 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Introduction
Phosphatidylethanolamine (PE)-conjugated nanoliposomes were developed, characterized, and investigated for their accumulation in liver, kidneys, and lungs in rats.
Methods
Drug-excipient interaction was studied using Fourier transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC), surface morphology by field emission scanning electron microscopy, elemental analysis by energy dispersive X-ray (EDX) analysis, zeta potential and size distribution using a Zetasizer and particle size analyzer, and in vitro drug release by dialysis membrane. In vivo accumulation of liposomes in tissues was also studied.
Results
No chemical reaction was observed between drug and excipients. EDX study confirmed PE-conjugation in liposomes. Doxorubicin-loaded liposomes (DOX-L) and PE-conjugated doxorubicin-loaded liposomes (DOX-PEL) were of smooth surface and homogenously distributed in nanosize range (32–37 nm) with a negative surface charge. Loading efficiencies were 49.25% ± 1.05% and 52.98% ± 3.22% respectively, for DOX-L and DOX-PEL. In vitro drug release study showed 69.91% ± 1.05% and 77.07% ± 1.02% doxorubicin released, from DOX-L and DOX-PEL, respectively, in nine hours. Fluorescence microscopic study showed that liposomes were well distributed in liver, lungs, and kidneys.
Conclusion
Data suggests that PE-conjugated nanoliposomes released the drug in a sustained manner and were capable of distributing them in various organs. This may be used for cell/ tissue targeting, attaching specific antibodies to PE.
Introduction
Paul Ehrlich initiated the era of development for targeted delivery when he envisaged a drug delivery mechanism that would target drugs directly to diseased cells. Since then, numerous attempts have been made to devise clinically effective targeted drug delivery systems. A number of carriers, utilized to carry drug at the target organs/tissues have been identified, including liposomes (Doxorubicin); niosomes; microspheres (Doxorubicin); nanospheres (Tamoxifen citrate); erythrocytes; and pharmacosomes.Citation1,Citation2 Among those various carriers, few drug carriers have reached the stage of commercial formulations, where the liposomes have shown strong potential for effective drug delivery to the site of action. Doxil®, Myocet®, Ambisome®, and Depocyt® are some of the examples of Food and Drug Administration (FDA) approved nanosize commercial products.Citation3
Cancer is a disease that is notoriously difficult to treat.Citation4 Cytotoxic drugs involved in treatment are designed to kill tumor cells, but generally also display unwanted toxicities as they lack tumor cell selectivity.Citation5 Liposomes, as carriers for anticancer drugs, have been shown to decrease significantly nonspecific toxicities and to deliver an increased amount of drug effectively to the tumor.Citation6 Conjugation of liposomes to a targeting ligand can potentially improve their selectivity for tumor cells. The accumulation of the liposomal drugs was shown to be still further improved by their specific targeting to the tumor, that is, by attaching certain tumor-specific molecules to the liposome surface.Citation7 Specific vector molecules, such as antibodies, peptides, folate, and transferrin,Citation8–Citation10 are capable of recognizing tumors. Monoclonal antibodies were found to recognize specific antigens from the majority of known tumors, such as antibodies against ovarian cancer, prostate cancer, or colorectal cancer.Citation11 Liposomes can also provide slow release of an encapsulated drug, resulting in sustained exposure to tumor cells and enhanced efficacy.Citation12
Doxorubicin (DOX) is an antineoplastic drug of the anthracycline class. General properties of drugs in this class include interaction with deoxyribonucleic acid (DNA) in a variety of different ways, including intercalation (squeezing between the base pairs), DNA strand breakage, and inhibition of activity of topoisomerase II.Citation13 Most of these compounds have been isolated from natural sources and antibiotics. However, they lack the specificity of the antimicrobial antibiotics and thus produce significant toxicity.
The anthracyclines are among the most important antitumor drugs available. Doxorubicin is widely used for the treatment of several solid tumors, while daunorubicin and idarubicin are used exclusively for the treatment of leukemia. Doxorubicin may also inhibit polymerase activity, affect regulation of gene expression, and produce free radical damage to DNA. Doxorubicin possesses an antitumor effect against a wide spectrum of tumors, either grafted or spontaneous.Citation14
Formulation scientists are engaged to exploit the technological advantages of nanosciences in drug delivery research. Significant effort has been devoted to develop nanosize formulations for controlled drug delivery since it offers a suitable means of delivering bioactive molecules. In this respect, a nanodimensional drug delivery system focuses on formulating bioactive molecules in biocompatible nano-systems such as drug nanocrystals, solid lipid nanoparticles, nanostructure lipid carriers, lipid drug conjugate nanoparticles, and nanoliposomes etc.Citation15 This system has multifaceted advantages in drug delivery.
The objective of this study was to develop and evaluate phosphatidylethanolamine (PE)-conjugated nanodimensional liposomes and to investigate the distribution of the nanoliposomes in some tissues in rats. PE-conjugated anti-body in nanoliposomes might be a useful ligand for targeted delivery of the drug.
Materials and methods
Materials
Doxorubicin (Doxorubicin hydrochloride) was obtained as gifts (Sun Pharma, Baroda, India). Soya-L-α-lecithin (SPC) (HiMedia Laboratories Pvt. Ltd, Mumbai, India), cholesterol (CHL) (Merck, Mumbai, India), phosphatidylethanolamine (PE) (Sigma-Aldrich, Bangalore, India), butylated hydroxy anisole (BHA) (Qualigens Fine Chemicals, Mumbai, India), and chloroform (Merck) were purchased. All other chemicals used were of analytical grade.
Procedure of liposome preparation
Liposomes were prepared by lipid layer hydration method.Citation16,Citation17 Weighed amounts of SPC, CHL, and BHA (1% w/v) were taken in 250 mL round bottom flasks and were dissolved in chloroform. They were mixed vigorously by shaking. The mixture was placed in a rotary vacuum evaporator fitted with an A3S aspirator (Eyela, Tokyo Rikakikai Co. Ltd., Tokyo, Japan) and a circulating bath (Spac-N Service, Kolkata, India) and rotated at 150 rpm at 37°C in a water bath to evaporate the solvent. The flask was kept in a vacuum desicator overnight for complete removal of residue of organic solvent. Doxorubicin (1 mg/mL) was dissolved in deionized water and poured into the flask containing lipid film. It was then hydrated at 60°CCitation18 in a water bath fitted with a rotary vacuum evaporator. The flask was rotated at 100 rpm until the lipid film dispersed in the aqueous phase. The dispersion was sonicated in a bath type sonicator at a frequency of about 30 ± 3 KHz (Trans-o-Sonic, Mumbai, India) at the same temperature. After sonication, the preparation was kept at room temperature for about one hour for vesicle formation and then the preparation was stored overnight at 4°C. The preparation was centrifuged at 16000 rpm for one hour and the sample was lyophilized.Citation17 In the case of PE-conjugated liposomes, weighed amounts of SPC, CHL, PE, and BHA were taken in 250 mL round bottom flasks and were dissolved in the mixture of chloroform and methanol (3:1). All other procedures were the same as described earlier. Fluorescent liposomes were prepared by the above described procedures, except fluorescein isothiocyanate (FITC) was dissolved in organic phase (mixture of chloroform and methanol).
Evaluation and characterization of doxorubicin-loaded liposomes and doxorubicin-loaded PE liposomes
Drug-excipients interaction study: Fourier transform infrared (FTIR) spectroscopy
The pure drug doxorubicin (DOX), CHL, SPC, PE, and a mixture of drug with CHL, SPC and a mixture of drug with CHL, SPC, and PE (dry powder and lyophilized formulation) were mixed separately, with infrared (IR) grade potassium bromide (KBr) in the ratio of 1:100. Corresponding pellets were prepared by applying 5.5 metric ton pressure with a hydraulic press. The pellets were scanned in an inert atmosphere over a wave number range of 4000–400 cm−1 in a Magna IR 750 series II FTIR instrument (Jasco, FTIR 4200, Japan).Citation17
Differential scanning calorimetry (DSC)
DSC measurement of SPC was performed with an instrument for measurement of thermotropic transition of phospholipids (Mettler TA4000, Toledo, OH). Empty aluminium pans were used as reference and samples were carefully placed in another aluminium pan. The measurement was done in an inert atmosphere within the temperature range of 30°C to 200°C, at 5°C per min. Differential thermal analysis (DTA) measurements were also performed simultaneously.
Field emission scanning electron microscopy (FESEM)
Morphology of liposomes was performed with the help of a JSM electron microscope (JEOL, Tokyo, Japan). Lyophilized liposome samples were reconstituted with deionized water and were spread on to a carbon tape over a stub. The samples were vacuum-dried and gold coating was applied using an ion sputtering device. The gold-coated samples were vacuum-dried and examined under the electron microscope.
Energy dispersive X-ray analysis (EDX analysis)
EDX is a technique used for identifying the elemental composition of the specimen, or an area of interest thereof. The EDX analysis system works as an integrated feature of a scanning electron microscope (SEM) (JEOL).
Size distribution study and zeta potential measurement
Size distribution and zeta potential of the different formulations were measured using a Zetasizer Nano ZS instrument and analyzed using DTS software (Malvern Instruments Limited, Malvern, UK) using M3-PALS technology (Malvern Instruments Limited), which enables accurate measurements of zeta potential in aqueous dispersions. Size distribution and zeta potential studies were conducted by dispersing the samples following the guidelines of the manufacturer of the Zetasizer.
Evaluation of doxorubicin loading
A weighed amount (5 mg) of liposomes was lysed with ethanol, centrifuged and the absorbance of supernatant was measured at 480 nm using an ultraviolet-visible (UV/VIS) spectrometer (Beckman Instruments, Fullerton, CA, USA). The same procedure was used for the batch without the drug. The absorbance due to drug was the difference between the readings obtained from the preparation with drug and without drug to avoid any minor error due to the excipients. The loading % and the loading efficiency were calculated using the following formulae:Citation19
In vitro drug release study
In a 250 mL conical flask, 50 mL of phosphate-buffered saline (PBS) was measured.Citation17 A weighed amount of lyophilized liposomes (5 mg) was reconstituted in 1 mL PBS and was taken into a dialysis bag (Himedia dialysis membrane-60, Mumbai, India). The two ends of the dialysis sac were tightly bound with cotton thread. The bag was hung inside the conical flask with the help of a glass rod so that the portion of the dialysis bag containing the formulation could dip into the buffer solution. The flask was kept on a magnetic stirrer. Stirring was maintained at 300 rpm with the help of a magnetic bead at room temperature. Sampling was done by withdrawing 1 mL from the released medium and 1 mL blank was added. The samples were analyzed in a spectrophotometer at 480 nm. The concentration was calculated from the standard curve.
Liposome-accumulation in liver, kidney, and lungs in rats
Fluorescein isothiocyanate-phosphatidylethanolamine-doxorubicin (FITC-PE-DOX) liposomes, PE-conjugated doxorubicin-loaded liposomes (DOX-PEL), and liposomes without drug were injected through the tail vein in different groups of rats (n = 6).Citation20 After one hour and three hours, rats were sacrificed and liver, lungs, and kidneys were separated out. The organs were fixed with 10% formalin solution and embedded in a paraffin block. The samples were sectioned with a thickness of 5 μm specimen and taken on to the slides. The slides were dipped into xylene to remove the wax. After air drying, the slides were observed under fluorescence microscope.
Statistical evaluation
The significance of the data was evaluated using Student’s t-test (two-tailed). P values < 0.05 were considered significant.
Results
Preformulation study
Drug-excipients interaction study by FTIR
In this study, we have initially used FTIR spectroscopy to determine any drug-excipient interaction at the level of functional groups. Spectra of CHL (), SPC (), DOX (), PE (), mixture of SPC, CHL, and DOX (), mixture of SPC, CHL, PE, and DOX (), and mixture of SPC, CHL, DOX, and PE in lyophilized formulation () were compared at their different reactive functional groups in terms of peak picking.
Figure 1 Fourier transform infrared spectroscopy (FTIR) spectra of A) cholesterol (CHL); B) soya-L-α-lecithin (SPC); C) doxorubicin (DOX); D) phosphatidylethanolamine (PE); E) mixture of SPC, CHL, and DOX; F) mixture of SPC, CHL, DOX, and PE; and G) lyophilized formulation (DOX-PEL).
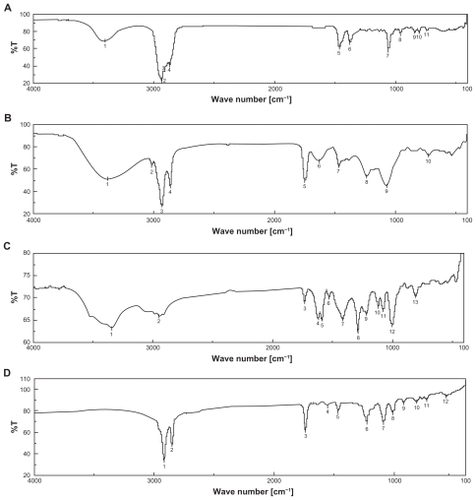
There were mild interactions observed in wave numbers between 3350 cm−1 and 3450 cm−1, between 1600 cm−1 and 1750 cm−1, and between 1050 cm−1 and 1250 cm−1. The range between wave numbers 3350 cm−1 and 3450 cm−1 is the characteristic stretching vibration of free and bonded hydroxyl (OH) and amine (NH2) groups. Peak variation in the range may be the effect of formation of weak hydrogen bonds.Citation24 In fact, cholesterol, SPC, and doxorubicin had bands in the range between 3500 cm−1 and 3200 cm−1 and the bands were very close to each other. Thus the band in the range could not be considered as a characteristic peak for the drug. Presence of doxorubicin, shown in , was evident from the observed bands at 1260–1000 cm−1 (carbonyl (CO) stretching of alcohol) and 900–675 cm−1 (out of plane OH bending).Citation21 Spectrum band 5 of SPC-CHL-PE-DOX appeared stronger in comparison to that of SPC-CHL-DOX. The peak was indicative of the presence of SPC in the mixture. However, probably due to the variation of quantity of SPC mixing with KBr, a sharper peak was obtained.
The range between wave numbers 1600 cm−1 and 1750 cm−1 is the strong intensity stretching vibration of CO, aryl ketone, α, β-unsaturation, and cyclo-pentanone, 1° NH2 and bending vibration range of medium to strong intensity NH2 scissoring (1° NH2)Citation25 and range between 1250 cm−1 and 1050 cm−1 is the strong intensity stretching vibration of CO, medium intensity stretching vibration of CN and medium intensity bending vibration of C-C-C bending, CH, CH2, CH3, and aromatic ring vibration. The drug, doxorubicin, has different reactive functional groups, such as free NH2 group, OH group, H, and CO. CHL has OH, H, and CH3 as reaction groups. SPC has CO, O, H, and reactive-NH2 group and PE has NH, O, and CO groups. Thus there may be physical interactions between functional groups of the drug and excipients, probably by formation of weak hydrogen bond or weak bond formation due to van der Waals force of attraction or dipole–dipole interaction, etc. Since all the characteristic peaks of the drug and excipients were present in the drug excipient mixture () and no predominant shifting of existing peaks or formation of new peaks was detected, this suggests that physical interactions took place only between the drug and excipients and that there was no chemical interaction between them. However, this has been further substantiated by DSC analysis of the drug, excipients, and their mixture. The physical interactions found here could be beneficial for the size and shape of the liposomes and drug release pattern from them.Citation22
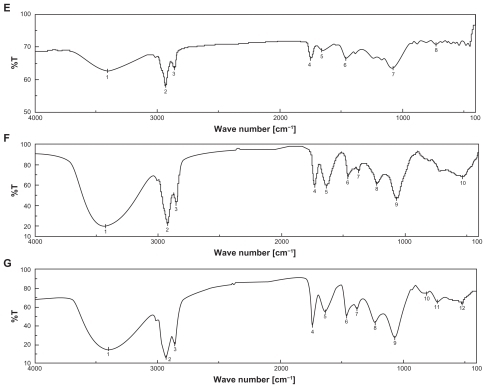
The data from the DSC experiments were obtained from the curves, by plotting heat flux against temperatures. SPC shows endothermic melting started at around 204°C (). In the case of the drug, endothermic melting was found to be at around 197°C (). CHL showed endothermic melting transition started at 40°C and at 149°C (). The initial endothermic peak could be responsible for loss of water and the next endothermic peak was for degradation. PE showed endothermic melting transition started at 105°C and had an endothermic peak at 125°C. One more peak close to 140°C was observed in the case of PE (). provides us with a DSC curve for the mixture of PE, CHL, DOX, and SPC. In this curve, all the individual endothermic transition peaks (a, b, c, and d respectively) are predominantly present. This suggests that there was no chemical interaction between the drug and excipients.
Figure 2 Differential scanning calorimetry (DSC) curve of A) cholesterol (CH L); B) soya-L-α-lecithin (SPC); C) phosphatidylethanolamine (PE); D) doxorubicin (DOX); and E) mixture of CHL, SPC, PE, and DOX.
Characterization of liposomes
After a thorough screening, based on physicochemical characteristics, two formulations (reported here) were selected and subjected to further studies. PE-nongrafted formulations showed better yield (50%) as compared to PE-grafted formulations. It was found that percentage drug loading in doxorubicin-loaded liposomes (DOX-L) was 2.46% and in PE-conjugated liposomes loaded with doxorubicin (DOX-PEL) was 2.65%. The loading efficiencies were 49.25% and 52.98% respectively ().
Table 1 % yield, % loading and loading efficiency
SEM photographs () show that the DOX-L and DOX-PEL had smooth surface with nano size dimension and were homogenously distributed. The average z-range of the liposomes (DOX-L and DOX-PEL) was 32–37 nm.
Figure 3 Field emission scanning electron microscopy (FESEM) of A) doxorubicin-loaded liposomes (DOX-L); and B) doxorubicin-loaded PE liposomes (DOX-PEL).

EDX analysis showed weight % and atomic % of various elements (C, O, and P) in various liposomes ( and ). The weight % of C, O, and P in DOX-L were 53.66, 42.06, and 4.28 respectively; and the values for DOX-PEL were 46.85, 47.58, and 5.57 respectively. The atomic % of C, O, and P in DOX-PEL were 61.75, 36.34, and 1.91 respectively; in DOX-PEL they were 55.30, 42.15, and 2.55 respectively. The differences in values of weight % and atomic % of elements were due to the presence of PE in DOX-PEL.
Figure 4 Energy dispersive X-ray (EDX) of A) doxorubicin-loaded liposomes (DOX-L); and B) doxorubicin-loaded PE liposomes (DOX-PEL).
Table 2 Weight % and atomic % of elements in various liposomes
About a 16% enhancement of z-average values (average diameter) was observed due to PE-grafting. The z-average of DOX-L was 32.67 nm and that of DOX-PEL was 37.84 nm (). The zeta potentials for DOX-L and DOX-PEL were −55.6 mV and −50.2 mV respectively (). Negative surface charge was due to ionization of free groups present on the surface of various liposomes. PE-conjugation was found to increase zeta potential by 10%. This may be due to the positive charge of PE.
Figure 5 Particle size distribution of A) doxorubicin-loaded liposomes (DOX-L); and B) doxorubicin-loaded PE liposomes (DOX-PEL).
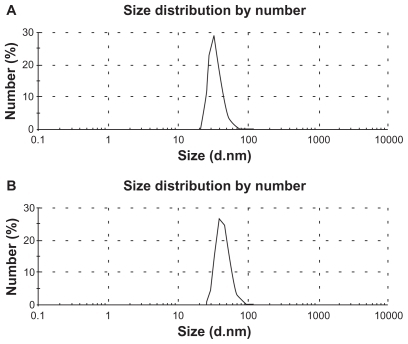
Table 3 Size distribution, PDI, and zeta potential of various liposomes
In vitro release study showed that 69.91% ± 1.05% and 77.07% ± 1.02% doxorubicin was released from DOX-L and DOX-PEL liposomes respectively, in nine hours (). Doxorubicin release was high in the first hour of study from both the formulations. This could be due to the release of the drug from the surface or near to the surface in bilayer. However, the drug then released very slowly. Much slower drug diffusion from the core of the formulation might be responsible for that. Drug release from DOX-L was very slow till until seven hours and again it increased until the end of the study. In the case of DOX-PEL, drug release was found to be very slow between the 1st and third hour and between the 4th and 5th hour. The reason for this is unknown. To evaluate the drug-release kinetic patterns, drug-release data were assessed using zero order, first order, Higuchi, Korsmeyer and Hixson–Crowell kinetic models.Citation25 Calculated R2 values for the kinetics were tabulated (). The corresponding plot (log cumulative percent drug release vs. time) (data not shown) for the Korsmeyer equation indicated a good linearity (R2 = 0.9375) for DOX-L, as compared to the others. The evaluation suggests that DOX-L formulations obeyed Korsmeyer kinetics which involves a coupling of the diffusion and erosion mechanism. For DOX-PEL, drug release followed Higuchi kinetics in a better way (R2 = 0.9747), suggesting diffusion is the only mechanism involved in the process for the period of study.
Figure 6 Release of doxorubicin from doxorubicin-loaded liposomes (DOX-L) and doxorubicin-loaded PE liposomes (DOX-PEL).
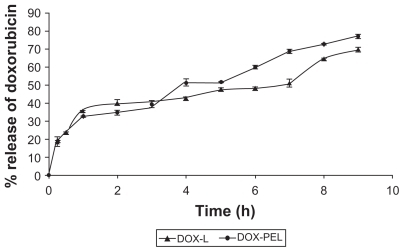
Table 4 In vitro release kinetics with R2 values for different formulations
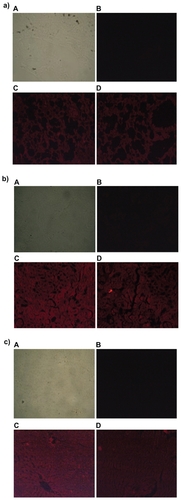
Since DOX-PEL was our main interest, we have studied the tissue accumulation (liver, kidneys, and lungs) of them in rats, treating with FITC-DOX-PEL as well as DOX-PEL. depicts the fluorescence microscopic photographs of liver, kidneys, and lungs of rats treated with FITC-DOX-PEL (after one and three hours of treatment). shows the fluorescence microscopic photographs of liver, kidneys, and lungs of rats treated with DOX-PEL. shows the fluorescence microscopic photographs of liver, kidneys, and lungs of rats treated with free DOX (1.5 mg/ kg), after one and three hours of treatment. Fluorescence intensities of FITC () and doxorubicin (–) were visualized. Fluorescence intensities were found to be more after three hours than after one hour in those tissues (). This indicates that the liposome accumulation was gradually enhanced. Signal of fluorescein isothiocyanate (FITC) refers to the existence of liposomes in liver, kidneys, and lungs and the results were further substantiated by visualizing the fluorescence from doxorubicin, a fluorophore. In , more signals (fluorescence) were observed in the tissues at one hour than at three. This indicates that the drug given intravenously was eliminated from those tissues quickly. As the negative and positive controls were similar for animal groups undergoing treatment of one and three hours, we have given one set of pictures for those controls in each case. Fluorescence microscopic study showed that liposomes were well distributed throughout the liver, lungs, and kidneys (). This study suggests that the experimental nanoliposomes might be useful to deliver the drug to those organs. Further, ligand molecule PE in the formulation may also be used to attach antibody conjugation to target specific cell type.
Figure 7 Fluorescence microscopic photographs of a) liver; b) kidneys; and c) lungs of rats treated with fluorescein isothiocyanate-phosphatidylethanolamine-doxorubicin (FITC-PE-DOX) liposomes (A = negative control; B = positive control; C = one hour after treatment; D = three hours after treatment).
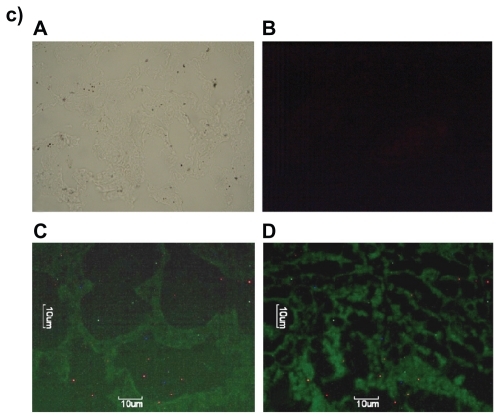
Figure 8 Fluorescence microscopic photographs of liver a), kidneys b) and lungs c); of rats treated with phosphatidylethanolamine-doxorubicin (PE-DOX) liposomes (A = negative control; B = positive control; C = one hour after treatment; D = three hours after treatment).
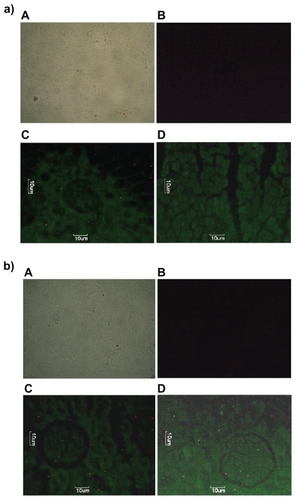
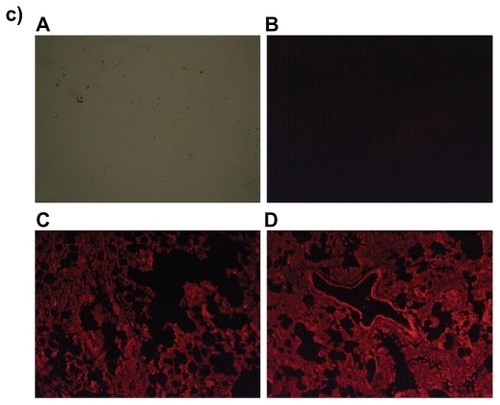
Figure 9 Fluorescence microscopic photographs of liver a), kidneys b) and lungs c); of rats treated with free DOX (A = negative control; B = positive control; C = one hour after treatment; D = three hours after treatment).
Discussion
Determination of drug-excipient interactions (if any), in a solid/semisolid dosage form, is one of the very important preformulation studies which indicates the stability of the drug in a formulation, the drug release pattern from it, and other physico-chemical properties, such as surface charge, shape, size, etc. related to the formulation.Citation23 There are various methods available for determination of drug-excipient interactions. Some of the popular and extensively-used methods to determine drug-excipient interactions are FTIR spectroscopy, DSC, and IR spectroscopy.Citation24,Citation25 FTIR spectroscopy showed that only physical interactions in some cases took place between the drug and excipients, which might facilitate drug loading in formulation and also have sustained release pattern of the drug from the liposomes. Various physical interactions have been reported to produce stable liposomesCitation26 and claimed to be responsible factors of drug release, as well as shape and size of liposomes.Citation27 Thus the physical interactions found here could be beneficial for the size and shape of the liposomes and responsible for drug release patterns from them. However, to confirm whether any chemical reaction took place, DSC study was conducted.
DSC is generally used to measure a number of characteristic properties of a sample. It is possible to observe fusion, crystallization, and even oxidation and other chemical reactions, along with the determination of glass transition temperature (Tg), crystallization temperature (Tc), and melting point (Tm) of a sample. Fluidity of lipid bi-layers depends on lipid or their combination used and their fluid gel transition temperature. The gel state (ordered) to fluidity (disordered) of lipid was observed by sensitive calorimetric instrument. The thermal transition of lipid bilayers was observed near 40°C. So the hydration temperature was kept above 40°C for formation of vesicles as reported earlier.Citation28 At 60°C, it showed the best vesicular formation in this study. Presence of individual endothermic transition peaks of drug-excipient formulation suggests that no chemical interaction took place between the drug and the excipient molecules.
When subjected to FESEM study, homogenous distribution of nanosize liposomes was seen. Size of DOX-L and DOX-PEL was below 100 nm and DOX-PEL liposomes were generally larger in size as compared to DOX-L. This may be due to conjugation of PE in the liposomes, since conjugation of molecules in bilayer lipids has been reported to enhance the size of the formulation.Citation29 Further, surface of the liposomes (DOX-L and DOX-PEL) was found to be smooth, suggesting no leakage on the formulation surface.
In EDX analysis, the difference in values of weight % and atomic % of elements was the proportional increase of the elements due to the presence of PE in DOX-PEL. This study suggests that PE was conjugated in liposomes.
Drug loading and drug loading efficiency were found to be 24.44 μg/mg and 49.25% respectively for DOX-L and they were 26.29 μg/mg and 52.98% respectively for DOX-PEL. The entrapment of drug molecules within lipid vesicles depends upon physico-chemical characteristics of drug, concentration of drug, ratio of drug to lipid, and the temperature at which liposome formation occurs.Citation19 Incorporation of cholesterol at low concentration into the lipid bilayers of liposomes leads to an increase in trans-membrane permeability, whereas incorporation of a higher amount of cholesterol (>30%) eliminates phase-transition and decreases the membrane permeability.Citation28 Cholesterol content in the experimental liposomes could also be a responsible factor for the amounts of drug entrapment in the present study. PE-grafting in liposome lipid bilayers was found to enhance the drug loading and loading efficiency to some extent. This could be due to the formation of comparatively bigger liposomes upon PE-grafting, which might have resulted in entrapment of a larger quantity of drug. However, the variation of those data compared between the PE-grafted and PE-nongrafted liposomes were statistically insignificant (P > 0.05).
The size and size distribution of liposomes depend on the method of size reduction, by which large lipid vesicles are reduced to small vesicles of nanometer scale. Ultra-sonication helps to reduce the size of the liposomes. In this case, nanosize liposomes may be formed as an effect of ultra-sonication. Ultra-sonication by bath sonication method, as compared to probe sonication method, has been reported to be more popular and efficient to reduce the size of the liposomes,Citation30 since the energy is disseminated from all directions continuously for a prolonged period in the lipid suspension. After bath-sonication, the formulations were kept for a minimum of one hour in the present study, to allow the fractured lipid bilayers to regain into small vesicles. Size and size distribution might depend on the ratio of SPC, cholesterol and PE used in the formulations, since variation in the ratio of the constituents was found to vary sizes (data not shown). Thus, the size range (32–37 nm) and narrow size distribution of liposomes, as assessed by polydispersity index (PDI) values could be due to the specific ratio of polymers and the manufacturing process parameters used for the formulations. PDI values suggest that homogeneous distributions of nanoliposomes occurred in the case of DOX-L and DOX-PEL. However, DOX-PEL had little wider range of size distribution. This may be due to the presence of PE. Zeta potentials more positive than +30 mV and more negative than −30 mV are normally considered stable for colloidal dispersion.Citation31 In our study, zeta potential values of both the formulations were more than −50 mV, which suggests that the reconstituted lyophilized nanoliposomes would form a stable suspension and thus would be easier for parenteral administration. However, PE-grafting in liposomes (DOX-PEL) showed less negative zeta potential value than that of DOX-L, due to the presence of PE (which is positively charged). Electrostatic and dynamic character of electrical double layer of liposomes is a dominant factor responsible for their recognition and uptake by cells. Electrostatic force in the liposome bilayer is easily modified by ion concentration in solution in which liposomes remain dispersed. Nature of surface charge of liposomes and counter-ion mobility (mobility) in the electrical double layer of liposomes are important factors for the relaxation phenomenon of electrical energy to cause double layer overlap.Citation32 Electrophoretic mobility of liposomes to a potential at a hydrodynamic plane of shear is called zeta potentialCitation33 and gives us information concerning charge beyond the hydro-dynamically stagnant layer. On the other hand, conductivity gives us information about the amount of mobile counter charges inside the stagnant layer. Therefore, electrical characterization of liposomes are quantified both by measuring stream of charge matter with charge (zeta potential) and without charge (conductivity). Presence of PE in DOX-liposomes did not predominantly vary both mobility and conductivity data, suggesting PE-conjugation would not behave differently in the case of DOX-PEL for in vivo recognition and cellular uptake.
PE-grafted liposomes showed comparatively higher amount of drug release in three hours as compared to the nongrafted ones. This may be due to the presence of PE in the liposomes. Presence of PE might vary the drug diffusion pathways. Presence of a rigid cholesterol nucleus along with the acyl chain of phospholipids is known to reduce the freedom of motion of acyl chain, which ultimately causes the membrane to condense, decreases its fluidity, and acts as a barrier to the entrapped drug.Citation34 Thus, presence of cholesterol molecules in lipid bilayers in the experimental liposomes might retard the drug release for a long period (nine hours). PE-grafting in the lipid–cholesterol liposomal membrane was Phosphatidylethanolamine-conjugated nanoliposomes found to release the drug in a lesser quantity (assessed by cumulative amount) in the first one to three hours than the PE-nongrafted ones. However, it enhanced the drug release from little more than three hours until the end of this study. This could be due to two reasons. Firstly, PE-grafted liposomes were bigger in size and the size might play a role to release more amount of drug with time due to larger surface area for drug diffusion. Secondly, presence of PE might loosen the compactness of structure due to the increase of size and provide easier and simpler diffusion pathways of drug.Citation35,Citation36 Drug release profile from DOX-L was best fitted with Korsmeyer kinetics (R2 = 0.9375), indicating involvement of anomalous diffusion and it may indicate that the drug release is controlled by more than one process. In contrast, DOX-PEL followed the Higuchi Kinetics (R2 = 0.9747), indicating drug diffusion from a matrix, without much involvement of other processes.Citation35,Citation36 This also suggests that PE-grafting in liposomes provided more structural stability as compared to the nongrafted one.
Doxorubicin-loaded PE-conjugated and nonconjugated nanoliposomes were found to accumulate in liver, kidneys, and lungs in rats. Since other organs were not assessed, we have restricted our discussion to these three organs only. Distribution of nanoliposomes had a similar distribution trend in liver, lungs, and kidneys. In the first hour, distribution was less and with time (three hours) it enhanced qualitatively (as assessed visually by fluorescence intensities) in all three organs. DOX-liposomes are properly characterized in terms of stability, loading, etc and since doxorubicin is fluorophore itself, we have also studied the FITC-free doxorubicin liposome accumulation in the tissues, by visualizing fluorescence of the drug in those tissues. Fluorescence of doxorubicin was also detected in liver, kidneys, and lungs of experimental animals. These data further support the finding of the tissue accumulation of FITC-labeled DOX-L, as assessed by visualizing the fluorescence of FITC-labeled DOX-L which also accumulated in liver, kidneys, and lungs. Stronger signals in tissues were also detected at three hours than those in one hour. Likewise, stronger signals were also seen in nine hours as compared to those at three hours (data not shown). The findings suggest the sustained drug release from liposomes. Taking the advantage of nanodimension and the cell membrane mimicking constituents (such as phospholipids and PE), they might easily have entered into the cellsCitation29 and gradually penetration was increased. Reports suggest that initially, after injection, the nanodimensional carriers are distributed in the blood and gradually make their passage to the organs.Citation29 Thus, the nanoliposomes were found to be suitable for easy drug distribution in the tissues and might provide sustained drug release there. Moreover, attaching antibodies (to PE) specific to particular cancer cells could specifically target the cell type with the nanoliposomes. Further studies are warranted in this area.
Conclusions
In the present study, PE-conjugated and nonconjugated liposomes were developed using a simple technique. They had an average size of 32–37 nm in a narrow size range and with a uniform distribution pattern. PE-grafting did not change the physicochemical properties of liposomes predominately, as compared to the PE-nongrafted formulations. Drug released from the formulations in a sustained manner in vitro. Drug and drug-loaded liposomes in liver, kidneys, and lungs of rats were observed. Further, taking the advantage of PE-grafting, this may be used for specific cells or tissue targeting, attaching specific antibodies or other targeting molecules.
Acknowledgment
This research work was funded by the Indian Council of Medical Research (ICMR), New Delhi, India; grant number 45/29/2009/PHA/BMS.
Disclosure
The authors report no conflicts of interest in this work.
References
- DavisMEChenZShinDMNanoparticle therapeutics: an emerging treatment modality for cancerNat Rev Drug Discov20087977178218758474
- LammersTHennikWEStormGTumor-targeted nanomedicines: principles and practiceBr J Cancer200899339239718648371
- ShahRBKhanMANanopharmaceuticals: Challenges and regulatory perspectivede VilliersMMAramwitPKwonGSNanotechnology in Drug DeliveryNew York, NYSpringer2009621647
- HaydenECGenomics boosts brain-cancer workNature2010463727927820090720
- GardnerSNFernandesMCytostatic anticancer drug developmentJ Exp Ther Oncol20044191815255288
- PapahadjopoulosDSteric stabilization. An overviewJanoffASLiposomes Rational DesignNew York, NYMarcel Dekker Inc1999112
- SapraPTyagiPAllenTMLigand-targeted liposomes for cancer treatmentCurr Drug Deliv20052436938116305440
- KurohaneKNambaYOkuNLiposomes modified with a synthetic Arg-Gly-Asp mimetic inhibit lung metastasis of B16BL6 melanoma cellsLife Sci200068327328111191643
- SchiffelersRMKoningGAten HagenTLFensMHSchraaAJJanssenAPAnti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicinJ Cont Rel20039112115122
- WillisMForssenELigand-targeted liposomesAdv Drug Deliv Rev199829324927110837594
- AgusDBBunnPAFranklinWGarciaMOzolsRFHER-2/neu as a therapeutic target in non-small cell lung cancer, prostate cancer, and ovarian cancerSemin Oncol2000276 Suppl 11S5363
- TardiPChoiceEMasinDRedelmeierTBallyMBMaddenTDLiposomal encapsulation of topotecan enhances anticancer efficacy in murine and human xenograft modelsCancer Res200060133389339310910044
- PommierYLeoEZhangHMarchandCDNA topoisomerase and their poisoning by anticancer and antibacterial drugsChem Biol201017542142320534341
- AlagkiozidisIFacciabeneACarpenitoCIncreased immunogenicity of surviving tumor cells enables cooperation between liposomal doxorubicin and IL-18J Transl Med2009710420003308
- PeerDKarpJMHongSFarokhzadOCMargalitRLangerRNanocarriers as an emerging platform for cancer therapyNature Nanotech20072751760
- BallieAJFlorenceATHumeLRMuirheadGTRogersonAThe preparation and properties of niosomes-non-ionic surfactant vesiclesJ Pharm Pharmacol198537128638682868092
- MukherjeeBPatraBLayekBMukherjeeASustained release of acyclovir from nano-liposomes and nano-niosomes: An in vitro studyInt J Nanomed200722213225
- ManconiMSinicoCValentiDLoyGFaddaAMNiosomes as carriers for tretinoin. I. Preparation and propertiesInt J Pharm20022341223724811839433
- XiangGWuJLuYLiuZLeeRJSynthesis and evaluation of a novel ligand for folate-mediated targeting liposomesInt J Pharm200835612293618242901
- EmanuelNKedarEBolotinEMSmorodinskyNIBarenholzYTargeted delivery of doxorubicin via sterically stabilized immunoliposomes: pharmacokinetics and biodistribution in tumor-bearing micePharm Res19961368618688792423
- ChouhanRBajpaiAKReal time in vitro studies of doxorubicin release from PHEMA nanoparticlesJ Nanobiotechnology20097519843333
- MukherjeeBMahapatraSGuptaRPatraBTiwariAAroraPA comparison between povidone-eudragit transdermal dexamethasone matrix patches based on in vitro skin permeationEur J Pharm Biopharm200559347548315760728
- BrittainHGPhysical Characterization of Pharmaceutical SolidsNew York, NYMarcel Dekker Inc1995
- MukherjeeBSantraKPattnaikGGhoshSPreparation, characterization and in-vitro evaluation of sustained release protein-loaded nanoparticles based on biodegradable polymersInt J Nanomed200834487496
- MukherjeeBRoyGGhoshSDevelopment of Denticap, a matrix based sustained release formulation for treatment of toothache, dental infection and other gum problemCurr Drug Deliv20096219920719450227
- CabanesABriggsKEGokhalePCTreatJARahmanAComparative in vivo studies with paclitaxel and liposome encapsulated paclitaxelInt J Oncol1998125103510409538125
- GokhalePCRadhakrishnanBHusainSRAn improved method of encapsulation of doxorubicin liposomes: pharmacological and toxicological and therapeutic evaluationBr J Cancer199674143488679456
- CorveraEMouristenOGSingerMAZuckermannMJThe permeability and effects of acyl chain length for phospholipid bilayers containing cholesterol; theory and experimentBiochim Biophys Acta199211072612701504071
- DadashzadehSValiAMRezaieMThe effect of PEG coating on in vitro cytotoxicity and in vivo disposition of topotecan loaded liposomes in ratsInt J Pharm20083531225125918082982
- MartinFJMedical Applications of LiposomesNew York, NYElsevier Science1998
- YuePFYuanHLYabgMPreparation, characterization and pharmacokinetic evaluation of puerarin submicron emulsionPDA J Pharm Sci Technol2008621324518402366
- WoodleMCCollinsLRSponslerEKossovskyNPapahadjopoulosDMartinFJSterically stabilized liposomes, reduction in electrophoretic mobility but not electrostatic surface potentialBiophys J19926149029101581503
- MatsumuraHVerbichSVDimitrovaMNSurface conductivity of counter ions on protein-adsorbed lipid liposomesPhysicochem Eng Asp200119213331336
- DamelRAKruyffBThe function of sterols in membraneBiochim Biophys Acta19764572109132184844
- KorsmeyerRWGurnyRDoelkerEBuriPPeppasNAMechanisms of solute release from porous hydrophilic polymersInt J Pharm19831512535
- HixsonAWCrowellJHDependence of reaction velocity upon surface and agitationInd Eng Chem Res1931238923931