
Abstract
Peptide self-assembly is one of the promising bottom-up approaches for creating synthetic supermolecular architectures. Noncovalent interactions such as hydrophobic packing, electrostatic interaction, and polypeptide chain entropy (ΔSC) are the most relevant factors that affect the folding and self-assembly of peptides and the stability of supermolecular structures. The GVGV tetrapeptide is an abundant repeat in elastin, an extracellular matrix protein. In this study, four GVGV-containing peptides were designed with the aim of understanding the effects of these weak interactions on peptide self-assembly. Transmission electron microscopy, circular dichroism spectroscopy, dynamic light scattering measurements, and rheometry assays were used to study the structural features of the peptides. Because self-assembling peptides with different amino acid sequences may significantly affect protein release, basic fibroblast growth factor (bFGF) was used as a model molecule and encapsulated within the P2 (RLDLGVGVRLDLGVGV) hydrogel to study the release kinetics. The results showed that the balance among hydrophobic effects, electrostatic interactions, and chain entropy determined the molecular state and self-assembly of the peptide. Moreover, encapsulation of bFGF within the P2 hydrogel allowed its sustained release without causing changes in the secondary structure. The release profiles could be tuned by adjusting the P2 hydrogel concentration. Cell Counting Kit-8 and Western blot assays demonstrated that the encapsulated and released bFGFs were biologically active and capable of promoting the proliferation of murine fibroblast NIH-3T3 cells, most likely due to the activation of downstream signaling pathways.
Introduction
Molecular self-assembly is a powerful approach for fabricating the sophisticated structures and materials from the bottom up.Citation1 Inspired by nature, a variety of biomolecules, including amino acids,Citation2–Citation4 peptides,Citation5–Citation8 proteins,Citation9,Citation10 DNA,Citation11 saccharides,Citation12 lipids,Citation13,Citation14 and their derivatives,Citation15–Citation17 have been used as versatile building blocks for the design of functional structures at the nanoscale and microscale through molecular self-assembly. Building blocks have unique physical and chemical properties, which, if harnessed effectively, can result in materials with highly functional characteristics.
Recently, peptide-based self-assembling materials have gained much attention in several fields for their diverse applications such as drug/protein release,Citation18,Citation19 regenerative medicine and tissue engineering,Citation20–Citation23 three-dimensional (3D) cell culture,Citation24,Citation25 and nanotechnology,Citation26 because their characteristics mimic the 3D structures of the extracellular matrix (ECM). Generally, these systems involve designing a peptide monomer consisting of a structural motif that folds to adopt a known secondary structure such as the α-helix,Citation15,Citation27 β-strand,Citation8,Citation23 and β-hairpin.Citation7,Citation19 Noncovalent interactions, including hydrophobic, hydrophilic, π-stacking, hydrogen bonding, and polypeptide chain entropy (ΔSC), play important roles in triggering the assembly of peptide nanostructures.Citation28 It is still a challenge for scientists and engineers to incorporate these interactions into the fabrication of functional peptide-based materials.
The 16-residue peptide RADARADARADARADA (RADA16-I) is an ionic self-complementary peptide consisting of four repeats of a four-amino acid sequence: hydrophobic–positively charged–hydrophobic–negatively charged.Citation8 This peptide can undergo spontaneous assembly into well-organized nanofibers and further forms scaffolds with pore sizes between 5 and 200 nm.Citation29 The scaffold has a similar physical structure to the natural ECM and has been used in several applications, including 3D cell culture, tissue recovery, and controlled protein release. To date, although the self-assembly dynamics of RADA16-I are quite clear, a comprehensive understanding is lacking about the effects of amino acid sequences and noncovalent intermolecular interactions on peptide self-assembly.
The GVGV tetrapeptide is an abundant repeat in elastin, an ECM protein that is responsible for the elastic and extensibility properties of vertebrate tissues. These GVGV repeats constitute the hydrophobic domain of tropoelastin, the monomeric precursor of elastin, whereas the hydrophobic domains confer properties of self-assembly and extensibility.Citation30 Poly-(GVGV), poly-(GVGVA), and poly-(GGVGV) are ambivalent sequences that form amyloid fibers under certain conditions,Citation31 whereas poly-(GVGVP) adopts a β-sheet structure in the folded state and can be used as building blocks to create elastin-like polypeptides (ELPs)Citation32,Citation33 and silk–elastinlike protein polymers (SELPs).Citation34,Citation35 In this study, the GVGV tetrapeptide was incorporated into the RADA16-I sequence to evaluate the effects of weak interactions on the folding and self-assembly of the peptide. shows the amino acid sequences of these peptides, which have different peptide chain entropies, charge distributions, and hydrophobic driving forces for self-assembly and peptide chain entropies. Generally, two RADA repeat sequences of RADA16-I were replaced by two GVGV to increase the peptide chain entropy (P1 [RADAGVGVRADAGVGV]). We also increased the hydrophobic nature of the P1 molecule by using leucine (L) instead of alanine (A; P2 [RLDLGVGVRLDLGVGV]). Finally, we changed the charge distribution pattern of the peptides by swapping the charged amino acids (P3 [RARAGVGVDADAGVGV] and P4 [RLRLGVGVDLDLGVGV]) to study the effects of the intramolecular electrostatic interaction on peptide self-assembly.
Figure 1 Amino acid sequences of the designed peptides 1–4 used in this study.
Note: The N- and C-termini of these peptides were blocked by acetylation and amidation, respectively.
The goal of this study was to develop biomaterials that can undergo in situ gelation; thus, we evaluated the possibility that GVGV-containing peptides may be able to serve as potential carriers for the controlled release of drugs or growth factors. To this end, basic fibroblast growth factor (bFGF) was chosen and encapsulated within the designed peptide hydrogel to study the release kinetics. In addition, the effects of this controlled released system on cell proliferation and the possible underlying mechanisms were also investigated.
Materials and methods
Peptide design and synthesis
All the peptides (P1–P4, purity 95%) were commercially synthesized by GL Biochem (Shanghai) Ltd (Shanghai, People’s Republic of China), stored at −20°C, and used without further purification. The N- and C-termini of these peptides were protected by acetylation and amidation, respectively. Stock solutions of peptides were prepared in sterile water and stored at 4°C before use.
Preparation of P2 hydrogel
For the preparation of the P2 hydrogel, the P2 stock solution was diluted to a desired concentration. Self-assembly of P2 was triggered with the addition of an equal volume of phosphate buffer (pH 7.2) or cell growth medium (pH 7.4) without fetal bovine serum (FBS), followed by 30 min of incubation to allow the hydrogel formation.
Transmission electron microscopy (TEM) measurement
For TEM examinations, the peptide stocks were diluted to 0.05% weight/volume (wt/vol) and incubated overnight at room temperature. Before observation, 5 μL of peptide solution was dropped on the surface of a copper grid covered by a perforated formvar film and negatively stained with 1% phosphotungstic acid. After drying in air for a few minutes, the grid was observed directly under the transmission electron microscope (H-7650; Hitachi, Tokyo, Japan).
Dynamic light scattering (DLS) measurement
Size distributions of nanostructures formed by the peptides were measured by using a Zetasizer Nano ZS instrument (Nano ZS90; Malvern Instruments, Malvern, UK). All the peptides were diluted to 0.05% wt/vol and sonicated for 15 min to break the weak interactions, followed by standing at room temperature for 4 h before measurements. For each sample, 1 mL of the peptide solution was added into a low-volume disposable cuvette and kept at equilibrium at 25°C for 3 min prior to measurement. Intensity data from each sample were recorded, and size-versus-fraction distribution plot was obtained.
Circular dichroism (CD) spectroscopy
Stock solutions of the peptides were diluted to 0.025% wt/vol (0.25 mg/mL), and far-UV CD spectra were recorded on a model 400 CD spectrophotometer (Aviv Biomedical, Inc., Lakewood, NJ, USA). To investigate the effects of pH on the self-assembly of the peptides, 50 mM potassium chloride buffer (pH 2), phosphate buffer (pH 6–8), and borate buffer (pH 9–10) were mixed with an equal volume of the peptide stock solutions, respectively. The software CDPro (Colorado State University, Fort Collins, CO, USA) was applied to estimate the secondary structure contents of the peptides. The peptide secondary structure fractions at various pH values were calculated by the SELCON3 method with comparison to the selected reference protein set (IBasis7 [SDP48]). For bFGF release experiment, freshly prepared bFGF solution was used to examine the possible secondary structure changes after 12 h of diffusion from 1% wt/vol (10 mg/mL) of P2 hydrogel. CD spectra were recorded between 195 and 250 nm at room temperature.
Rheology measurement
Time and dynamic frequency sweep rheology experiments 1% wt/vol of P2 hydrogel were performed at room temperature on a rheometer (HAAKE RheoStress 1; Thermo Fisher Scientific Inc., Waltham, MA, USA) with a cone and plate geometry system (cone diameter 2 cm, angle 1°, truncation 51 μM). In brief, the P2 solution was mixed with an equal volume of phosphate buffer (pH 7.2), and an aliquot of 100 mL of the mixture was quickly placed on the plate. The time sweep experiment was performed at constant strain amplitude (0.5%) and constant frequency (1 Hz) for 16 min to monitor the hydrogel storage modulus (G′) and loss modulus (G″). Meanwhile, the hydrogel strength was measured via frequency sweep measurement over a range of frequencies of 10–0.01 Hz at fixed strain amplitude (0.5%) after 30 min incubation with the phosphate buffer.
bFGF release experiment
Various concentrations of P2 solution were mixed with the bFGF solution (pH 7.2). The final concentration of bFGF was 5 μM. Then, 50 μL of the mixtures was transferred into the wells of a 96-well plate before the formation of hydrogels. Subsequently, 350 μL of the phosphate buffers was carefully pipetted into the hydrogels. The release experiments were carried out at room temperature, where 150 μL of the supernatants was collected and replaced by the same volume of phosphate buffer at discrete time points. The collected supernatants were frozen until required and were then assayed for protein content by using Micro BCA Assay Kit (Thermo Fisher Scientific Inc.) with a standard curve of BSA (0–40 μg/mL) on a plate reader at an absorption wavelength of 562 nm. These data allowed for the direct determination of released protein amounts as a function of time.
Cell culture in hydrogel
NIH-3T3 cell line (obtained from American Type Culture Collection [ATCC], Manassas, VA, USA) was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific) supplemented with 10% FBS (Thermo Fisher Scientific). Prior to 3D culture, bFGF (Thermo Fisher Scientific) was added (40 ng/mL) into the media and cells were trypsinized and resuspended in bFGF containing media at a concentration of 5×105 cells/mL. The cell suspension was mixed with an equal volume of P2 stock solution to trigger the hydrogel formation (1% wt/vol), and 40 μL of the mixture was pipetted into each cell of the 96-well plate. Then, 150 μL of the bFGF (20 ng/mL) containing media was carefully added and followed by 30 min incubation in a 5% CO2 incubator at 37°C. After incubation, the upper bFGF containing media was replaced by bFGF-free media with FBS.
Cell proliferation assay
The Cell Counting Kit-8 (CCK-8; Sigma-Aldrich Inc., St. Louis, MO, USA) was used to analyze cell proliferation on days 0, 2, 4, and 6. For traditional two-dimensional (2D) petri dish culture and 3D hydrogel culture, NIH-3T3 cells were seeded at 5×103 cells/well and 104 cells/well in 96-well plates, respectively. To perform the CCK-8 assay, each sample was carefully washed with phosphate buffer three times and incubated with 10 μL of CCK-8 agent in 150 μL of serum-free DMEM for 2 h at 37°C. The absorbances at 450 nm were measured using a plate reader (μQuant; BioTek, Winooski, VT, USA). The 2D petri dishes without cells and 3D hydrogels without cells were subjected to the same process, to use as blanks. The data of both 2D culture samples and 3D hydrogel constructs were normalized to day 0. Three parallel samples were analyzed in each group.
Western blot analysis
Cells were collected and lysed in cold radioimmunoprecipitation assay buffer (Thermo Fisher Scientific) containing protease inhibitor cocktail (Sigma-Aldrich). Cell lysates were boiled and subjected to 10% gradient sodium dodecyl sulfate polyacrylamide gel electrophoresis. The proteins were transferred from gels to a polyvinylidene difluoride (PVDF) membrane (Merck Millipore, Billerica, MA, USA). The membranes were blocked by immersion for 1 h at room temperature in 5% nonfat skim milk/Tris-buffered saline and Tween 20 (TBST) and then incubated with primary antibodies against extracellular signal-related kinase (ERK) 1/2 (1:500; Abcam Inc., Cambridge, MA, USA), phospho-extracellular signal-regulated kinase 1/2 (p-ERK1/2, 1:1,000; Cell Signaling Technology, Danvers, MA, USA), p27 (1:300; Thermo Fisher Scientific), cyclin D1(1:1,000; Cell Signaling Technology), and actin (1:1,000; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) overnight at 4°C. After washed in TBST, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:3,000; Thermo Fisher Scientific) for 1 h and then developed using an electrochemiluminescence blotting reagent (GE Healthcare, Piscataway, NJ, USA).
Results and discussion
Peptide self-assembly in aqueous solution
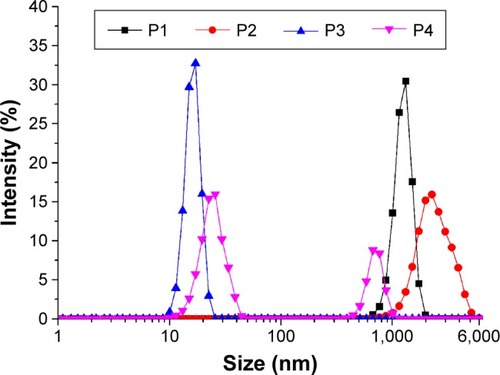
TEM images of the peptide samples in aqueous solution are shown in . P1 and P2 formed fibrillar assemblies with widths of 4.9±0.8 nm and 4.2±0.6 nm, respectively, and lengths ranging from hundreds of nanometers to several micrometers in aqueous solution (). Meanwhile, P3 formed a globular aggregate with a diameter of 21.0±5.0 nm (). Both types of assemblies could be observed repeatedly in P4 solution, regardless of the peptide concentration. The nanofibers formed by P4 were wider (6.0±1.3 nm) but significantly shorter (<1 μM) than those formed by P2 (). These results reveal the distinct innate self-assembly abilities of peptides with different sequence properties. DLS is a powerful tool for studying the ability to form nanostructures in solution. In this study, size distribution obtained by DLS experiments revealed the poly-morphological size distributions of the nanostructures formed by the designed peptides (). Combined with the TEM results, observation of a monodisperse population ranging in size from 670 to 6,000 nm for P1 and P2 indicated the dominant distribution of the nanofiber structures. The average size distribution of P2 was larger than that of P1. With regard to P3, the population observed at a range of 10–23 nm suggested the formation of globular aggregates. Interestingly, two peaks with dimensions ~10–50 nm and 400–1,000 nm could be found for P4. These peaks likely represent the two populations of globular aggregates and nanofibers seen in , as the globular aggregates were generally smaller in diameter than the nanofibers. Data from the DLS experiments were in good accordance with the TEM results and were not influenced by pH changes (data not shown). It should be noted that the hydrodynamic diameter obtained from the DLS measurements was not equivalent to the actual structural diameter because nonspherical nanostructures such as vesicles, tubes, and fibers do not follow the Stokes–Einstein equation.Citation36 In these cases, DLS can be used as a means for confirming the presence of nanostructures and for studying the relative size distributions of nanostructures.Citation37
Figure 2 TEM images of the nanostructures formed from P1 (A), P2 (B), P3 (C), and P4 (D) in Milli-Q water.
Note: All the peptides were diluted with Milli-Q water to 0.05% wt/vol (~0.3 mM) and negatively stained with phosphotungstic acid before observation.
Abbreviations: P1, RADAGVGVRADAGVGV; P2, RLDLGVGVRLDLGVGV; P3, RARAGVGVDADAGVGV; P4, RLRLGVGVDLDLGVGV; TEM, transmission electron microscopy; wt/vol, weight/volume.
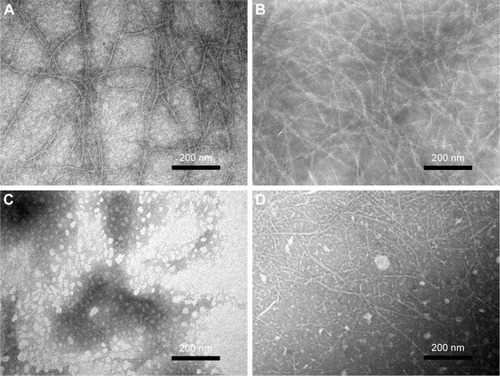
Figure 3 Size distribution of nanostructures formed by P1 (A), P2 (B), P3 (C), and P4 (D) as determined by DLS measurements.
Notes: All the peptides were diluted to 0.05% wt/vol and sonicated for 15 min followed by standing at room temperature for 4 h before measurements.
Abbreviations: DLS, dynamic light scattering; P1, RADAGVGVRADAGVGV; P2, RLDLGVGVRLDLGVGV; P3, RARAGVGVDADAGVGV; P4, RLRLGVGVD-LDLGVGV; wt/vol, weight/volume.
Secondary structure of the designed peptides
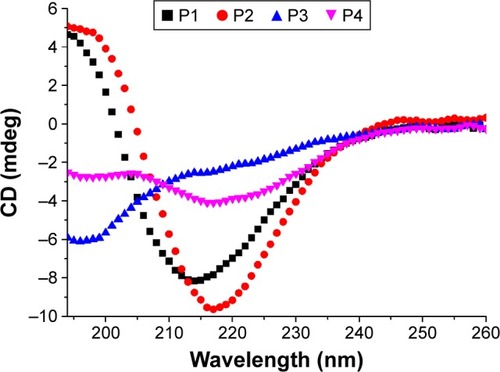
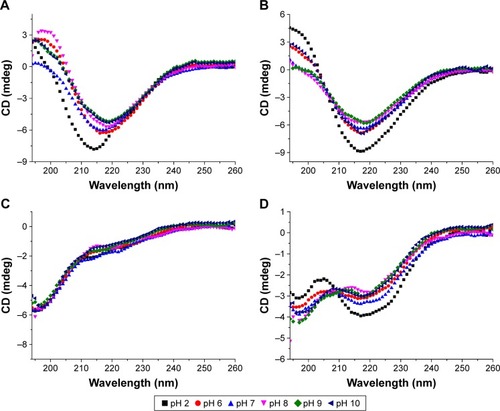
CD spectra of 0.125 mg/mL of peptides 1–4 in Milli-Q water are shown in . A significant difference in secondary structure was seen among these peptides. The CD spectra of P1 and P2 demonstrated a sharp minimum at ~215 nm, indicating the predominant presence of β-sheets, which are crucial for peptide self-assembly and nanofiber formation. However, P3 displayed typical unordered structures with a minimum at 196 nm. Interestingly, CD signals of P4 at ~216 and ~196 nm indicated the presence of both β-sheets and unordered structural components. The β-sheet content of P4 was much lower than that of P1 and P2. According to CDPro software analysis, the β-sheet contents of P1, P2, and P4 were 34.6%, 40.8%, and 26%, respectively (regular β-strand [s (r)]; distorted β-strand [a partial but far-from-complete distortion of the regular β-strand, due to lack of some hydrogen bonds] [S (d)]) (). These results are in accordance with the TEM images, suggesting that higher β-sheet content and lower unordered structures are required for peptide self-assembly and nanofiber formation.
Table 1 Estimated structure fractions of the designed peptides in aqueous solution
Figure 4 Secondary structures of the designed peptides 1–4 (0.125 mg/mL) in Milli-Q water.
Notes: CD spectra of peptides 1–4 were recorded at room temperature. The CD spectrum of P2 shows a typical β-sheet conformation, whereas P1 and P3 display unordered structures. With regard to P4, the CD spectrum ranging from 200 to 220 nm reflects the coexistence of β-sheets and unordered structures.
Abbreviations: CD, circular dichroism; P1, RADAGVGVRADAGVGV; P2, RLDLGVGVRLDLGVGV; P3, RARAGVGVDADAGVGV; P4, RLRLGVGVDL-DLGVGV.
We also investigated the effects of pH on the self-assembly of the designed peptides. The results showed that the secondary structures of the peptides were not significantly altered at different pH levels (). All the peptides had two positively charged arginines and two negatively charged aspartic acid residues at a neutral pH. Arginine and aspartic acid had a pKa of 12.5 and 3.9, respectively, in proteins. Similar to RADA16-I, only slight changes in the structural stability of the peptides were observed at all the pH values tested. With regard to P1 and P2, the difference in microdegree at ~218 nm between each pH point was very small (), and all the spectra indicated a stable β-sheet structure. This implies that the overall β-sheet structure was not significantly altered at different pH values; even the negatively charged aspartic acid residues are neutralized at pH 2.0. This stability may be due to complementary electrostatic interactions and hydrogen bonds that are present when peptides self-assemble into β-sheet structures.
Figure 5 CD spectra of P1 (A), P2 (B), P3 (C), and P4 (D) in solutions from pH 2–10 at room temperature.
Note: The peptides were incubated in potassium chloride buffer (pH 2), phosphate buffer (pH 6–8), and borate buffer (pH 9–10), respectively, to give a final concentration of 0.125 mg/mL.
Abbreviations: CD, circular dichroism; P1, RADAGVGVRADAGVGV; P2, RLDLGVGVRLDLGVGV; P3, RARAGVGVDADAGVGV; P4, RLRLGVGVDLDLGVGV.
Rheological properties of the hydrogel formed by P2
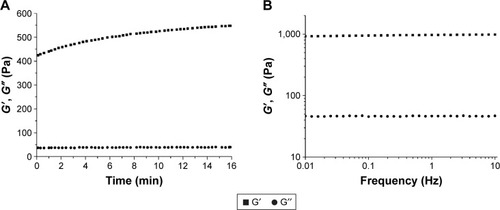
To test the gel formation abilities of the peptides, P1 and P2 were subjected to rheological measurements. The results showed that the hydrogel formation process from 1% wt/vol of P2 took place in a few minutes once the P2 solution was mixed with an equal volume of phosphate buffer (pH 7.2), whereas P1 remained viscous and failed to form a hydrogel. Time sweep and frequency sweep measurements were performed at room temperature to further study the elastic property of the hydrogel formed by P2. First, the mixture was treated with a constant strain amplitude of 0.5% at a constant frequency (1 Hz), after which the G′ values of the P2 hydrogel increased over time, whereas the G″ values did not change with time (). The increasing G′ value reflects the spontaneous growth of P2 nanofibers under self-assembly conditions. The frequency sweep measurements of the hydrogel displayed a frequency-independent modulus elastic response because the G′ and G″ values were relatively constant and the G′ values were much greater than zero (). Furthermore, we found that the G′ values of P2 hydrogel increased to nearly 1,200 Pa after 1 month of incubation (data not shown). These results clearly demonstrate the gel-like properties and stability of the P2 hydrogel in aqueous solution.
Figure 6 Rheology measurements of the hydrogel formed by P2 at 1% wt/vol.
Notes: Time sweep (A) and frequency sweep (B) for the P2 hydrogel. The hydrogel was made by mixing with an equal volume of phosphate buffer (pH 7.2), and the frequency sweep measurement was performed after a 30-min incubation at room temperature.
Abbreviations: G′, storage modulus; G″, loss modulus; P2, RLDLGVGVRLDLGVGV; wt/vol, weight/volume.
Effects of weak interactions on ionic complementary peptide self-assembly and hydrogel formation
The ionic self-complementary peptide forms a β-sheet with a hydrophobic face and a hydrophilic face once it self-assembles into high-aspect-ratio nanofibers. The hydrophobic effect is the main driving force for peptide self-assembly, but this assembly is hindered by like-charge repulsion between the hydrophilic faces.Citation38 Hence, the peptides remain swollen until the hydrophobic effect dominates over this repulsion. In our case, with regard to P1, the incorporation of two GVGV repeats into RADA16-I resulted in an increase in hydrophobic effects and a decrease in like-charge repulsion. P1 could undergo spontaneous assembly into well-defined nanofibers with an internal β-sheet structure during aggregation (). The secondary P1 structure was quite stable at various pH values; however, it was viscous and failed to form a hydrogel even though its net charge was almost zero in phosphate buffer (pH 7.2; data not shown). It has been reported that bringing the peptide net charge to zero should eliminate the kinetic barrier to assembly and facilitate gelation.Citation7,Citation16,Citation38 This contradiction can be explained by the increasing peptide chain entropy, because incorporating glycine increases the entropic cost of constraining the peptide backbone during β-sheet formation.Citation31 Peptide self-assembly is favored by the hydrophobic effect but opposed by peptide chain entropy since the assembly events dramatically decrease the number of accessible conformations. Hence, we propose that the balance among electrostatic repulsion, chain entropy, and hydrophobic effects determine the molecular state and assembly of the designed peptides and even formation of the hydrogel. To further validate this hypothesis, the side chain hydrophobicity was increased by substitution of As with Ls (P2). The β-sheet content of P2 was increased () compared with that of P1, and hydrogel formation occurred when charges on the arginine and aspartic acid side chains were screened in phosphate buffer (). This can be explained by the fact that the side chain hydrophobicity of P2 is larger than that of P1. As a result, increasing the hydrophobicity level leads to hydrogel formation because formation and the associated mechanical properties are influenced by the level of peptide hydrophobicity.Citation39,Citation40 This phenomenon can also be seen in P3 and P4. The increasing hydrophobicity of the side chain indeed increased the driving force for assembly, although only partial P4 molecules tended to adopt the β-sheet structure and subsequently self-assemble into fiber-like microstructures. In addition, in some cases, the intramolecular electrostatic interaction played a role in the folding and self-assembly of the peptides. Swapping an arginine with an aspartic acid residue results in a change in the charge arrangement (P3), that is, from a positive–negative–positive–negative pattern to a positive–positive–negative–negative pattern. For example, at an intermediate pH, P3 appeared to have a stronger intramolecular electrostatic interaction between positively charged arginine residues and negatively charged aspartic acid residues than P1. The unique characteristic of glycine lacking a side chain allowed the GVGV sequences to twist without steric hindrance. Consequently, the P3 molecule may be able to bend to form a hairpin-like structure driven by the intramolecular electrostatic interaction. The resulting bent molecules may continue to grow into larger globular assemblies ().
In vitro release of bFGF through the hydrogel
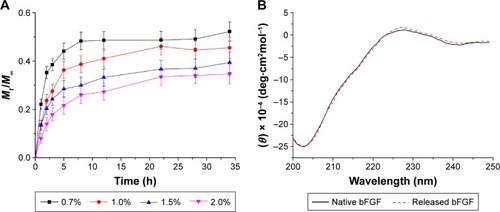
Self-assembling peptides have been increasingly studied as 3D matrices for numerous biomedical applications, including tissue engineering and controlled drug/protein release. The physicochemical properties of these nanofibers may affect not only the release rate but also protein conformation and function.Citation41 Self-assembling peptides with different amino acid sequences may significantly affect protein release.Citation18 To test this theory, bFGF was used as a model molecule and was encapsulated within the P2 hydrogel. bFGF is a recognized mitogen for multiple cell types, including fibroblasts, and it also promotes cell proliferation by inducing progression of the cell cycle from the G0/G1 to S phase.Citation42 Its release profile through P2 hydrogels of different peptide nanofiber densities is presented in . The hydrogels consisted of self-assembling P2 with concentrations of 0.7, 1.0, 1.5, and 2.0 wt/vol%. Two distinct stages, including the initial burst stage and plateau stage, were observed. With regard to the 1% wt/vol concentration of P2, ~36% of bFGF was released through the hydrogel in the first 5 h, although the release decreased over time and seemed to plateau after 22 h. The overall release rate increased concomitant with an increase in peptide concentration, which also led to a higher nanofiber density that hindered the release of bFGF molecules. These results show that the P2 hydrogel can be used as a potential carrier for protein release; the release of bFGF could be tuned by adjusting the concentration of the P2 hydrogel and the growth factors encapsulated within. The interactions between proteins and between proteins and peptides and their assemblies may lead to structural disruptions during peptide self-assembly and nanofiber formation and during the release process. Structural disruptions in proteins often result in aggregation and decreased biological activity. Far-UV light CD spectroscopy was used to obtain further insights into the conformational state of the released protein. The results showed that the secondary structure of native bFGF was quite similar to that reported earlierCitation43 (). The secondary structure of the released bFGF closely resembled that of native bFGF, suggesting that the interaction of proteins with the nanofiber hydrogel did not induce changes in the secondary structure, which is crucial for maintaining the activity of bFGF.
Figure 7 In vitro release of bFGF through the self-assembling P2 hydrogel.
Notes: (A) Release profiles for bFGF through P2 hydrogels of different peptide nanofiber densities. The P2 concentrations are 0.7, 1.0, 1.5, and 2.0 wt/vol%. Release experiments were performed in PBS (pH 7.4) at room temperature. Data points represent the average of five samples. (B) CD spectra of native and released bFGF solutions through the hydrogel (1% wt/vol) in PBS (pH 7.4). Spectra were recorded at room temperature. Data points represent the average of four samples. Mt and M∞ are the cumulative mass of the diffusing compound released from the hydrogel after t and infinite time (∞), respectively.
Abbreviations: bFGF, basic fibroblast growth factor; CD, circular dichroism; P2, RLDLGVGVRLDLGVGV; PBS, phosphate-buffered saline; wt/vol, weight/volume.
Encapsulated bFGF promotes the proliferation of NIH-3T3 cells
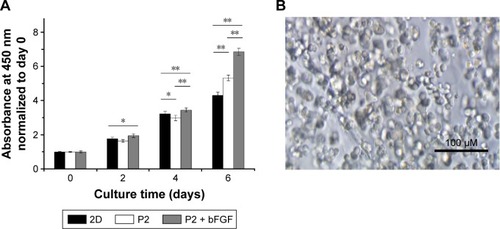
Next, we used the NIH-3T3 mouse embryo fibroblast cell line to determine if the released bFGF could elicit its biological functions in vitro. To determine the effects of controlled bFGF release on cell proliferation, cells were subjected to traditional 2D culture or were embedded within the P2 hydrogel with or without 20 ng/mL bFGF. As shown in , the cells survived and continued to divide in the P2 hydrogel during the course of the experiment. The NIH-3T3 cells began to aggregate into clusters without losing their spherical morphology after 4 days of culture. To evaluate the cell proliferation in 3D hydrogel and 2D culture, a CCK-8 assay was used to test the metabolic activity on days 0, 2, 4, and 6. The results showed that, compared with day 0, cells embedded in P2 hydrogel alone showed 1.6-fold proliferation on day 2, 3.0-fold proliferation on day 4, and 5.3-fold proliferation on day 6, whereas cells in hydrogel/bFGF constructs showed 2.0-fold proliferation on day 2, 3.5-fold proliferation on day 4, and 6.9-fold proliferation on day 6. The number of cells cultured on the 2D petri dish on days 2, 4, and 6 was 1.8-, 3.2-, and 4.3-fold greater than that at day 0. The proliferation rate was highest in the hydrogel/bFGF constructs and was significantly higher when cultured in P2 hydrogel compared to the 2D petri dish (). These data confirm that the encapsulated bFGF remained biologically active after release. Considering that there was no addition of bFGF during the experiment, encapsulation within the P2 hydrogel led to a continuous release of biologically active bFGF. These data collectively suggest that encapsulation within the P2 hydrogel does not change the ability of bFGF to stimulate the proliferation of NIH-3T3 cells and the biological activity of bFGF can be maintained when encapsulated within the hydrogel.
Figure 8 Effect of hydrogel-released bFGF on NIH-3T3 cell proliferation.
Notes: (A) Proliferation curve of cultured NIH-3T3 cells as determined by the CCK-8 test. The P2 hydrogel alone and a traditional 2D petri dish were used as the controls. The data from both 2D culture samples and 3D hydrogel constructs were normalized to day 0. One asterisk (*) indicates a P-value smaller than 0.05 (P<0.05). Two asterisks (**) indicate a P-value smaller than 0.01 (P>0.01). Data points represent the average of three samples. (B) Optical micrograph of NIH-3T3 cells embedded in the bFGF-releasing hydrogel after 4 days of culture.
Abbreviations: 2D, two-dimensional; 3D, three-dimensional; bFGF, basic fibroblast growth factor; CCK-8, Cell Counting Kit-8; P2, RLDLGVGVRLDLGVGV.
Encapsulated bFGF can activate downstream signaling pathways
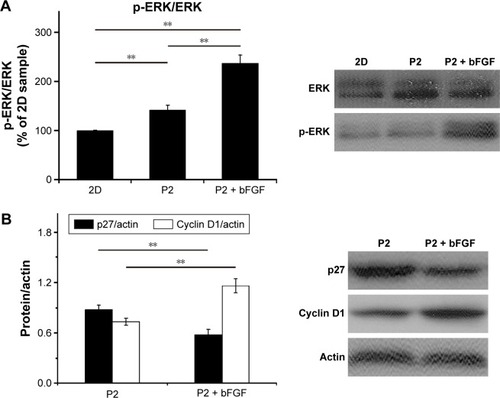
Our data show that the proliferation of NIH-3T3 cells was promoted by encapsulated bFGF and suggest that encapsulated bFGF can be released from the hydrogel into the local milieu with biological activity. To determine if the encapsulated bFGF can activate downstream signaling pathways, ERK, p27, and cyclin D1 levels were analyzed. ERK is a subfamily member of mitogen-activated protein kinases (MAPKs), which have been implicated in diverse cellular processes, including proliferation, differentiation, and migration.Citation44 Once activated, ERK translocates from the cytoplasm to the nucleus, where it phosphorylates various nuclear targets, resulting in cell proliferation.Citation45,Citation46 We found that the encapsulated bFGF led to an increased p-ERK/ERK ratio after 2 days of culture (). Interestingly, the p-ERK/ERK ratios in the P2 hydrogel culture groups were significantly higher than those in the 2D culture groups. These results are in accordance with data from the CCK-8 analyses and can be explained by the fact that the P2 hydrogel has 3D nanofiber structures similar to that of the natural ECM. Thus, the hydrogel supports attachment of NIH-3T3 cells and enhances the ECM–cell interactions, resulting in stimulation of cell proliferation. These findings indicate that combined treatment with P2 hydrogel and bFGF might be superior to either one alone for improving NIH-3T3 cell proliferation. Cyclin D1 is a protein required for progression through the G1 phase of the cell cycle, and p27 is a negative regulator of the cell cycle that restricts the G1/S phase transition and inhibits cell proliferation.Citation47 ERK activity contributes to the induction of cyclin D1 and downregulation of p27.Citation45,Citation48 Here, we examined the protein levels of cyclin D1 and p27 and found that encapsulated bFGF led to increased levels of cyclin D1 and reduced levels of p27 in NIH-3T3 cells (), indicating that growth factor signaling pathways were not disrupted by hydrogel encapsulation. Together, these experiments show that encapsulated and released bFGF is biologically active and can activate downstream signaling pathways.
Figure 9 Growth factor signaling after bFGF was encapsulated within the P2 hydrogel.
Notes: Western blot analysis of ERK, p-ERK, p27, and cyclin D1 after 2 days of culture and quantitative analysis of the protein levels. Two asterisks (**) indicate a P-value smaller than 0.01 (P<0.01).
Abbreviations: 2D, two-dimensional; bFGF, basic fibroblast growth factor; ERK, extracellular signal-regulated kinase; p-ERK, phospho-extracellular signal-regulated kinase; P2, RLDLGVGVRLDLGVGV.
Conclusion
In this study, the GVGV tetrapeptide, an elastin-derived sequence, was incorporated into the RADA16-I sequence to evaluate the effects of weak interactions on the folding and self-assembly of the peptide. The results demonstrated that the balance among the hydrophobic effects, intermolecular electrostatic interactions, and chain entropy determined the molecular state and assembly of the designed peptides. The intramolecular electrostatic interaction also contributed to the secondary structure formation. Among the four designed peptides, P2 could undergo a structural transition and form an elastic hydrogel when exposed to phosphate buffer. Encapsulation of bFGF resulted in a low, steady release from the hydrogel into the local milieu in a biologically active form, and the release rate was related to the peptide concentration. The self-assembly of the peptide and encapsulation within the hydrogel did not induce changes in the secondary structure of bFGF. In addition, the released bFGF promoted the proliferation of NIH-3T3 cells and activated downstream signaling pathways, such as ERK pathways. Taken together, our data suggest that the P2 hydrogel has the potential to be used in fine-tune growth factor delivery.
Acknowledgments
We thank Accdon for its linguistic assistance during the preparation of this manuscript. This work was supported by the National Natural Science Foundation of China (31360232), the Key Project of Guizhou Province Science and Technology Department (Qian ke he J zi LKZ[2013]01), and the Natural Science Research Project of Education Department of Guizhou Province (Qian jiao he KY zi [2015]418).
Disclosure
The authors report no conflicts of interest in this work.
References
- ChenCLRosiNLPeptide-based methods for the preparation of nanostructured inorganic materialsAngew Chem Int Ed2010491119241942
- Adler-AbramovichLAronovDBekerPSelf-assembled arrays of peptide nanotubes by vapour depositionNat Nanotechnol200941284985419893524
- WangMDuLWuXXiongSChuPKCharged diphenylalanine nanotubes and controlled hierarchical self-assemblyACS Nano2011564448445421591732
- ChenYTangCZhangJGongMSuBQiuFSelf-assembling surfactant-like peptide A6K as potential delivery system for hydrophobic drugsInt J Nanomedicine20151084725670898
- SchneiderJPPochanDJOzbasBRajagopalKPakstisLKretsingerJResponsive hydrogels from the intramolecular folding and self-assembly of a designed peptideJ Am Chem Soc200212450150301503712475347
- DongHParamonovSEAulisaLBakotaELHartgerinkJDSelf-assembly of multidomain peptides: balancing molecular frustration controls conformation and nanostructureJ Am Chem Soc200712941124681247217894489
- LiuYYangYWangCZhaoXStimuli-responsive self-assembling peptide made from antibacterial peptideNanoscale20135146413642123739953
- ZhangSHolmesTLockshinCRichASpontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membraneProc Natl Acad Sci U S A1993908333433387682699
- van EldijkMBMcGannCLKiickKLvan HestJCElastomeric polypeptidesTop Curr Chem20123107111621826606
- XiaX-XXuQHuXQinGKaplanDLTunable self-assembly of genetically engineered silk–elastin-like protein polymersBiomacromolecules201112113844385021955178
- UmSHLeeJBParkNKwonSYUmbachCCLuoDEnzyme-catalysed assembly of DNA hydrogelNat Mater200651079780116998469
- LiaoSWYuT-BGuanZDe novo design of saccharide−peptide hydrogels as synthetic scaffolds for tailored cell responsesJ Am Chem Soc200913148176381764619908839
- ConnCEDrummondCJNanostructured bicontinuous cubic lipid self-assembly materials as matrices for protein encapsulationSoft Matter201391334493464
- ZhangLChanJMGuFXSelf-assembled lipid−polymer hybrid nanoparticles: a robust drug delivery platformACS Nano2008281696170219206374
- O’LearyLERFallasJABakotaELKangMKHartgerinkJDMulti-hierarchical self-assembly of a collagen mimetic peptide from triple helix to nanofibre and hydrogelNat Chem2011382182821941256
- HartgerinkJDBeniashEStuppSIPeptide-amphiphile nanofibers: a versatile scaffold for the preparation of self-assembling materialsProc Natl Acad Sci U S A20029985133513811929981
- WongVWRustadKCGalvezMGEngineered pullulan-collagen composite dermal hydrogels improve early cutaneous wound healingTissue Eng Part A2011175–663164420919949
- GelainFUnsworthLDZhangSSlow and sustained release of active cytokines from self-assembling peptide scaffoldsJ Control Release2010145323123920447427
- LindseySPiattJHWorthingtonPBeta hairpin peptide hydrogels as an injectable solid vehicle for neurotrophic growth factor deliveryBiomacromolecules20151692672268326225909
- Ellis-BehnkeRGLiangY-XYouS-WNano neuro knitting: peptide nanofiber scaffold for brain repair and axon regeneration with functional return of visionProc Natl Acad Sci U S A2006103135054505916549776
- WangZLinMXieQElectrospun silk fibroin/poly (lactide-co-ε-caprolactone) nanofibrous scaffolds for bone regenerationInt J Nanomedicine201611148327114708
- Tysseling-MattiaceVMSahniVNieceKLSelf-assembling nanofibers inhibit glial scar formation and promote axon elongation after spinal cord injuryJ Neurosci200828143814382318385339
- LooYWongY-CCaiEZUltrashort peptide nanofibrous hydrogels for the acceleration of healing of burn woundsBiomaterials201435174805481424636214
- ZhangSDesigner self-assembling peptide nanofiber scaffolds for study of 3-D cell biology and beyondAdv Cancer Res20089933536218037409
- SilvaGACzeislerCNieceKLSelective differentiation of neural progenitor cells by high-epitope density nanofibersScience200430356621352135514739465
- LakshmananAZhangSHauserCAShort self-assembling peptides as building blocks for modern nanodevicesTrends Biotechnol201230315516522197260
- SmithAMBanwellEFEdwardsWRPandyaMJWoolfsonDNEngineering increased stability into self-assembled protein fibersAdv Funct Mater200616810221030
- MartRJOsborneRDStevensMMUlijnRVPeptide-based stimuli-responsive biomaterialsSoft Matter20062822835
- GelainFBottaiDVescoviAZhangSDesigner self-assembling peptide nanofiber scaffolds for adult mouse neural stem cell 3-dimensional culturesPLoS One20061e11917205123
- VrhovskiBWeissASBiochemistry of tropoelastinEur J Biochem199825811189851686
- RauscherSBaudSMiaoMKeeleyFWPomèsRProline and glycine control protein self-organization into elastomeric or amyloid fibrilsStructure200614111667167617098192
- LeeJMacoskoCWUrryDWElastomeric polypentapeptides cross-linked into matrixes and fibersBiomacromolecules20012117017911749169
- CostaRRCustódioCATesteraAMStimuli-responsive thin coatings using elastin-like polymers for biomedical applicationsAdv Funct Mater2009192032103218
- AnumoluRGustafsonJAMagdaJJCappelloJGhandehariHPeaseLF3rdFabrication of highly uniform nanoparticles from recombinant silk-elastin-like protein polymers for therapeutic agent deliveryACS Nano2011575374538221696150
- CappelloJCrissmanJDormanMGenetic engineering of structural protein polymersBiotechnol Prog1990631982021366613
- von MaltzahnGVautheySSantosoSZhangSPositively charged surfactant-like peptides self-assemble into nanostructuresLangmuir2003191043324337
- WuCLiMKwanSCMLiuGJLaser light scattering characterization of a novel polymer nanofiberMacromolecules1998312175537554
- ZhangSLockshinCCookRRichAUnusually stable β-sheet formation in an ionic self-complementary oligopeptideBiopolymers19943456636728003624
- CaplanMRMoorePNZhangSKammRDLauffenburgerDASelf-assembly of a β-sheet protein governed by relief of electrostatic repulsion relative to van der Waals attractionBiomacromolecules20001462763111710192
- LeonEJVermaNZhangSLauffenburgerDAKammRDMechanical properties of a self-assembling oligopeptide matrixJ Biomater Sci Polym Ed1998932973129556763
- KoutsopoulosSUnsworthLDNagaiYZhangSControlled release of functional proteins through designer self-assembling peptide nano-fiber hydrogel scaffoldProc Natl Acad Sci U S A2009106124623462819273853
- JonesSMKazlauskasAGrowth-factor-dependent mitogenesis requires two distinct phases of signallingNat Cell Biol20013216517211175749
- HuangZYeCLiuZSolid-phase N-terminus PEGylation of recombinant human fibroblast growth factor 2 on heparin-sepharose columnBioconjug Chem201223474075022433083
- WetzkerRBöhmerF-DTransactivation joins multiple tracks to the ERK/MAPK cascadeNat Rev Mol Cell Biol20034865165712923527
- ChambardJ-CLeflochRPouysségurJLenormandPERK implication in cell cycle regulationBiochim Biophys Acta2007177381299131017188374
- Moreno-LaysecaPStreuliCHSignalling pathways linking integrins with cell cycle progressionMatrix Biol20143414415324184828
- MalumbresMBarbacidMCell cycle, CDKs and cancer: a changing paradigmNat Rev Cancer20099315316619238148
- VillanuevaJYungYWalkerJLAssoianRKERK activity and G1 phase progression: identifying dispensable versus essential activities and primary versus secondary targetsMol Biol Cell20071841457146317314399