 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Solid lipid nanoparticles (SLNs) have been used for carrying different therapeutic agents because they improve absorption and bioavailability. The aim of the study was to prepare lipidic nanoparticles containing cyclosporine (CyA) by the emulsification-diffusion method and to study their physicochemical stability. Glyceryl behenate (Compritol® ATO 888) and lauroyl macrogolglycerides (Gelucire® 44/14) were used as carrier materials. Nanoparticles with good stability were obtained with Gelucire®, while it was difficult to obtain stable systems with Compritol®. Systems with Gelucire® were characterized by particle size, Z-potential, differential scanning calorimetry (DSC), scanning electron microscopy (SEM), entrapment efficiency and in vitro release. Particle size and Z-potential were evaluated for at least three months. With a high CyA content (≥60 mg) in Gelucire® SLNs, variations in size were greater and particle size also increased over time in all batches; this effect may have been caused by a probable expulsion of the drug due to the lipid’s partial rearrangement. While the Z-potential decreased 10 mV after three months, this effect may be explained by the superficial properties of the drug that make the molecules to be preferably oriented at the solid-liquid interface, causing a change in the net charge of the particle. SEM confirmed size and shape of the nanoparticles. DSC studies evidenced that CyA affects the lipid structure by a mechanism still unknown. The entrapment efficiency was higher than 92%, and CyA release from SLNs was relatively fast (99.60% in 45 min).
Introduction
In the 90s, a new type of carrier was developed, the so-called solid lipid nanoparticles (SLNs) which have been studied and modified from the original concept into more complex systems (eg, nanostructured lipid carriers or lipid-drug conjugate),Citation1,Citation2 but basically the usage of lipid materials to form the carrier has presented many advantages over other kinds of materials. Advances in this field of investigation are reported in literature mainly related to the method of production (eg, electrospray technique,Citation3 coacervation),Citation4 the type of drug being encapsulated (eg, water soluble drugs,Citation5 gene delivery),Citation6 or the enhancement of bioavailability and absorption (eg, pentoxifylline,Citation7 digoxin).Citation8
SLNs are submicron-sized carriers composed of a lipid solid matrix stabilized by a surfactant. Such systems have some advantages over other colloidal carriers (nanoemulsions, microparticles, polymeric nanoparticles, liposomes, etc), such as low toxicity, high drug payload, capability of including lipophilic and hydrophilic drugs, drug targeting, controlled release (fast or sustained), and occlusive properties.Citation9
SLNs can be produced by many methods, although the following are the most common: i) hot high pressure homogenization (HPH) technique and cold HPH technique; ii) solvent emulsification/evaporation (SEE); iii) high shear homogenization (HSH) and/or ultrasound, and iv) microemulsion.Citation10
The most popular method is HPH; however, with both techniques (hot and cold), the drug, lipid, or other components may suffer chemical degradation due to the need of melting the lipid and/or to the increment of sample temperature during the process. Class I (highly toxic) or Class II (toxic) International Conference on Harmonization solvents are required to obtain SLNs by the SEE method, thus, its use is limited because of toxicological implications. With HSH and/or ultrasound, there is a broad particle size distribution and the product can be contaminated during the ultrasonication step. These factors may lead to particle growth and physical instability. The microemulsion method has the disadvantage of requiring high concentrations of surfactants and co-surfactants, and a great quantity of water should be eliminated from the final product.
Recently, Quintanar-Guerrero et alCitation10 proposed a modification of the emulsification-diffusion method (EDM) to prepare SLNs. In this modification, the author used a lipid instead of a preformed polymer. In general, a pre-emulsion is prepared; the internal phase contains the drug and the lipid which are solubilized in a partially water-miscible organic solvent, and the external phase is an aqueous solution of the stabilizer. This step can be performed with a cold or hot solvent, depending on the lipid’s solubility in the organic phase. Then, the solvent diffuses into the external phase when water is added to the system, causing the formation of nanoparticles (NP). The rest of the solvent can be eliminated by the use of techniques such as cross-flow filtration, or under reduced pressure.
In addition to the advantages mentioned before, one of the most interesting features of SLNs is that they can improve the absorption of various drugs, making them an attractive alternative for the administration of substances with poor aqueous solubility and absorption.Citation11−Citation13
Cyclosporine (CyA) is a neutral, highly lipophilic undecapeptide obtained from Tolypoclaudium inflatum and Cylindrocarpus lucidum, and is used as an immunosuppressant drug to prevent transplant rejection. It is a Class II drug that has been studied in many pharmaceutical formulations due to its absorption and bioavailability problems.Citation14−Citation19 In recent years, SLNs containing CyA have been prepared using different preparation methods, optimized and studied by some research groups around the world.Citation20−Citation23 In this work EDM was used to prepare stable CyA-containing SLNs, showing an alternative to encapsulate this drug for the reason that CyA has exhibited absorption and bioavailability problems since it was launched into the market, and such lipidic carriers may represent a good alternative to improve these parameters.Citation2,Citation24,Citation25 In the preformulation phase of this study, we performed the EDM at room temperature (RT) for Gelucire® 44/14, and at controlled temperature (65°C, CT) for Compritol® 888 ATO. Only Gelucire® gave stable NP, so that systems with this lipid were characterized by their stability, particle size, Z-potential, morphology, thermal behavior, and entrapment efficiency. In the last phase of the study, in vitro drug release was evaluated, which is the first step of a further bioavailability study. Because of its adequate stability, this nanocarrier can be useful in further investigations for oral administration.
Materials and methods
Materials
CyA was a contribution from Moléculas Finas, SA de CV (DF, Mexico). Glyceryl behenate (Compritol® ATO 888) and lauroyl macrogolglycerides (Gelucire® 44/14) were provided by Lubrizol (Mexico City, Mexico). Polyvinyl alcohol (PVAL, Elvanol® 5105) was obtained from Comercial Argento (DF, Mexico).
Distilled water was obtained from a Milli-Q station. Analytical grade ethyl acetate and methyl ethyl ketone (Fermont, Monterrey, NL, Mexico) were selected according to their low toxicity. All other chemical reagents were of analytical grade.
Method of production of SLN
According to previous data (Quintanar-Guerrero, 2005),Citation10 stable SLNs were obtained with Gelucire® 44/14 or Compritol ® 888 ATO. Furthermore, the low toxicity and high biocompatibility of these materials are well known.
Preparation of SLNs at RT
The lipid used in this case was Gelucire® 44/14, since it is soluble at RT in ethyl acetate, which was used as the internal phase in the pre-emulsification step. This partially water-miscible solvent was selected because in previous studies, stable particles of nanometric size were obtained. In this approach, at RT the amount of CyA was evaluated as variable, since the optimal amount of lipid, the type and amount of stabilizer, and the stirring rate have been defined in a previous study.Citation10,Citation26
The solvent and water were mutually saturated for 10 min at RT in order to generate equilibrium between the two liquids. Variable amounts of CyA (20, 25, 30, 40, 60, 120, and 200 mg) and 400 mg of lipid (Gelucire® 44/14) were weighted.
The drug and lipid were dissolved in 20 mL of water-saturated solvent, which were emulsified with 40 mL of solvent-saturated aqueous solution containing 5% w/v of PVAL, using a mechanical stirrer (Caframo® RZR-1; Ontario, Canada; propeller: PR 31; Heidolph®, Schwabach, Germany) at 1,800 rpm for 10 min. Then, 160 mL of water (dilution medium) were added to allow the solvent to diffuse into the external phase, thus causing the aggregation of the lipid and the subsequent formation of SLNs. Finally, the solvent was eliminated from the sample by vacuum distillation at 30°C and 70 mmHg. The remaining stabilizer and possible un-entrapped drug was removed by ultracentrifugation at 30,000 rpm for 50 min (Beckman® Optimal LE-80K, CA, USA).
In this same way, empty SLNs and batches containing only CyA were prepared. All batches were produced in triplicate and the average size was immediately measured.
Preparation of SLNs at CT
In this case, we used Compritol® for particle preparation, and heating was required to dissolve the lipid; therefore, keeping both phases at a CT (65°C that is lower than lipid’s melting point) during the entire process was a crucial factor to obtain a system with good characteristics. In general, 200 mg of lipid were weighted and dissolved in 20 mL of water-saturated solvent (methyl ethyl ketone). Variable amounts of CyA (5, 6, 8, and 10 mg) were added. This phase was emulsified with 40 mL of solvent-saturated aqueous solution containing 5% w/v PVAL, using a mechanical stirrer at 1,400 rpm for 10 min. Afterwards, 160 mL of water were added in order to cause the solvent diffusion to the continuous phase and to start the lipid aggregation and formation of NPs. Vacuum distillation was used to eliminate the solvent. PVAL and/or any remaining drug were removed in the same way as described above. Also empty SLN were prepared. Each batch was prepared in triplicate, and the average size was evaluated.
Characterization of the systems
Particle size analysis
The average size was determined by the dynamic light scattering technique (Coulter® N4, CA, USA). Each measurement was obtained at a 90° fixed-angle for 180 s, at 25°C, and the laser light wavelength (He/Ne, 10 mW) was set at 678 nm. A digital correlator was used to analyze the scattering intensity data under a unimodal analysis mode. All dispersions were diluted with distilled water until the light scattering signal (reported as counts per second) of the particles in suspension was within the instrument’s sensitivity range. All batches prepared were measured in triplicate. Also, the macroscopic aspect of the systems was followed for six months in order to detect any instability signs and these results were corroborated with the information obtained in every analysis by dynamic light scattering technique.
Z-potential
Dispersions were diluted (1:10) with filtered water (0.22 μm pore). The electrophoretic mobility was transformed into Z-potential by applying the Smoluchowski approximation. Measurements were made in triplicate (Malvern Instruments ® NS ZEN 3600, Worcestershire, UK) at 25°C in a capillary cell.
Scanning electron microscopy (SEM)
After removing the excess stabilizer in the sample by two centrifugations (30,000 rpm/50 min) and following particle resuspension in water, a few drops of the dispersion were placed on a slab and dried under vacuum at RT. A Sputter Coater® JFC-1100 (JEOL, Tokyo, Japan) was used to coat the dried samples with gold (~20 nm thickness), placing them onto stubs. Finally, the samples were observed under a JSM-6400® SEM (JEOL, Tokyo, Japan).
Differential scanning calorimetry (DSC)
DSC measurements were carried out on the following samples: a) CyA; b) Gelucire® 44/14; c) PVAL; d) physical mixtures of CyA-Gelucire® (40, 80, and 200 mg of CyA with 400 mg of Gelucire® in all cases); e) physical mixtures of CyA-PVAL (20, 80, and 200 mg of drug with 100 mg of PVAL in all cases); f) empty SLNs; g) batches containing only CyA (without lipid); and h) batches with variable amounts of CyA (20, 60, and 200 mg) and a constant amount of Gelucire® (400 mg). Batches containing only CyA were prepared in order to check if drug nanocrystals were formed. All samples were dried and stored in a desiccator at RT and standard humidity. Quantities between 3–6 mg of each sample were weighted. DSC studies were performed in a DSC Q10® (TA Instruments, DE, USA), using aluminum open pans. The heating rate was 10°C/min, with a temperature range of 0–230°C and a 50 mL/min nitrogen flow. Indium was used as the reference standard to check the calibration of the device.
Stabilizer quantification
The remaining amount of PVAL in SLNs was determined by a colorimetric method, based on the formation of a stable complex with iodine in the presence of boric acid.Citation27 After lyophilizing an SLN dispersion (30 mg of drug with 400 mg of Gelucire®), about 10 mg of the sample were weighted and 5 mL of ethanol were added. The sample was stirred for 24 h at RT. Then, 3 mL of distilled water were added and stirring was continued for 3 h. Samples were filtered through a membrane (0.22 μm) and the volume was adjusted to 10 mL with distilled water. Then 5 mL were taken from this solution and mixed with 2 mL of 0.65 M boric acid and 1 mL of an iodine solution (0.05 M iodine and 0.15 M potassium iodide). The analysis was performed in triplicate. The absorbance of the resulting samples was measured at 640 nm (Varian® Cary IE 9531003 Spectrophotometer, Australia) and this result was interpolated in a calibration curve for PVAL (linear within the range of 5–50 μg/mL, R2 = 0.9993). A solution composed of 5 mL of water, 2 mL of boric acid solution, and 1 mL of iodine solution was used as blank.
Drug loading and entrapment efficiency
Three batches with 400 mg of Gelucire® 44/14 and 30 mg of CyA were prepared at RT as described above and lyophilized, the reason for preparing this batch is because particle size had little variation and Z-potential had the highest value. A combination of 20 mg of lyophilized NP and 2 mL of ethanol were put into a beaker and were subsequently agitated for 24 h. After filtering the samples, 6,000 nL were applied on a reverse-phase plate to perform the determination by high performance thin layer chromatography (HPTLC); mobile phase acetonitrile-water (65:35) + 1% triethylamine, saturation time 15 min, absorbance-reflection mode, deuterium lamp, λ = 240 nm). The results were interpolated in a calibration curve which was linear within the range of 2–10 μg, R2 = 0.9950. The analysis was made in triplicate.
Equations (1) and (2) were applied to calculate drug loading (DL) and entrapment efficiency (EE):
It is important to introduce correction factor 1/(1-fraction of residual PVAL) to avoid an overestimation of the EE, due to the fact that during the SLN formation process, a certain amount of stabilizer remains on the carrier’s surface. The initial CyA content is the theoretical CyA loading in the batch.
In vitro release
A batch with 50 mg of CyA and 400 mg of Gelucire® was prepared as described previously and placed in a glass flask containing 10 mL of sodium lauryl sulfate (0.5% w/v aqueous solution). The stoppered flask was kept at 37°C in a water bath. Then, 2 mL samples were withdrawn from the medium at 5, 15, 30, 45, and 60 min, and then at 2, 3, and 4 h, replacing the medium with a fresh one at the same temperature and introducing it through the same filter holder; in this way sink conditions were maintained during the test. The samples were withdrawn through a syringe adapted to a filter holder (Swinnex® 13; Millipore, MA, USA) using IsoporeTM membrane filters (0.1 μm, VCTP; Millipore). Residual water was removed from each sample under reduced pressure, and then 2 mL of ethanol were added. Following stirring (1 h), the samples were filtered and quantified by HPTLC, as mentioned before.
Turbidity measurement during in vitro release
Turbidity (τ) was measured in each sample taken during the release experiment, as a complementary test to follow up changes in particle size of SLNs in the medium with surfactant. The sample was placed in a quartz cell and was analyzed at 360 nm. Absorbance is related to τ by the next equation:Citation28
where l is the optical length (1 cm), and A is the absorbance.
Results
With both methods (at RT and CT), it was possible to obtain SLNs, unless there was a statistical difference in size for empty NP, P < 0.05 ().
Table 1 Mean particle size of empty SLNs prepared at RT (Gelucire® = 400 mg) and at CT (Compritol® = 200 mg), with 5% w/v PVAL; n = 3
During particle size evaluation on batches with Compritol®, a relatively rapid destabilization of the systems was evident, even with a lower CyA content (5 mg), leading to the presence of macroscopic agglomerates. The initial Z-potential (t = 0) was under 30 mV in all batches with Compritol®, and was associated with a low dispersion stability. Also, a higher particle size variation in SLN batches compared with Gelucire ® was observed. Based on these results, it was concluded that Compritol® is not adequate to produce CyA-SLNs, due to poor stability related problems.
summarizes the composition and some characterization results obtained for all the systems prepared with Gelucire®.
Table 2 Composition of the formulations, percentage (w/w) of CyA with respect to the weight of lipid, mean particle size, Z-potential and probable day of aggregation of SLNs prepared at RT (Gelucire®) with 5% w/v PVAL
In the case of SLNs prepared with Gelucire®, the macroscopic evaluation of the systems showed that the time of appearance of abundant and bigger agglomerates was shorter as the drug content in the dispersion increased (≥60 mg CyA) ().
SEM showed that all batches evaluated had a solid matrix structure without crystal formation ().
Figure 1 Scanning electron micrographs of loaded Gelucire® 44/14 SLNs with different theoretical loadings (bar = 1 μm). A) CyA-Gelucire® (20:400 mg; 5% CyA) t = 0; B) CyA-Gelucire® (40:400 mg; 10% CyA) t = 0; C) CyA-Gelucire® (30:400 mg; 7.5% CyA) t = 0; D) CyA-Gelucire® (30:400 mg; 7.5% CyA) t = 4 months.
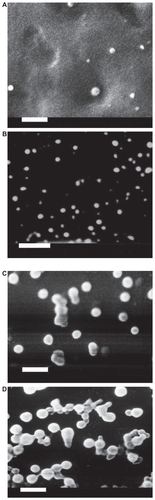
It was evident that alongside time the particles started a process of aggregation (), which could be confirmed with measurements of particle size (dynamic light scattering and visual appearance).
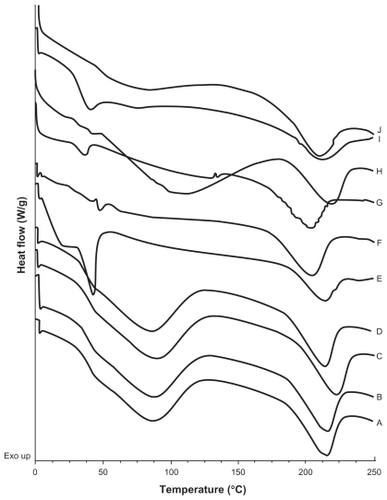
A calorimetric analysis was performed on free samples of Gelucire®, CyA, PVAL, physical mixtures (CyA-Gelucire and PVAL-CyA), and loaded and unloaded SLNs in order to determine the physical state of the drug and lipid, and to find out whether there were any interactions between them. show the thermograms obtained. shows the thermal events of raw materials. Enthalpy of fusion of the drug is at 128.5°C. The first peak of Gelucire® at 40.6°C is attributable to the fusion of the major proportion of glycerides, and the endotherm at 136.88°C corresponds to minority components of the mixture of glycerides. For PVAL the thermal events corresponding to its glass transition are shown. The physical mixtures analyzed are shown in . For physical mixtures CyA-Gelucire® we observed any peak corresponding to crystal melting of drug, and no changes are observed in enthalpy fusion of Gelucire® (around 40°C in the three physical mixtures). An effect of drug amount is observed in physical mixtures CyA-PVAL; as drug increases in the physical mixture it is more evident its melting point. Thermal events of all batches analyzed are shown in . None of them showed the peak corresponding to crystal melting of the drug and Gelucire® peak (40.6°C) showed less sharpness at t = 0 with high drug amounts in the SLN batches, suggesting that CyA somehow affected lipid’s structure.
Figure 2 DSC thermograms of A) CyA; B) Gelucire® 44/14; C) PVAL.
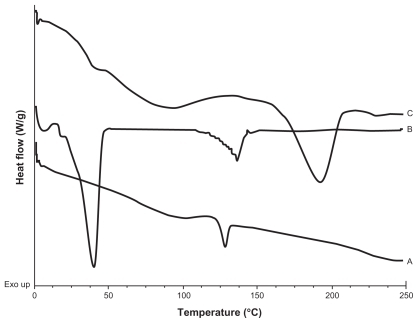
Figure 3 DSC thermograms of A) Physical mixture of CyA-Gelucire® (40:400 mg w/w); B) Physical mixture of CyA-Gelucire® (80:400 mg w/w); C) Physical mixture of CyA-Gelucire® (200:400 mg w/w); D) Physical mixture of CyA-PVAL (20:100 mg w/w); E) Physical mixture of CyA-PVAL (80:100 mg w/w); and F) Physical mixture of CyA-PVAL (200:100 mg w/w).
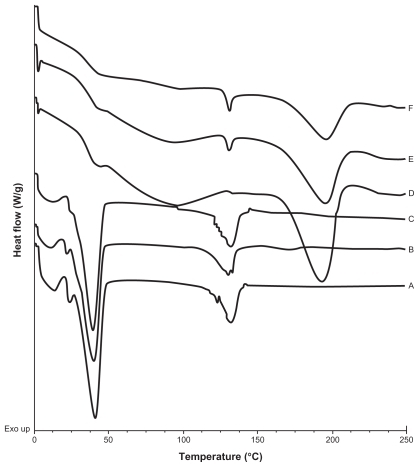
Figure 4 DSC thermograms of A) and B) batches prepared with CyA only, t = 0 and t = 5 months respectively; C) and D) empty SLNs; t = 0 and t = 5 months respectively; E) and F) SLNs with 5% CyA (drug-lipid ratio 20:400 (w/w)), t = 0 and t = 5.5 months, respectively; G) and H) SLNs with 15% CyA (drug-lipid ratio 60:400 (w/w)), t = 0 and t = 5.5 months, respectively; I) and J) SLNs with 50% CyA (drug-lipid ratio 200:400 (w/w)), t = 0 and t = 5.5 months, respectively.
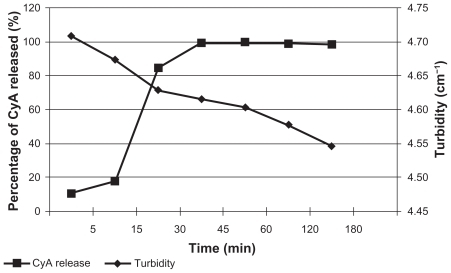
The in vitro release results and the turbidity measurement are shown in .
Figure 5 In vitro release of CyA and turbidity changes during this experiment.
The remaining PVAL quantified in the formulation Gelucire®-CyA (400:30 mg) was 4.37% ± 0.08 (n = 3). The residual PVAL for polymeric NPs is reported to be proximately 2%–4%,Citation29 so it can be concluded that purification process of the dispersion was adequate.
In the results for encapsulation efficiency are shown. In all systems these values were greater than 92%.
Discussion
As was stated before, a relatively rapid destabilization was evident during particle size evaluation on batches with Compritol ®, even with a lower CyA content (5 mg), leading to the presence of macroscopic agglomerates at the second day of analysis. Comparing the quantity of drug that Compritol® SLNs can carry with that of Gelucire®, it can be assumed that chemical composition (higher content of hydrophobic and longer chain length, >83% C22) and the high degree of crystallinity of the former could lead to drug expulsion from the NP easily. In this respect, Varia et alCitation22 corroborate that chemical composition of the lipid has an important effect to incorporate drugs in SLNs; a lipid which is formed mainly by structurally similar molecules cannot accommodate big quantities of drug compared with a lipid which is a mixture of mono-, di- and triglycerides, and has the ability to offer many imperfections to incorporate the drugs.
When comparing the mean particle size of NP prepared with Gelucire® with that of NP prepared with Compritol® (), the former had a smaller size. This can be attributed to the complex mixtures of mono-, di-, and triglycerides that formed both materials; however, Gelucire® has a higher content of short hydrophilic chains [C8, C10, C12 (30%–50%), C14 (5%–25%), C18 (5%–35%)] than Compritol® [C16, C18, C20 (<10%), C22 (>83%), C24 (<3%)]. For this reason, the higher content of hydrophobic and longer chains in Compritol® may somehow hinder its dispersion in aqueous media, compared with Gelucire®.
The initial Z-potential (t = 0) was under 30 mV in all batches with Compritol®, and was associated with a low dispersion stability. Also, the higher particle size variation in SLN batches compared with Gelucire® was determinant to conclude that Compritol® was not adequate to produce CyA-SLNs, due to poor stability related problems.
The presence of bigger and abundant agglomerates in systems prepared with Gelucire® was more evident with DL > 10% and also the time of appearance of these agglomerates were developed in shorter time (). This effect may have been caused by a probable expulsion of the drug due to the lipid’s partial rearrangement. It is well known that glycerides have three main polymorphs: α, β’ and β. Assuming that our NP were made from a material containing mono-, di-, and triglycerides (proportions: [C8, C10, C12 (30%–50%), C14 (5%–25%), C18 (5%–35%)]), it is possible to believe that, during storage, a part of the lipid matrix transformed from one polymorph into another, reducing the spaces in the matrix and causing the drug to come out. Several reports have described this behavior.Citation1,Citation30–Citation32 Once the CyA molecules were outside or on the nanoparticle’s surface, they started to interact inter- or intramolecularly, producing associations between them and causing a physical stability loss, although SEM studies did not confirm crystal deposition on SLN surface.
For the dispersions with 25 mg of CyA, the Z-potential was higher than 30 mV (), which is related to a complete electrostatic stabilization of particles in suspension, with a maximum value of 39.50 mV for a CyA content of 30 mg. When the dispersion had more than 30 mg of drug, the Z-potential did not show a specific trend. This effect may be explained by the superficial properties of the drug that make the molecules to be preferably oriented at the solid-liquid interface, causing a change in the net charge of the particle. We may have expected a direct increase of the charge for a higher drug content, but this behavior was not observed, probably because CyA can be located inside and/or outside the NP; in this respect, Lechuga-Ballesteros et alCitation33 have recently reported that CyA can form a liquid crystal structure in repeated layers.
After three months of storage, the Z-potential decreased by almost 10 mV for all dispersions prepared with Gelucire®. This fact can be explained by the partial and gradual arrangement of the lipid, and by the ejection of small amounts of CyA. In the latter case, the CyA molecules outside the NP may induce the formation of the aforementioned structures, thus changing the net charge of the particle. Also, the production method may provide another explanation. Following dispersion production, the molecules have not a specific arrangement, they are randomly arrayed all over the nanoparticle; however, CyA molecules may acquire a more ordered distribution in space over timeCitation33 which can change the surface charge.
SEM showed that all batches evaluated had a solid matrix structure without crystal formation () and alongside time showed the aggregation of NP (); this fact can be explained by changes in the particles’ charge by the ejection of small amounts of the drug or by the rearrangement of CyA molecules to a more ordered state after production of SLNs.
Thermograms are shown in . An endotherm at 128.5°C was shown for pure CyA; this event was not observed for batches containing only CyA (40 mg) at t = 0 and t = five months (), and it may be related to a low drug amount in the sample, because in the CyA-PVAL physical mixtures analyzed, the endotherm corresponding to the melting process of the drug could only be observed from 80 mg of CyA (). Melting point of drug in these physical mixtures did not change (around 128°C).
DSC thermograms of empty SLNs at t = 0 and t = five months did not show any change in thermal events (). It is possible that hydroxyl groups of glycerides can form hydrogen bonds with hydroxyl groups of PVAL, avoiding lipid crystallization. In the batches of SLN containing 5, 15, and 50% of drug (20, 60, and 200 mg), there was no endothermic peak corresponding to crystal melting, even at five months, suggesting the presence of a molecular dispersion of the drug and lipid. Furthermore, it can be assumed that CyA maintained an amorphous or disordered crystalline phase of a molecular dispersion or a solid solution state in the lipid matrix after the process, as reported previously by our group for triclosan-containing polymeric NP,Citation29 and for CyA-loaded SLNs.Citation21
Thermograms of CyA-PVAL physical mixtures with 80 and 200 mg of drug () showed the endothermic peak corresponding to the melting of CyA; thus, the absence of an endotherm in the thermogram of the batch containing 200 mg of drug () supports our assumption regarding the formation of a molecular dispersion.
Another important event is that observed for the endotherm associated to Gelucire® melting. At t = 0, this endotherm was sharper than at five months (); in fact, it was not clearly detected at this time point. During the SLN formation process, the solvent diffuses from the emulsion’s globules; the drug and lipid aggregate randomly, possibly leaving regions of free lipid with a high order or crystalline state. The drug and lipid may interact over time, and some CyA molecules may be inserted between the glyceride chains,Citation21 reducing the crystalline state of the lipid. On the other hand, it was noteworthy that the Gelucire ® peak (40.6°C) showed less sharpness at t = 0 with high drug amounts in the SLN batches, suggesting first that CyA was encapsulated into the NPs and second, that the drug had an important impact on the lipid’s structure. This behavior is similar to that reported in model lipidic membranes, in which the peptide is considered as a “defect” in the lipid bilayer, which can be randomly distributed in the hydrocarbon region, affecting lipid interactions due to the electrostatic charges and to the geometry of the guest molecule.Citation34,Citation35
An immediate release was observed (); 99.60% of CyA was released from the SLN at 45 min, indicating the presence of an amorphous structure of the lipid and maybe the existence of CyA molecules on the NP surface, which allows a rapid dissolution and possibly higher loadings.
Turbidity measurement () is an indirect way to determine changes of particle size in the dispersion once it enters the biological fluids. In this experiment, turbidity decreased over time due to the reduction of entire nanospheres owing to the action of the dissolution medium.
The great advantage of using Gelucire®, composed of a mixture of mono-, di-, and triglycerides with different chain lengths, is that the matrix may have more imperfections in which higher quantities of drug can accommodate, in contrast with the reduced spaces provided by structurally similar molecules (for example tripalmitin and tristearin). This feature was also important to achieve high EE, >92% (). Moreover, the possibility of an amorphous state in the lipidic matrix could be responsible for the amount of drug encapsulated. High EE were also found using different preparation methods of SLNs.Citation20,Citation22,Citation23
Conclusion
With the EDM, it was possible to obtain Gelucire® NP with a good stability. Although NP with Compritol® were also prepared, the significant instability issues at the first stages of storage made this material inadequate to obtain such carriers.
As the CyA content increased in Gelucire® NP, variations in size were more evident and particle size increased over time in all batches. The Z-potential decreased 10 mV after three months in SLNs prepared with Gelucire®. The formation of molecular dispersions was evident, and it was shown that CyA affected the lipid’s structure by a mechanism still unknown, probably by the ability of these molecules to form a liquid crystal with a repeated-layer structure. High EE was achieved (>92%). CyA release from SLNs was relatively fast (99.60% in 45 min), in association with a rapid dissolution due to the presence of many imperfections in lipid structure. We are still working on the evaluation of these carriers, since they may have a great impact on CyA’s bioavailability profile as a result of a dissolution improvement because of the imperfections of the lipidic matrix.
Acknowledgment
The authors are grateful to Mr Rodolfo Robles for his technical assistance with the scanning electron microscope.
Disclosure
The authors report no conflicts of interest in this work.
References
- MullerRHRadtkeMWissingSANanostructured lipid matrices for improved microencapsulation of drugsInt J Pharm200224212121128
- MullerRHKeckCMChallenges and solutions for the delivery of biotech drugs – a review of drug nanocrystal technology and lipid nanoparticlesJ Biotech200411313151170
- TrottaMCavalliRTrottaCBussanoRCostaLElectrospray technique for solid lipid-based particle productionDrug Dev Ind Pharm201036443143819788405
- BattagliaLGallarateMCavalliRTrottaMSolid lipid nanoparticles produced through a coacervation methodJ Microencaps20102717885
- SinghSDobhalAKJainAPanditJKChakrabortySFormulation and evaluation of solid lipid nanoparticles of a water soluble drug: zidovidineChem Pharm Bull201058565065520460791
- del Pozo-RodríguezADelgadoDSolinisMASolid lipid nanoparticles as potential tools for gene therapy: in vivo protein expression after intravenous administrationInt J Pharm20103851215716219819319
- VarshosazJMinayianMMoazenEEnhancement of oral bioavailability of pentoxifylline by solid lipid nanoparticlesJ Liposome Research201020211512319694503
- HuLJiaHLuoZLiuCXingQImprovement of digoxin oral absorption in rabbits by incorporation into solid lipid nanoparticlesPharmazie201065211011320225654
- MehnertWMaderKSolid lipid nanoparticles. Production, characterization, and applicationsAdv Drug Deliv Rev20014723165196
- Quintanar-GuerreroDTamayo-EsquivelDGanem-QuintanarAAllémannEDoelkerEAdaptation and optimization of the emulsification-diffusion technique to prepare lipidic nanospheresEur J Pharm Sci200526221121816046105
- Tamayo-EsquivelDGanem-QuintanarAMartinezALNavarrete-RodriguezMRodriguez-RomoSQuintanar-GuerreroDEvaluation of the enhanced oral effect of omapatrilat-monolein nanoparticles prepared by the emulsification-diffusion methodJ Nanosci Nanotechnol2006691031343138
- Yi FanLDa WeiCLi XiangRXiu LiZJingQSolid lipid nanoparticles for enhancing vinpocetine’s oral bioavailabilityJ Control Release20061141535916828192
- NanZQinengPGuihuaHWenfangXYannaChXiuzhenHLectin-modified solid lipid nanoparticles as carriers for oral administration of insulinInt J Pharm20063271215315916962267
- KovarikJMMuellerEAvan BreeJBTetzloffWKutzKReduced inter- and intra-individual variability in cyclosporine pharmacokinetics from a microemulsion formulationJ Pharm Sci19948334444468207699
- MolpeceresJGuzmanMAberturasMRChaconMBergesLApplication of central composite designs to the preparation of poly-caprolactone nanoparticles by solvent displacementJ Pharm Sci19968522062138683450
- TarrBDYalkowskySHEnhanced intestinal absorption of cyclosporine in rats through the reduction of emulsion droplet sizePharm Res19896140432717516
- SanchezAVila-JatoJLAlonsoMJDevelopment of biodegradable microspheres and nanospheres for the controlled release of cyclosporine AInt J Pharm19939923263273
- GuzmanMMolpeceresJGarciaFAberturasMRRodriguezMFormation and characterization of cyclosporine-loaded nanoparticlesJ Pharm Sci19938254985028360826
- Al-AngaryAABayomiMAKhidrSHAl-MeshalMAAl-DardiriMCharacterization, stability, and in vivo targeting of liposomal formulations containing cyclosporinInt J Pharm19951142221225
- MullerRHRungeSRavelliVMehnertWThunemannAFSoutoEBOral bioavailability of cyclosporine: solid lipid nanoparticles (SLN®) versus drug nanocrystalsInt J Pharm20063171828916580159
- MullerRHRungeSARavelliVThunemannAFMehnertWSoutoEBCyclosporine-loaded solid lipid nanoparticles (SLN®): drug-lipid physicochemical interactions and characterization of drug incorporationEur J Pharm Biopharm200868353554417804210
- VariaJKDodiyaSSSawantKKCyclosporine A loaded solid lipid nanoparticles: optimization of formulation, process variable, and characterizationCurr Drug Del2008516469
- GokceEHSandriGBonferoniMCCyclosporine A loaded SLNs: evaluation of cellular uptake and corneal cytotoxicityInt J Pharm20083641768618725276
- HanafyASpahn-LangguthHVergnaultGPharmacokinetic evaluation of oral fenofibrate nanosuspensions and SLN in comparison to conventional suspensions of micronized drugAdv Drug Deliv Rev200759641942617566595
- ManjunathKVenkateswarluVPharmacokinetics, tissue distribution, and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administrationJ Control Release2005107221522816014318
- Tamayo-EsquivelD2005Preparación de dispersiones lipídicas de talla submicrónica por medio del método de emulsificación difusiónTesis de MaestríaUniversidad Nacional Autónoma de México
- AllemannEDoelkerEGurnyRDrug loaded poly(lactic acid) nanoparticles produced by a reversible salting-out process, purification of an injectable dosage formEur J Pharm Biopharm19933911318
- Quintanar-GuerreroDAllemannEDoelkerEFessiHA mechanistic study of the formation of polymer nanoparticles by the emulsion-diffusion techniqueColloid Polym Sci19972757640647
- Pinon-SegundoEGanem-QuintanarAAlonso-PerezVQuintanar-GuerreroDPreparation and characterization of triclosan nanoparticles for periodontal treatmentInt J Pharm20052941221723215814226
- MullerRHMaderKGohlaSSolid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the artEur J Pharm Biopharm200050116117710840199
- WestesenKBunjesHKochMHJPhysicochemical characterization of lipid nanoparticles and evaluation of their drug loading capacity and sustained release potentialJ Control Release19974823223236
- HeurtaultBSaulnierPPechBPrustJEBenoitJPPhysicochemical stability of colloidal lipid particlesBiomaterials200324234283430012853260
- Lechuga-BallesterosDAbdul-FattahAStevensonCLBennettDBProperties and stability of a liquid crystal form of Cyclosporine – the first reported naturally occurring peptide that exists as a thermotropic liquid crystalJ Pharm Sci20039291821183112950000
- GrassoDMilardiDLa RosaCImpellizzeriGPappalardoGThe interaction of a peptide with a scrambled hydrophobic/hydrophilic sequence (Pro-Asp-Ala-Asp-Ala-His-Ala-His-Ala-His-Ala-Ala-Ala-His-Gly) (PADH) with DPPC model membranes: a DSC studyThermochim Acta2002390127378
- PanickerLInfluence of the leprosy drug, dapsone, on the model membrane dipalmitoyl phosphatidylethanolamineThermochim Acta20064472123130