with consequent EMH.Citation48,Citation49 Efforts are underway to thoroughly study other candidate factors involved in the development of fibrosis in patients with MPNs. Results of a recent study suggest that the risk of patients with PV developing PPV-MF is increased significantly by high (>50%) JAK2V617F allele burden.Citation50
Results of a recent study suggest that the bone marrow fibrosis grade (determined according to the European consensus on grading bone marrow fibrosis)Citation51 may have prognostic value in patients with PMF,Citation52 and thus may deserve attention during a patient’s risk assessment when allogeneic hematopoietic stem cell transplantation (allo-SCT) is considered. However, although allo-SCT may provide rapid regression of bone marrow fibrosis in some patientsCitation53 and at present is the only potentially curative therapy,Citation54 it is also associated with high risks of relapse, morbidity, and mortality.Citation55–Citation57
Transformation into secondary acute leukemia occurs in a small minority of patients with MPNs; it typically involves the myeloid lineages (secondary acute myeloid leukemia [sAML]) but, rarely, lymphoid transformation (secondary acute lymphoid leukemia) also may occur. Patients who develop acute leukemia have a median survival time of less than 3 months.Citation58 Leukemic transformation is rare in patients with non-fibrotic MPNsCitation59 but common in patients with MF.Citation3,Citation58,Citation60 The 10-year risk of leukemic transformation for patients with PMF has been estimated at 12%–31%, depending on the presence of thrombocytopenia and/or unfa-vorable karyotype.Citation61 A peripheral blood blasts count of ≥2% has been identified as an independent predictor of poor leukemia-free survival.Citation62 The pathogenetic events leading to leukemic transformation are poorly understood, but the presence of IDH (isocitrate dehydrogenase) mutations has been shown to be a significant independent prognostic factor of leukemic transformation.Citation35 In contrast, the presence of JAK2V617F is not essential for nor prognostic of leukemic transformation,Citation63,Citation64 with various case reports indicating that JAK2V617F allele burden may diminish or completely disappear during leukemic transformation of JAK2V617F-positive chronic-phase MF.Citation63,Citation65–Citation67 It has been shown that exposure to select cytoreductive agents may increase the risk of leukemic transformation, possibly by causing additional genetic and/or epigenetic lesions.Citation68,Citation69
Clinical presentation and complications of MF
The median age of patients with MF at the time of diagnosis is approximately 65 years.Citation3,Citation60 In the USA, MF appears to affect slightly more men than women.Citation60 Until recently, it was estimated that up to 21% of all patients with MF are asymptomatic at the time of diagnosis, which is often made by the determination of an abnormal blood count, usually indicating anemia, or an abnormal peripheral blood smear demonstrating leukoerythroblastosis, or after a routine physical examination revealing splenomegaly and sometimes hepatomegaly.Citation70 However, the results of recently published, Internet-based symptom surveys among patients with MPNs showed that the great majority (ie, more than 80%) of patients with MF experienced at least one disease-related symptom at the time of the survey.Citation71,Citation72 The most common presenting symptoms are constitutional symptoms and the consequences of ineffective hematopoiesis.Citation2,Citation73 Fatigue is extremely common; it is often relentless, chronic, and profoundly debilitating, and is also seen in patients who do not have anemia.Citation71
Constitutional symptoms, including fever, night sweats, pruritus, and weight loss, are thought to result from the abnormal production of cytokines.Citation13 Such symptoms not only tend to have a negative impact on a patient’s quality of life (QOL)Citation71 but also have been firmly associated with a poor prognosis for overall survival.Citation3 Both hepatomegaly and splenomegaly are considered fundamentally characteristic of MF and to be largely, although not exclusively, due to EMH. Splenomegaly may result from splenic sequestration of immature myeloid cells or other ill-defined mechanisms. Indeed, palpable splenomegaly (of any degree, from merely palpable to massive) has been noted in more than 80% of non-selected patients with MFCitation3 and can lead to a range of physical complaints, including generalized abdominal discomfort, left upper quadrant/subcostal pain, and early satiety. As splenomegaly worsens, it is not uncommon to witness patients developing severe generalized abdominal pain and sometimes an “acute abdomen”-like clinical picture; some patients may also experience splenic infarcts.Citation74
Progressive bone marrow fibrosis leads to a “myelophthisic” phenotype with worsening cytopenias, particularly thrombocytopenia and anemia.Citation70,Citation75,Citation76 The latter typically results in fatigue, weakness, palpitations, compensatory tachycardia, bone pain, and dyspnea (either exertional or at rest), and may lead to (or exacerbate) symptoms of tissue hypoxia in patients with vasculopathies, such as athero-sclerotic cardio-, cerebro-, and renovascular disease, or in those with pre-existing heart failure (combination of hypo-oxygenation and hypoperfusion).Citation77 Anemia also can negatively affect pulmonary function parameters in patients with various pre-existing lung diseases.Citation78 The above considerations are clinically meaningful and important because patients with MF tend to have significant general medical comorbidities owing to their (on average) advanced age. The consequences of thrombocytopenia, which may include bleeding of any grade/severity, can be compounded by acquired platelet dysfunction and sometimes a state of low-grade disseminated intravascular coagulation, resulting occasionally in a complex thrombohemorrhagic picture.Citation79 These latter manifestations may be catastrophic and are potentially life-threatening, particularly in the presence of portal hypertension. Moreover, MF is associated with an increased susceptibility to infections (viral, bacterial, and atypical),Citation70,Citation76 even if there is no evidence of frank leukopenia/neutropenia.Citation80
Hepatomegaly, which in various studies has been observed in 39%–65% of patients with MF at diagnosis,Citation3,Citation4,Citation76 may result in abnormal liver function tests, coagulopathy, and increased abdominal complaints.Citation81 MF is the most common cause of massive splenomegalyCitation82 and, with increased hepatic blood flow (or intrahepatic venous obstruction/stasis), marked splenomegaly may result in portal hypertension.Citation83 This complication is noted in about 7% of patients with MFCitation84 and may present with ascites and esophageal or gastric varices; bleeding in these varices may lead to catastrophic hemorrhage. Splanchnic (portal, mesenteric, or splenic) vein thrombosis and associated complications (eg, Budd–Chiari syndrome) also may occur in MF, although they do so less often in MF than in PV or ET.Citation85 Interestingly, although hyperplasia of Kupffer cells has been detected in MF, neither hepatic stellate cell activation nor hepatic parenchymal fibrosis are features of this malignancy.Citation86 In addition, EMH can result in a wide range of complications other than hepatosplenomegaly, depending on the specific organ involved, with potentially life-threatening consequences. EMH affecting the central nervous system may lead to intracranial hypertension, which in turn may result in chronic headaches, delirium, photophobia, papilledema, gait instability, alterations in the level of consciousness and, rarely, coma, paralysis, and/or death. In cases in which EMH develops in a paraspinal location, spinal cord compression may ensue, occasionally presenting as acute cord or spinal root/nerve compression syndrome.Citation87 Involvement of lymph nodes by this process can lead to generalized lymphadenopathy and, in advanced cases, lymphoceles. Pleural infiltration by EMH may result in pleural effusions and hemothorax.Citation88
EMH in the gastrointestinal tract manifests itself as exacerbation of already existing abdominal pain and/or intestinal lumen obstruction. Rarely, obstructive uropathy may result from EMH infiltration of the kidneys, ureters, and the bladder neck,Citation84 which may lead to renal failure. Other very rare secondary complications of EMH include gastric outlet obstruction, bile duct obstruction, acalculous cholecystitis from gallbladder infiltration, arthritis from synovial involvement, and renal colic from intrarenal or intra- or periureteric obstruction. Skin manifestations of EMH are quite rare and may include erythematous plaques, nodules, ulcers, bullae, myeloid leukemic-like infiltrates, and even neutrophilic dermatosis (Sweet’s syndrome).Citation87,Citation89 The risk of thromboembolic events in patients with MF (particularly PMF) appears to be considerably lower than that in patients with PV or ET. Nonetheless, cardiovascular, thromboembolic, and hemorrhagic complications are common in patients with MF,Citation70,Citation76 and several investigators reported thrombohemorrhagic events to be among the principal causes of death in patients with MF.Citation3,Citation4 The risk of leukemic transformation (mainly to sAML) appears to increase with time, and patients with sAML have a grim prognosis.Citation58,Citation61 Very seldom, leukemic transformation can present with granulocytic sarcomas (also known as “chloromas” in the earlier literature) at any anatomical site.Citation90,Citation91
Prognosis and MF-associated complications
MF is clearly associated with substantial and increasingly burdensome morbidity, which has a very significant negative impact on patients’ QOL and is associated with a substantial long-term risk (which increases over time) of developing potentially life-threatening complications. Because the median age of patients with MF is around 65 years at diagnosis, disease-associated complications are often compounded by concurring medical conditions such as diabetes, hypertension, atherosclerotic or pulmonary disease, and obesity. Current estimates suggest an overall survival of about 5 years for patients with intermediate-risk disease, and less than 2 years for those with high-risk disease.Citation61 Some patients, in particular those with low-risk disease, may survive for longer periods, but as the disease progresses, they often experience relentlessly progressive debilitating symptoms, inexorably worsening QOL, and increasing disability. Therefore, it is prudent to assess individual patients carefully and devise a treatment plan commensurate with each individual patient’s risk stratification and symptoms. The only treatment that can accord long-term remission and possible cure is allo-SCT. This treatment is available to a small minority (around 3%) of eligible patients with MF and carries significant risks and complications.Citation54,Citation74
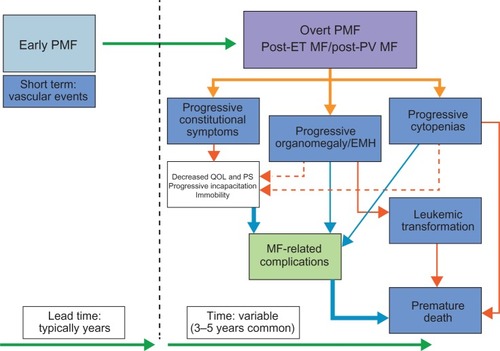
Over the past two decades, efforts have been made to identify clinical and laboratory parameters that are independently associated with prognosis in MF.Citation3,Citation4,Citation61 The principal candidate prognostic parameters have been age at diagnosis, constitutional symptoms, abnormal karyotype, anemia, thrombocytopenia, low reticulocyte count, peripheral blood CD (cluster of differentiation) 34+ progenitor cells, monocytosis, and peripheral blood blasts. Interestingly, the presence of hepatosplenomegaly has not been included in these prognostication systems. The most widely accepted prognostic scale so far is based on the 2009 International Working Group for Myelofibrosis Research and Treatment initiative and is known as the “International Prognostic Scoring System” (IPSS).Citation3 The IPSS validated the following parameters at diagnosis to be associated with a worse prognosis: age >65 years, presence of constitutional symptoms, hemoglobin <10 g/dL, leukocyte count >25 × 109/L, and ≥1% circulating blasts.Citation3 Compared with the IPSS, the Dynamic International Prognostic Scoring System gives more prognostic weight to the presence of disease-related anemia and is valid not only at diagnosis but at any time during disease progression.Citation4 Recent efforts have established the additional value of abnormal karyotype as an independent negative prognostic factor for overall and leukemia-free survival.Citation61 Conversely, patients with a normal karyotype seem to have a very low risk of transformation to acute leukemia. Nonetheless, even for these patients, substantial residual risk of shortened overall survival remains, suggesting that multiple and diverse clinical events (other than leukemic transformation) contribute to the morbidity and, indeed, the mortality associated with MF. Some of the mechanisms responsible for the emergence of complications in patients with MF (as delineated here) are summarized in .
Figure 3 Mechanisms of the emergence of complications in patients with myelofibrosis (MF).
Abbreviations: EMH, extramedullary hematopoiesis; ET, essential thrombocythemia; PMF, primary myelofibrosis; PS, performance status; PV, polycythemia vera; QOL, quality of life.
Management of MF-associated complications
Until recently, best available pharmacotherapy for MF consisted of the use of conventional agents that generally had limited and non-lasting efficacy and were often poorly tolerated, including cytoreductive agents such as hydroxyurea (hydroxycarbamide), and immunomodulatory agents.Citation92 Thus, treatment of MF was essentially palliative, without durably effective options for the alleviation of major clinical manifestations such as splenomegaly and MF-associated symptoms. The discovery of the essential role of dysregulated JAK-STAT signaling in the pathobiology of MF led to the development of JAK inhibitors as novel targeted therapies. The recent approval of ruxolitinib in MF in many countries – including the USA, where it is approved for the treatment of intermediate- or high-risk MF – is a validation of JAK inhibitor therapy in MF and a milestone in the development of targeted therapies for this disease. In light of the complexity of MF pathogenetics, experimental therapies targeting additional disease mechanisms are likely to have future roles in MF disease management.Citation92 is a summary of current management options for MF-associated complications that highlights the benefits of JAK-targeted therapy.
Table 2 Management of complications associated with myelofibrosis (MF)
Ruxolitinib is currently the only approved treatment for patients with MF that has been shown in pivotal randomized clinical trials to be highly effective in alleviating symptom burden and splenomegaly.Citation11,Citation12 In addition, ruxolitinib has been shown to reduce hepatomegaly in patients with prior splenectomy,Citation93 to mitigate cachexia-related weight loss and hypocholesterolemia in MF,Citation32 and to reduce the levels of cytokines driving systemic inflammation in this malignancy.Citation11,Citation13 Preliminary data for experimental therapies suggest that a number of JAK inhibitors currently in development also have the capacity to reduce splenomegaly.Citation94–Citation96 Although hydroxyurea may reduce splenomegaly in some patients with mostly non-massive splenomegaly, its benefit is usually of short duration and tolerability is poor.Citation97 Results of a randomized Phase III study showed that best available therapy, including the use of hydroxyurea in 47% of the patients, was significantly less effective than ruxolitinib in reducing MF-associated splenomegaly or improving several cardinal indices of health-related QOL.Citation12 Splenectomy is an option for patients with refractory symptomatic splenomegaly and/or portal hypertension but should only be considered if the qualifying patient has an adequate life expectancy.Citation98 Palliative splenic irradiation may be indicated for patients with highly symptomatic splenomegaly and adequate platelet count; however, the benefits are usually transient and, in some cases, profound and prolonged cytopenias may develop.Citation98
Currently available therapies have no or limited efficacy in the treatment of MF-related anemia. Thus, management of anemia largely depends on supportive care and the use of androgens or erythropoietin-stimulating agents. Immunomodulators, such as thalidomide and lenalidomide, may be effective in select patients but are generally poorly tolerated in the long term. A recent study of pomalidomide in patients with MF and significant anemia found that dosing was severely limited by tolerability, and treatment at tolerable doses was only moderately effective.Citation99 In addition, the manufacturer of pomalidomide recently announced in a press release that a Phase-3 double-blind, placebo-controlled study of pomalidomide in persons with myeloproliferative-neoplasm-associated myelofibrosis and red blood cell (RBC)-transfusion-dependence myelofibrosis and RBC-transfusion-dependence (RESUME)Citation100 did not meet its primary end point.Citation101 JAK inhibitors, with the possible exception of CYT387,Citation96 appear to have no beneficial effect on MF-related anemia, and, currently, no therapies in development clearly demonstrate efficacy in mitigating MF-related thrombocytopenia. Treatment of additional complications of MF, such as thrombosis, bleeding, portal hypertension, infections, chronic inflammation, and sAML consists mostly of supportive care and/or palliative therapies that are specific for the type of complication.
Because of the essential role of JAK2-STAT signaling in definitive hematopoiesis, decreases in blood cell counts are an expected effect of JAK2 inhibition. Thus, thrombocytopenia, and to a lesser extent anemia, are the most common adverse events associated with ruxolitinib therapy. However, the frequency and severity of these treatment-related cytopenias can be reduced by careful individual patient management. Consequently, the rates of grade 3 and 4 events of thrombocytopenia or anemia tend to be highest within the first 8–12 weeks of therapy,Citation11 and, in the two Phase III studies, cytopenias were rarely a cause for discontinuation of ruxolitinib therapy.Citation11,Citation12 Effective management of ruxolitinib-related thrombocytopenia requires initial dose titration, monitoring of platelet counts, and appropriate dose adjustments based on serially tested platelet counts.Citation10 Treatment-related decreases in hemoglobin level usually occur within the first 8–12 weeks after treatment initiation. However, hemoglobin levels generally recover with appropriate dose modifications and/or the use of RBC transfusions, on average returning to near baseline values by week 24 of therapy.Citation11
Conclusion
Many efforts since the seminal description of the mutations affecting JAK2 and the enhanced understanding of the universal presence of a dysregulated JAK-STAT signaling pathway in MF (even in the absence of a JAK2 mutation) have led to a wide array of therapeutic agents being studied clinically in this malignancy, ranging from adenosine triphosphate-competitive inhibitors of JAKs (eg, ruxolitinib) to immunomodulatory agents (eg, lenalidomide) and other novel approaches.Citation92 Current experience confirms the notion that treating patients with intermediate- and high-risk MF (either primary or secondary) with the JAK1 and JAK2 inhibitor ruxolitinib has an overall favorable benefit–risk profile. Robust clinical trial dataCitation11–Citation13 have led to the drug receiving regulatory approval in health authorities in many countries, including the USA,Citation10 Canada,Citation102 and the European Union.Citation103 The drug has a notable clinical impact on the management of patients with intermediate- or high-risk MF, with the majority achieving durable symptomatic responses and reduction of splenomegaly. Moreover, recent updates from two prospective, randomized, Phase III studies showed that patients with MF treated with ruxolitinib had improved survival over placebo (COntrolled MyeloFibrosis study with ORal JAK inhibitor Treatment: The COMFORT-I Trial [COMFORT-I])Citation14,Citation104 and best available therapy (COntrolled MyeloFibrosis study with ORal Janus-associated kinase [JAK] inhibitor Treatment-II: The COMFORT-II Trial [COMFORT-II]),Citation15,Citation105 suggesting an overall survival benefit. The median follow-up period for data reported to date remains relatively short for either trial (approximately 2 years), and there are indeed numerous challenges, both clinical and scientific, that need further elucidation, including the precise mode of action of ruxolitinib in this malignancy.
Interestingly, recent data from an exploratory analysis of bone marrow trephine biopsies in patients with MF treated at the MD Anderson Cancer Center who participated in the Phase I/II trial of ruxolitinibCitation10,Citation106 raise the possibility that JAK inhibitor therapy with ruxolitinib for a minimum of 2 years may retard the progression of bone marrow fibrosis in some patients.Citation107 Long-term hydroxyurea therapy had no comparable benefit in a European cohort of patients with MF. Data from randomized controlled studies are needed to substantiate a positive effect of ruxolitinib therapy on bone marrow fibrosis.
In conclusion, given the clinical efficacy and safety profile of ruxolitinib treatment in intermediate- and high-risk MF and with several other candidate agents of the same class currently under development,Citation92 the focus of the treating clinician should now be the prompt identification and effective preventive management of MF-associated complications.
Disclosure
TIM consults for Incyte Corporation and is on the speakers’ bureau for Bristol-Myers Squibb. SV has received research support from Incyte, Bristol-Myers Squibb, AstraZeneca, NS Pharma, Roche, Celgene, Gilead, Infinity, Exelixis, YM Bioscience, S*Bio, Geron, and Lilly. NJS and KV are employees of Incyte Corporation.
Acknowledgments
Assistance with editing an advanced draft of the manuscript was provided by Roland Tacke, PhD, of Evidence Scientific Solutions, and funded by Incyte Corporation.
References
- LevineRLGillilandDGMyeloproliferative disordersBlood200811262190219818779404
- GregorySAMesaRAHoffmanRShammoJMClinical and laboratory features of myelofibrosis and limitations of current therapiesClin Adv Hematol Oncol201199 Suppl 2211622362131
- CervantesFDupriezBPereiraANew prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and TreatmentBlood2009113132895290118988864
- PassamontiFCervantesFVannucchiAMDynamic International Prognostic Scoring System (DIPSS) predicts progression to acute myeloid leukemia in primary myelofibrosisBlood2010116152857285820947690
- MehtaJWangHIqbalSUMesaREpidemiology of myeloproliferative neoplasms in the United StatesLeuk Lymphoma Epub7292013
- AksoyMMalignancies due to occupational exposure to benzeneHaematologica19806533703736778789
- TondelMPerssonBCarstensenJMyelofibrosis and benzene exposureOccup Med (Lond)199545151527703476
- BoschXCampistolJMMontoliuJRevertLMyelofibrosis and focal segmental glomerulosclerosis associated with toluene poisoningHum Toxicol1988743573613410485
- MesaRAGreenABarosiGVerstovsekSVardimanJGaleRPMPN-associated myelofibrosis (MPN-MF)Leuk Res2011351121320684988
- Jakafi® (Ruxolitinib) tablets, for oral use [prescribing information]Wilmington, DEIncyte Corporation Available from: http://www.incyte.com/products/uspi_jakafi.pdfAccessed September 9, 2013
- VerstovsekSMesaRAGotlibJA double-blind, placebo-controlled trial of ruxolitinib for myelofibrosisN Engl J Med2012366979980722375971
- HarrisonCKiladjianJJAl-AliHKJAK inhibition with ruxolitinib versus best available therapy for myelofibrosisN Engl J Med2012366978779822375970
- VerstovsekSKantarjianHMesaRASafety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosisN Engl J Med2010363121117112720843246
- VerstovsekSMesaRAGotlibJEfficacy, safety and survival with ruxolitinib treatment in patients with myelofibrosis: results of a median 2-year follow-up of COMFORT-IHaematologica Epub2013913
- CervantesFKiladjianJJNiederwieserDLong-term safety, efficacy, and survival findings from Comfort-II, a phase 3 study comparing ruxolitinib with best available therapy (BAT) for the treatment of myelofibrosis (MF)Blood (ASH Annual Meeting Abstracts)201212021 Abstract 801
- VardimanJWThieleJArberDAThe 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changesBlood2009114593795119357394
- LatailladeJJPierre-LouisOHasselbalchHCFrench INSERM and the European EUMNET Networks on MyelofibrosisDoes primary myelofibrosis involve a defective stem cell niche? From concept to evidenceBlood200811283026303518669872
- PrchalJFAxelradAABone marrow responses in polycythemia veraN Engl J Med19742902413824827655
- AdamsonJWFialkowPJMurphySPrchalJFSteinmannLPolycythemia vera: stem-cell and probable clonal origin of the diseaseN Engl J Med197629517913916967201
- FialkowPJFaguetGBJacobsonRJVaidyaKMurphySEvidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cellBlood19815859169197296002
- XuMBrunoEChaoJThe constitutive mobilization of bone marrow-repopulating cells into the peripheral blood in idiopathic myelofibrosisBlood200510541699170515471948
- RöderSSteimleCMeinhardtGPahlHLSTAT3 is constitutively active in some patients with Polycythemia rubra veraExp Hematol200129669470211378264
- UgoVMarzacCTeyssandierIMultiple signaling pathways are involved in erythropoietin-independent differentiation of erythroid progenitors in polycythemia veraExp Hematol200432217918715102479
- VainchenkerWDelhommeauFConstantinescuSNBernardOANew mutations and pathogenesis of myeloproliferative neoplasmsBlood201111871723173521653328
- BaxterEJScottLMCampbellPJCancer Genome ProjectAcquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disordersLancet200536594641054106115781101
- JamesCUgoVLe CouédicJPA unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia veraNature200543470371144114815793561
- KralovicsRPassamontiFBuserASA gain-of-function mutation of JAK2 in myeloproliferative disordersN Engl J Med2005352171779179015858187
- LevineRLWadleighMCoolsJActivating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosisCancer Cell20057438739715837627
- CrossNCGenetic and epigenetic complexity in myeloproliferative neoplasmsHematology Am Soc Hematol Educ Program2011201120821422160036
- OkuSTakenakaKKuriyamaTJAK2 V617F uses distinct signalling pathways to induce cell proliferation and neutrophil activationBr J Haematol2010150333434420553273
- Quintás-CardamaAKantarjianHCortesJVerstovsekSJanus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyondNat Rev Drug Discov201110212714021283107
- MesaRAVerstovsekSGuptaVImprovement in weight and total cholesterol and their association with survival in ruxolitinib-treated patients with myelofibrosis from COMFORT-IBlood (ASH Annual Meeting Abstracts)201212021 Abstract 1733
- TefferiANovel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1Leukemia20102461128113820428194
- VainchenkerWConstantinescuSNJAK/STAT signaling in hematological malignanciesOncogene201332212601261322869151
- TefferiAJimmaTSulaiNHIDH mutations in primary myelofibrosis predict leukemic transformation and shortened survival: clinical evidence for leukemogenic collaboration with JAK2V617FLeukemia201226347548021912393
- VannucchiAMBiamonteFEpigenetics and mutations in chronic myeloproliferative neoplasmsHaematologica201196101398140221972209
- SchaubFXLooserRLiSClonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasmsBlood2010115102003200720061559
- RumiEHarutyunyanAElenaCIdentification of genomic aberrations associated with disease transformation by means of high-resolution SNP array analysis in patients with myeloproliferative neoplasmAm J Hematol2011861297497921953568
- HummelJMKleteckaMCSanksJKConcomitant BCR-ABL1 translocation and JAK2(V617F) mutation in three patients with myeloproliferative neoplasmsDiagn Mol Pathol201221317618322847163
- PikmanYLeeBHMercherTMPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasiaPLoS Med200637e27016834459
- ScottLMTongWLevineRLJAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosisN Engl J Med2007356545946817267906
- PassamontiFElenaCSchnittgerSMolecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutationsBlood2011117102813281621224469
- Abdel-WahabOPardananiABernardOAUnraveling the genetic underpinnings of myeloproliferative neoplasms and understanding their effect on disease course and response to therapy: proceedings from the 6th International Post-ASH SymposiumAm J Hematol201287556256822460584
- WalzCAhmedWLazaridesKEssential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in miceBlood2012119153550356022234689
- ConstantinescuSNVainchenkerWSmall-molecule inhibitors in myeloproliferative neoplasms: are we aiming for the right targets?Hematology Am Soc Hematol Educ Program2012201255356023233634
- Le Bousse-KerdilèsMCMartyréMCSamsonMCellular and molecular mechanisms underlying bone marrow and liver fibrosis: a reviewEur Cytokine Netw2008192698018632420
- TefferiAPathogenesis of myelofibrosis with myeloid metaplasiaJ Clin Oncol200523338520853016293880
- KreipeHBüscheGBockOHusseinKMyelofibrosis: molecular and cell biological aspectsFibrogenesis Tissue Repair20125Suppl 1S2123259436
- Le Bousse-KerdilèsMCPrimary myelofibrosis and the “bad seeds in bad soil” conceptFibrogenesis Tissue Repair20125Suppl 1S2023259918
- PassamontiFRumiEPietraDA prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complicationsLeukemia20102491574157920631743
- ThieleJKvasnickaHMFacchettiFFrancoVvan der WaltJOraziAEuropean consensus on grading bone marrow fibrosis and assessment of cellularityHaematologica20059081128113216079113
- GianelliUVenerCBossiAThe European Consensus on grading of bone marrow fibrosis allows a better prognostication of patients with primary myelofibrosisMod Pathol20122591193120222627739
- KrögerNKvasnickaMThieleJReplacement of hematopoietic system by allogeneic stem cell transplantation in myelofibrosis patients induces rapid regression of bone marrow fibrosisFibrogenesis Tissue Repair20125Suppl 1S2523259545
- GuptaVHariPHoffmanRAllogeneic hematopoietic cell transplantation for myelofibrosis in the era of JAK inhibitorsBlood201212071367137922700718
- PatriarcaFBacigalupoASperottoAGITMOAllogeneic hematopoietic stem cell transplantation in myelofibrosis: the 20-year experience of the Gruppo Italiano Trapianto di Midollo Osseo (GITMO)Haematologica200893101514152218728030
- BallenKKShresthaSSobocinskiKAOutcome of transplantation for myelofibrosisBiol Blood Marrow Transplant201016335836719879949
- ZangDYDeegHJAllogeneic hematopoietic cell transplantation for patients with myelofibrosisCurr Opin Hematol200916214014619468277
- MesaRALiCYKetterlingRPSchroederGSKnudsonRATefferiALeukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 casesBlood2005105397397715388582
- KundrandaMNTibesRMesaRATransformation of a chronic myelo-proliferative neoplasm to acute myelogenous leukemia: does anything work?Curr Hematol Malig Rep201271788622170483
- TefferiALashoTLJimmaTOne thousand patients with primary myelofibrosis: the mayo clinic experienceMayo Clin Proc2012871253322212965
- GangatNCaramazzaDVaidyaRDIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion statusJ Clin Oncol201129439239721149668
- TefferiAPardananiAGangatNLeukemia risk models in primary myelofibrosis: an International Working Group studyLeukemia20122661439144122289985
- Lopes da SilvaRRibeiroPLourençoAWhat is the role of JAK2(V617F) mutation in leukemic transformation of myeloproliferative neoplasms?Lab Hematol2011171121621421540
- HelbigGWieczorkiewiczAWoŸniczkaKWiśniewska-PiatyKRusekAKyrcz-KrzemieńSThe JAK2V617F tyrosine kinase mutation has no impact on overall survival and the risk of leukemic transformation in myelofibrosisMed Oncol20122942379238422383244
- KreftASpringerELipkaDBKirkpatrickCJWild-type JAK2 secondary acute erythroleukemia developing after JAK2-V617F-mutated primary myelofibrosisActa Haematol20091221363819713696
- WuYYHungHMChenTSChaoTYHoCLDecreased JAK2 V617F allele burden level in a myelofibrosis with myeloid metaplasia patient with leukemic transformationLeuk Res200832111783178618455791
- TheocharidesABoissinotMGirodonFLeukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutationBlood2007110137537917363731
- NielsenIHasselbalchHCAcute leukemia and myelodysplasia in patients with a Philadelphia chromosome negative chronic myeloproliferative disorder treated with hydroxyurea alone or with hydroxyurea after busulphanAm J Hematol2003741263112949887
- KiladjianJJChevretSDosquetCChomienneCRainJDTreatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980J Clin Oncol201129293907391321911721
- VarkiALottenbergRGriffithRReinhardEThe syndrome of idiopathic myelofibrosis. A clinicopathologic review with emphasis on the prognostic variables predicting survivalMedicine (Baltimore)19836263533716633248
- MesaRANiblackJWadleighMThe burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patientsCancer20071091687617123268
- JohanssonPMesaRScherberRAssociation between quality of life and clinical parameters in patients with myeloproliferative neoplasmsLeuk Lymphoma201253344144421883029
- Abdel-WahabOILevineRLPrimary myelofibrosis: update on definition, pathogenesis, and treatmentAnnu Rev Med20096023324518947294
- BarbuiTBarosiGBirgegardGPhiladelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNetJ Clin Oncol201129676177021205761
- MichielsJJClinical, pathological and molecular features of the chronic myeloproliferative disorders: MPD 2005 and beyondHematology200510Suppl 121522316188676
- HasselbalchHIdiopathic myelofibrosis: a clinical study of 80 patientsAm J Hematol19903442913002195869
- ScheinbergPYoungNSHow I treat acquired aplastic anemiaBlood201212061185119622517900
- OuelletteDRThe impact of anemia in patients with respiratory failureChest20051285 Suppl 2576S582S16306056
- PrenticeCRAcquired coagulation disordersClin Haematol19851424134423899441
- WolachBGavrieliRManorYLishnerMLeukocyte function in chronic myeloproliferative disordersBlood Cells Mol Dis19982445445519887281
- PereiraABrugueraMCervantesFRozmanCLiver involvement at diagnosis of primary myelofibrosis: a clinicopathological study of twenty-two casesEur J Haematol19884043553613366226
- O’ReillyRASplenomegaly at a United States County Hospital: diagnostic evaluation of 170 patientsAm J Med Sci199631241601658853064
- Alvarez-LarránAAbraldesJGCervantesFPortal hypertension secondary to myelofibrosis: a study of three casesAm J Gastroenterol2005100102355235816181389
- TefferiAMyelofibrosis with myeloid metaplasiaN Engl J Med2000342171255126510781623
- AngerBRSeifriedEScheppachJHeimpelHBudd-Chiari syndrome and thrombosis of other abdominal vessels in the chronic myeloproliferative diseasesKlin Wochenschr198967168188252796252
- Bioulac-SagePRouxDQuintonALamouliatteHBalabaudCUltrastructure of sinusoids in patients with agnogenic myeloid metaplasiaJ Submicrosc Cytol19861848158213783800
- KochCALiCYMesaRATefferiANonhepatosplenic extramedullary hematopoiesis: associated diseases, pathology, clinical course, and treatmentMayo Clin Proc200378101223123314531481
- NadrousHFKrowkaMJMcClureRFTefferiALimKGAgnogenic myeloid metaplasia with pleural extramedullary leukemic transformationLeuk Lymphoma200445481581815160962
- AltomareGCapellaGLFrigerioESweet’s syndrome in a patient with idiopathic myelofibrosis and thymoma-myasthenia gravis-immunodeficiency complex: efficacy of treatment with etretinateHaematologica199681154588900854
- ChanACKwongYLLamCCGranulocytic sarcoma of megakaryoblastic differentiation complicating chronic idiopathic myelofibrosisHum Pathol19962744174208617486
- FrohnaBJQuintDJGranulocytic sarcoma (chloroma) causing spinal cord compressionNeuroradiology19933575095118232876
- AtallahEVerstovsekSEmerging drugs for myelofibrosisExpert Opin Emerg Drugs201217455557023186315
- BenjaminiOJainPEstrovZKantarjianHMVerstovsekSTherapeutic effects of ruxolitinib in patients with myelofibrosis without clinically significant splenomegalyBlood2012120132768276923019204
- SantosFPKantarjianHMJainNPhase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosisBlood201011561131113620008298
- PardananiAGotlibJRJamiesonCSafety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosisJ Clin Oncol201129778979621220608
- PardananiALabordeRRLashoTLSafety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosisLeukemia20132761322132723459451
- VannucchiAMManagement of myelofibrosisHematology Am Soc Hematol Educ Program2011201122223022160038
- MesaRAHow I treat symptomatic splenomegaly in patients with myelofibrosisBlood2009113225394540019332765
- DaverNShastriAKadiaTModest activity of pomalidomide in patients with myelofibrosis and significant anemiaLeuk Res Epub7242013
- Celgene CorporationPhase-3 double-blind, placebo-controlled study of pomalidomide in persons with myeloproliferative-neoplasm-associated myelofibrosis and RBC-transfusion-dependence myelofibrosis and RBC-transfusion-dependence (RESUME)ClinicalTrialsgov [website on the Internet]Bethseda, MDUS National Library of Medicine2010 [updated November 13, 2012]. Available from: http://clinicaltrials.gov/show/NCT01178281. NLM identifier: NCT01178281Accessed September 9, 2013
- Celgene CorporationCelgene reports first quarter 2013 operating and financial results [web page on the Internet]Summit, NJCelgene Corporation nd. Available from: http://ir.celgene.com/phoenix.zhtml?c=111960&p=irol-newsArticle&ID=1811033&highlight=myelofibrosisAccessed August 19, 2013
- JAKAVI® the first medication to receive health canada approval to treat patients with myelofibrosis [press release]CNW Group201275 Available from: http://www.newswire.ca/en/story/1003655/-pr-jakavi-the-first-medication-to-receive-health-canada-approval-to-treat-patients-with-myelofibrosisAccessed July 17, 2013
- NovartisAGNovartis drug JAKAVI® first medication to receive European Commission approval to treat patients with myelofibrosis [media release]BaselNovartis AG2012828 Available from: http://www.novartis.com/newsroom/media-releases/en/2012/1636508.shtmlAccessed July 17, 2013
- Incyte CorporationCOntrolled MyeloFibrosis Study With ORal JAK Inhibitor Treatment: The COMFORT-I TrialClinicalTrialsgov [website on the Internet]Bethseda, MDUS National Library of Medicine2009 [updated May 13, 2013]. Available from: http://clinicaltrials.gov/show/NCT00952289. NLM identifier: NCT00952289Accessed September 9, 2013
- Novartis PharmaceuticalsCOntrolled MyeloFibrosis study with ORal Janus-associated kinase (JAK) inhibitor Treatment-II: The COMFORT-II TrialClinicalTrialsgov [website on the Internet]Bethseda, MDUS National Library of Medicine2009 [updated March 12, 2013]. Available from: http://clinicaltrials.gov/show/NCT00934544. NLM identifier: NCT00934544Accessed September 9, 2013
- Incyte CorporationOpen label ruxolitinib (INCB018424) in patients with myelofibrosis and post polycythemia vera/essential thrombocythemia myelofibrosisClinicalTrialsgov [website on the Internet]Bethseda, MDUS National Library of Medicine2007 [updated October 9, 2012]. Available from: http://clinicaltrials.gov/show/NCT00509899. NLM identifier: NCT00509899Accessed September 9, 2013
- KvasnickaHMThieleJBueso-RamosCEExploratory analysis of the effect of ruxolitinib on bone marrow morphology in patients with myelofibrosisJ Clin Oncol201331Suppl Abstract 7030
- VaddiKSarlisNJGuptaVRuxolitinib, an oral JAK1 and JAK2 inhibitor, in myelofibrosisExpert Opin Pharmacother201213162397240723051187