 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Introduction
During recent years, there has been growing interest in use of topical vehicle systems to assist in drug permeation through the skin. Drugs of interest are usually those that are problematic when given orally, such as piroxicam, a highly effective anti-inflammatory, anti-pyretic, and analgesic, but with the adverse effect of causing gastrointestinal ulcers. The present study investigated the in vitro and in vivo pharmacodynamic activity of a newly synthesized palm oil esters (POEs)-based nanocream containing piroxicam for topical delivery.
Methods
A ratio of 25:37:38 of POEs: external phase: surfactants (Tween 80:Span 20, in a ratio 80:20), respectively was selected as the basic composition for the production of a nanocream with ideal properties. Various nanocreams were prepared using phosphate-buffered saline as the external phase at three different pH values. The abilities of these formulae to deliver piroxicam were assessed in vitro using a Franz diffusion cell fitted with a cellulose acetate membrane and full thickness rat skin. These formulae were also evaluated in vivo by comparing their anti-inflammatory and analgesic activities with those of the currently marketed gel.
Results
After eight hours, nearly 100% of drug was transferred through the artificial membrane from the prepared formula F3 (phosphate-buffered saline at pH 7.4 as the external phase) and the marketed gel. The steady-state flux through rat skin of all formulae tested was higher than that of the marketed gel. Pharmacodynamically, nanocream formula F3 exhibited the highest anti- inflammatory and analgesic effects as compared with the other formulae.
Conclusion
The nanocream containing the newly synthesized POEs was successful for trans-dermal delivery of piroxicam.
Introduction
Piroxicam is a nonsteroidal, anti-inflammatory, and analgesic agent. It is a water-insoluble drug with an acidic pKa value of 5.3. Structurally, the pyridine ring that is attached to the amide group also provides a pKa value of 1.86. Therefore, it is possible that piroxicam can act as a zwitterionic drug at a certain pH value. As a weak acid, piroxicam ionizes at pH 7.4 and at physiologic pH.Citation1 It has a log P value of 1.8.Citation2
It is well known that, when drug molecules are transported through the skin, they undergo two processes, starting with drug penetration through the stratum corneum followed by drug diffusion into the deeper tissues. The rate and extent of drug transport through the stratum corneum depends on the size, log P, ionic strength, hydrogen bonding ability, and physicochemical properties of the vehicle.Citation3
The correlation between the physicochemical properties of nonsteroidal anti-inflammatory drugs (NSAIDs) and the extent of their absorption from the skin has been confirmed by many investigations.Citation4,Citation5 These studies have found that the log P value of NSAIDs seems to be the most important factor compared with their pKa value and molecular weight. This can be related to the relatively comparable pKa and molecular weight of all NSAIDs and their highly variable log P values. Piroxicam with a log value of P 1.8 has shown a higher plasma concentration after topical administration in rats compared with NSAIDs of log P value ≥ 3.5. Drugs with a log P value of around 2 are suggested to be good candidates for topical delivery.Citation6
The enhancement of transdermal piroxicam transport has been extensively investigated. Various techniques have been used, including ion pairing,Citation7 pretreatment of the skin with permeation enhancer,Citation8,Citation9 inclusion into a carrier system with drug ionization,Citation10–Citation12 and application of permeation enhancement.Citation13,Citation14
Microemulsions and nanoemulsions have many advantages for use as topical drug delivery systems. Firstly, the main advantage of these dosage forms is that a large amount of drug can be incorporated into the formulation due to the increased solubilization capacity, thereby enhancing thermodynamic activity in the skin. Secondly, the permeation rate of the drug can be enhanced by using a micro/nanoemulsion due to the synergistic effect of various components to enhance drug delivery across the skin. Thirdly, the main ingredients, ie, oil, water, and surfactant mixtures, or surfactant-cosurfactant mixtures, can be combined synergistically to enhance drug flux.Citation15
Palm oil esters (POEs)Footnotea are constituents of a modified palm oil synthesized by reaction with olyel alcohol to produce long ester chains of the original triglyceride palm oil. It has been reported that POEs have a skin hydration activity of 40.7% within 90 minutes of application.Citation16 Skin hydration is known to enhance transdermal drug delivery.
In the present study, we investigated the suitability of newly prepared domestic POEs to act as an internal phase for topical delivery of piroxicam in a nanocream formulation. We further compared the effect of the external phase on the permeation of piroxicam from these nanocream-based POEs. We also compared the potential of the formulated topical nanocream to deliver piroxicam with that of the currently marketed formula. Experimentally, we assessed the in vitro transfer of the prepared piroxicam nanocream through an artificial cellulose acetate membrane and full thickness rat skin using the marketed gel formula as a reference. Furthermore, the in vivo anti-inflammatory and analgesic activities of the tested and the commercially available preparation were measured and compared.
Materials and methods
Materials
Potassium dihydrogen phosphate and sodium hydroxide were supplied by R M Chemicals (Essex, UK), while the orthophosphoric and hydrochloric acids were supplied by BDH (London, UK). In addition, acetonitrile was purchased from JT Baker (Phillipsburg, NJ), and the POEs was a gift from the Universiti Putra Malaysia, Selangor, Malaysia. Tween 80, Tween 85, Span 20, Span 85, and sodium benzoate were purchased from Sigma-Aldrich (Munich, Germany), while piroxicam was supplied by Noveltek Life-science (Tanggu, China). The cellulose acetate membrane of 0.2 μm pore size and 13 mm in diameter was purchased from Sterlitech (Kent, WA). The ingredients of the reference formulation, as stated in the product leaflet, were 0.5% anhydrous piroxicam, Carbopol®, propylene glycol, ethanol, and triethanolamine.
Nanocream preparation
All formulations were prepared using the continental method in which the oil and surfactant mixtures were mixed thoroughly for 15 minutes at 750 rpm in a beaker. A low-shear mixer with three blades (marine-type propeller) was used to mix and emulsify the formulae. Piroxicam was added to the mixture and mixed for another 30 minutes until it was completely dissolved. An aqueous external phase containing sodium benzoate was added gradually to the above mixture in the beaker. The formula was subsequently mixed for another 30 minutes. The droplet size of the nanocream preparations were measured by photon correlation microscopy using a Nanophox particle size analyzer (Sympatec Gmbh, Clausthal-Zellerfeld, Germany) and Malvern zeta sizer 1000 HAS (Malvern Works, Malvern, UK). The polydispersity of the formulae were measured using a Malvern zeta sizer 1000 HAS. Also, transmission electron microscopy (TEM), Leo 912 AB Eftem (LEO Electron Optics, Oberkochen, Germany) was used to confirm the previously measured droplet size and to determine the structural appearance of the system.Citation17 Additionally, the stability of the final formula was assessed under different environmental conditions for three months.Citation18
Nanocreams of 0.5% w/w piroxicam were prepared using POEs as the oil phase, phosphate-buffered saline (0.2M), pH 4.0, pH 6.0, and pH 7.4 as the aqueous phases, and a mixture of Tween 80:Span 20 (80:20) HLB 13.72 as the surfactant mixture. A ratio of 25:37:38 was selected from the triangular-phase diagrams previously constructed as the basic composition of oil:aqueous phase:surfactant for the production of nanocream with the best properties (data not shown).Citation17 Phosphate-buffered salines of pH 4, pH 6, and pH 7.4 were used as the external aqueous phases for formulae F1, F2, and F3, respectively. Sodium benzoate 0.01% was used as a preservative in all formulae.Citation17
In vitro drug transport through cellulose acetate membrane
The in vitro drug transport through the artificial cellulose acetate membrane (molecular weight cutoff 1000 Da) was carried out using a horizontally static type Franz diffusion cell. Franz diffusion cells are characterized by an effective diffusion surface area of 0.636 cm2 and a receptor cell volume of 5 mL. The static receptor cell was filled with 5 mL phosphate-buffered saline (pH 7.4) and was stirred with a small magnetic bar at a speed of 500 rpm for uniform mixing.Citation19 The receptor compartment was maintained at 37 ± 0.5°C using a circulating water bath.
Formulae F2, F3, and reference containing 0.5 mg of piroxicam in amounts of 100 mg were placed on the cellulose membrane surface facing the donor compartment, and 400 μL samples were withdrawn from the receptor compartment at predetermined time points of hours 0.16, 0.33, 0.5, 1, 2, 3, 4, 5, 6, and 8. The 400 μL sample withdrawn was replaced by fresh phosphate-buffered saline (pH 7.4) and maintained at 37 ± 0.5°C.Citation20 The drug content in the collected samples was determined by a validated high-pressure liquid chromatography method. All experiments for each sample were carried out in triplicate. The chromatographic conditions used for analysis were as follows: the mobile phase consisted of 5 mM disodium hydrogen phosphate adjusted to pH 3 with concentrated orthophosphoric acid, methanol, acetonitrile, and glacial acetic acid at ratios of 27:20:52:1, respectively. The flow rate was 1 mL/min, and samples were detected by an ultraviolet-visible detector at a wavelength of 350 nm. The retention time of piroxicam was 4.7 minutes. The limit of detection was 0.035 μg/mL and the limit of quantification was 0.0625 μg/mL.
In vitro drug transport across full thickness rat skin
In vitro drug transport through full thickness rat skin was achieved using the horizontally static-type Franz diffusion cell under the experimental conditions mentioned earlier. Male Wistar rats weighing 200–250 g were used in this study. All experimental procedures were in accordance with the Guidelines of the Animal Ethical Committee of Universiti Sains Malaysia and had its approval. Rats were anesthetized using sodium pentobarbitone 60 mg/kg intraperitoneally. The abdominal skin was shaved using electric and hand razors then removed surgically. The adherent subcutaneous fat was then carefully cleaned. In order to remove the extraneous debris and leachable enzymes, the skin was immersed in normal saline solution (0.9% NaCl) for two hours. The cleaned skin was washed with distilled water, wrapped in aluminum foil, and stored in a deep freezer at −20°C until further use. Following skin excision, rats were euthanized using an overdose of pentobarbitone. The frozen excised full thickness skin was brought to room temperature, and thereafter mounted between the donor and receptor compartments of the diffusion cell, with the stratum corneum side facing the donor compartment and the dermal side facing the receptor compartment.Citation21,Citation22 All other experimental steps were sequentially followed as mentioned above. Triplicate trials were conducted on all samples tested.
Permeability parameter calculation
Steady-state flux
Flux is defined as the rate of diffusion or transport of a sub-stance across a permeable membrane. After drug permeation has reached steady state, the steady-state flux was calculated using the following equation:
Where dM is the amount of drug that permeates through a unit cross section area, S, per unit time, t.
The slope of the steady-state portion of the permeation curve created by plotting the cumulative amount of drug permeated in micrograms versus time in hours is the flux.Citation23
Permeability coefficient
The permeability coefficient through the membrane (Kp) was determined according to the following equation:
Where H is the thickness of membrane and., C0 is the initial drug concentration.
Enhancement ratio
This factor was calculated to find the relative enhancement in the flux of formulations in respect to the reference enhancement ratio. The enhancement ratio was estimated according to the following equation:
Anti-inflammatory activity
The anti-inflammatory activity of the tested preparations and the commercially available preparation were measured and compared. Male Sprague-Dawley rats weighing 180–200 g were used in this experiment. Measurements of the in vivo anti-inflammatory and analgesic activities of the formulae conformed with the guidelines and practices of the Animal Ethics Committee of Universiti Sains Malaysia, and had its approval. The anti-inflammatory action was evaluated using the carrageenan-induced hind paw edema method with a slight modification.Citation24 Rats were randomly selected and divided into four groups of six animals each. These groups were divided, according to the formulae administered, into control (vehicle base), F2, F3, and reference gel groups. The animals were housed in polypropylene cages at 25 ± 1°C and 60 ± 5% relative humidity, with free access to food and water.
One day prior to application of the trial formulation, the hair on the dorsal surface of the rat was shaved. F2, F3, reference gel, and control formulae were applied on the shaved dorsal surface by gentle rubbing for 15 seconds. After five hours, 0.1 mL of 1% w/v suspension of carrageenan in normal saline was injected into the subplantar region of the right hind paw of all control and treated rats. Edema volume, in terms of thickness, was measured in all four groups at hours 2, 4, and 6 after carrageenan injection using a micrometer (Ozaki Ltd, Tokyo, Japan).Citation25 The induced thickness was measured by placing the foot of the rat between the anvil and spindle of the micrometer.
Mathematically, the degree of swelling can be expressed as:
Where Ct is hind paw thickness at hours 2, 4, and 6 after injection of carrageenan, and C0 is the initial hind paw thickness before injection of carrageenan.
Analgesic activity
This study was conducted similarly to the anti-inflammatory study, except that the pain threshold response of the rat right hind paw was measured instead of edematous thickness. The pain threshold was measured prior to and at hours 2 and 4 intervals after injection of carrageenan using a portable pain threshold device (YMF-P1) equipped with a data acquisition system (developed by the Department of Pharmacology, School of Pharmaceutical Sciences, Universiti Sains Malaysia, Penang, Malaysia). The pressure (g) applied to the edematous hind paw caused the rat to withdraw its hind paw. The vocalization or struggle of the rat was recorded as the pain threshold, as described by Yam et al.Citation26 Change in pain threshold (Δg) was calculated as the difference in pain threshold before and after injection of carrageenan.
Statistical analysis
Statistical analyses of steady-state flux across skin, amount of drug transferred through the cellulose membrane in vitro, and the analgesic and anti-inflammatory effects of the various formulations were performed using one-way analysis of variance (ANOVA). A statistically significant difference was accepted at P < 0.05.
Results and discussion
Formulation and characterization
Droplet size measurements were found to be less than 140 nm for the F1, F2, and F3 formulae as determined by photon correlation microscopy. Droplet size was also measured for formula F3 by TEM.Citation17 Formula F3 was found to be stable for a three-month period at 40°C, 25°C, and 5°C (data not shown).Citation18 Formulation F1 was excluded from this study because it showed drug precipitation within seven days of preparation.Citation18 Only the F2 and F3 formulae were assessed for their in vitro activity. The polydispersity index of formulations F2 and F3 were 0.037 ± 0.006 and 0.052 ± 0.009, respectively.
Drug transfer across cellulose acetate membrane
A drug must be released from its vehicle prior to penetration and partition into the skin. For certain formulations, drug release from the topical preparation is the rate-limiting step for drug absorption. Therefore, to ascertain that drug release from the vehicle was not the rate-limiting step for absorption, diffusion studies through an artificial synthetic membrane using the Franz diffusion cell has been proposed by earlier researchers. The membrane used must be inert and porous so as to allow drug passage in accordance with molecular weight. When drug molecules have a molecular weight as small as the pores of the synthetic membrane, they are able to pass through it.Citation27,Citation28
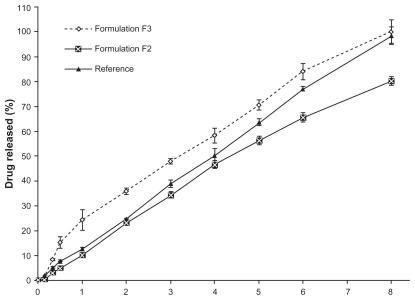
Drug transfer rates through the cellulose membranes of both nanocream formulae were compared with the transfer rate of the commercially available 0.5% piroxicam gel. shows a considerably higher and faster drug transfer rate across the membrane for nanocream F3 than for nanocream F2. It can be observed that drug transfer through the membrane is affected by the pH of the external phase of the nanocream. This difference in drug transfer may be attributed to the difference in the solubility of piroxicam at different pH conditions. Piroxicam with its weak acidic properties is more soluble in pH 7.4 buffer than in pH 6 buffer. Due to its high solubility at pH 7.4, partitioning of piroxicam from the oil phase to pH 7.4 buffer would be higher, hence leading to a higher drug transfer rate.
Figure 1 Comparative mean in vitro cellulose acetate membrane transport profiles of piroxicam from formulations F2, F3 and reference gel.
Note: Mean ± S.D., N = 3.
Diffusion of the drug incorporated into an oil-in-water cream or nanocream system is affected by partitioning of the drug between the internal oil phase and the external water phase. This means that the drug must diffuse from the internal phase to the external phase where the drug molecules are free to be released. Hence, by increasing drug solubility in the external phase, ie, enhancement of drug partitioning from the internal oil phase to the external aqueous phase, it is possible to observe the driving force for enhanced drug release.Citation29
Because the solubility of piroxicam in the mixed surfactants system of Tween 80 and Span 20 is higher compared with that in the POEs and buffers pH 6 and 7.4, the piroxicam molecules would then be mainly integrated at the oil water interface.Citation30 Therefore, it is expected that drug diffusion to the external phase would be faster. The solubility of piroxicam in pH 7.4 buffer is about 10 times higher than that in pH 6 buffer.Citation17 Presumably, partitioning of piroxicam from the oil to the pH 7.4 buffer would also be about 10 times higher than that in pH 6 buffer. This higher solubility and partitioning would be the driving force for the drug to be transferred through the membrane at a faster rate. shows that 100% of the drug was transferred from formulation F3 within eight hours, while only 80% of the drug was transferred from formulation F2 over the same time period.
Transfer of the drug through the membrane from formula F3 was faster, even during the initial hours, than that from formula F2. This may be due to the presence of a higher amount of soluble piroxicam in the external phase (pH 7.4 buffer) of the nanocream.Citation31 In the context of the reference gel, such gels can be prepared by various techniques. The simplest method involves dispersion of the desired quantity of polymer in the selected media and keeping the mixture undisturbed over a certain period of time. This is followed by adding the drug initially dissolved in a suitable medium. In the case of the reference system, it is expected that piroxicam is dissolved in the cosolvent system (propylene glycol and ethanol) that is subsequently mixed with the previously prepared carbopol dispersion.
The reference showed a comparable release profile to that of F3 at eight hours, but was slower during the initial hours of release, as indicated by the difference in T50% of drug release presented in . There was a statistically significant difference (P < 0.05) in T50% between formulation F3 and the reference gel. This difference may be due to the swelling of polymer in the reference formula, which swells in the presence of diffusion fluid, thereby slowing down the release process. Generally, polyacrylic acid polymers have a wide range of applications in modified-release and external gel preparations. This can be attributed to their versatile properties. These resins exert their action via different mechanisms. They can be pH-sensitive, which makes them soluble under specific body conditions, or absorb water, swell, and subsequently form a gel network.Citation32 Carbopol, in particular, is widely used as a gelling and thickening agent. Its gelling capabilities are highly dependent on the neutralization process of the carboxylate groups available in the polymer backbone. The higher the pH value of the neutralizing agent, the greater the thickening ability. Phosphate-buffered saline (pH 7.4) was used to assess the in vitro release profile. This media causes the carbopol to swell and form a gel network that may impede or reduce the release of piroxicam from the reference formula.Citation33
Table 1 Mean T50% drug transport across the cellulose acetate membrane from different formulations
At eight hours, the gel formulation had lost its strength and 96% of the drug was released. The high drug transfer rate from the reference formulation could be the result of free passage of piroxicam dissolved in the medium through the pores of the cellulose membrane. The release from the reference was higher than that from formulation F2. This may be because, as explained earlier, in case of a nanocream the drug needs to be partitioned from the inner layer to the outer layer before it is released. Presumably piroxicam is present in the palisade layer on the inner side of the droplet, which may result in controlled release of the drug. As the partitioning of the drug into phosphate-buffered saline (pH 6) from the oil phase was lower, the release is expected to be slower.
Drug transport through rat skin
In vivo measurement of drugs administered transdermally is a complex process. This is because some drugs exhibit very low plasma concentrations, rendering them impossible to be detected using analytical techniques. Problems with obtaining ethical approval for in vivo skin studies, as well as time constraints and expense, have motivated researchers to find an alternative technique for measuring drug permeation through skin.Citation34 Hence, in vitro drug permeation through skin was introduced as an alternative way to measure drug permeation. Unless the compound is highly lipophilic, the stratum corneum is the limiting step for transdermal drug absorption. Because drugs are mainly absorbed from the dermis layer, the amount of drug that can be transported through the stratum corneum can be used to represent the amount of drug that enters the systemic circulation.Citation35
Accordingly, topical drugs that exert their effects in deeper tissues are best assessed using full thickness skin. Undeniably, human skin is the best model for obtaining in vitro permeability data. However, lack of availability of human skin has caused researchers to shift their attention to animal skin as an alternative model. The Wistar rat skin model has been used extensively, in view of the fact that its stratum corneum thickness, as well as water permeability, is similar to that of human skin.Citation36
In our study, drug transport prior to steady state was similar for all the formulations during the first three hours, as can be seen in . From , it can also be observed that the flux of piroxicam during the steady state of formula F3 was significantly higher (P < 0.05) than that of formula F2 and that of the reference formulation. The most widely accepted mechanism by which the nanocream enhances the flux is that permeation of the drug-loaded nanocream occurs directly via the droplets to the stratum corneum without nanocream fusion at the stratum corneum.Citation37 This indicates that enhancement of the effect of the nanocream is caused by the nanosized droplets dispersed in the continuous phase, which can move easily into the stratum corneum and transport the drug through the skin barrier. The oil can enter the hydrophobic tail of the stratum corneum bilayer, disturb it by creating separate domains, and induce highly permeable pathways in the stratum corneum.Citation38 Another possible reason for the observed enhancement may be a synergistic effect of the nanocream components.Citation15
Figure 2 Comparative mean in vitro rat skin permeation profiles of piroxicam from formulations F2, F3 and reference gel.
Note: Mean ± S.D., N = 3.
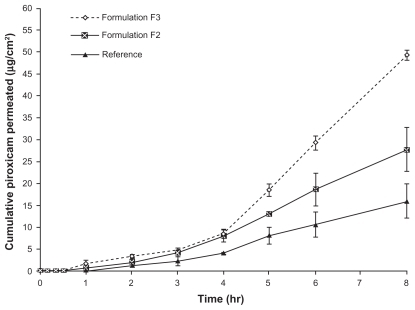
Table 2 Permeability parameters of the different formulations
POEs has been found to have an occlusive effect that can induce hydration of the skin combined with the hydration effect of water, thereby resulting in increased skin porosity which improves absorption.Citation16 A possible explanation for this is the presence of an aqueous “pore pathway” consisting of discontinuous domains within the lipid bilayers of the stratum corneum, which may expand and interconnect to form a continuous “pore pathway” under high hydration conditions.Citation37 Accordingly, when piroxicam was prepared in a microemulsion form comprising different oil phases, oleic acid was the oil of choice for improvement of drug solubility and skin permeability through excised rat skin.Citation39
The surfactants used, ie, Tween 80 and Span 20, have been found to act as permeation enhancers in different skin models.Citation40–Citation42 These surfactants can disturb the lipid bilayers of the stratum corneum, thereby making it a less effective barrier to drug permeation. Correspondingly, surfactants with polyoxyethylene chains have been found to enhance permeation of piroxicam through various skin models by reversible disturbance of the stratum corneum layer.Citation43 The small droplet size of the nanocream may also be correlated with an increased permeation rate. This is because of an increase in surface contact between the skin and the small droplets containing the drug. Kreilgaard has described the topical enhancing permeability effect of droplet size by comparing formulations containing the same constituents but different droplet sizes.Citation44
When the flux of formulations F2 and F3 was compared, because both formulations contain equal amounts of oil and surfactants, an increase in the pH of the external phase of the nanocream seemed to have a high enhancement effect on drug permeation through the skin. This result is consistent with the results of previously reported piroxicam permeation studies.Citation45,Citation46 When piroxicam was incorporated into an oil- in-water ointment base and the pH of the system was adjusted with sodium carbonate from 3.2 to 9.2, there was an increase in percutaneous absorption of piroxicam through rabbit skin.Citation45 Similarly, a gradual increase in pH of topical preparations containing different NSAIDs leads to an approximately10-fold increase in their flux. There is an assumption that higher absorption of ionized NSAIDs is related to an ion-pairing effect.Citation46 It is supposed that ionic drugs can form an ion pair with external skin components, such as skin fatty acids, and the resulting ion pair would be more lipophilic.Citation47,Citation48 Piroxicam could also form an ion pair with the buffer components of the external phase of the nanocream, ie, sodium hydroxide and potassium dihydrogen phosphate, resulting in higher lipophilicity and better penetration. Accordingly, the complexity of combining piroxicam with diethanolamine to produce ion pairs has also been investigated by pairing of phenolic groups of piroxicam and an amine salt, thereby improving drug flux through rat skin. Skin permeation enhancement is possible because of increased piroxicam solubility.Citation49 The net effect of drug solubility in the formulation and its permeation and flux through porcine epidermis has also been studied.Citation50 It was found that the incorporation of piroxicam into β-cyclodextrin or hydroxypropyl β-cyclodextrin increased piroxicam flux by several-fold when compared with passive diffusion. On the other hand, the incorporation of a piroxicam-cyclodextrin complex into a microemulsion system has also been performed, showing that the microemulsion system had a reservoir effect on drug release. This is possibly due to the higher tendency of piroxicam to dissolve in the oil phase (isopropyl myristate), leading to lesser partitioning of the drug to the external phase.Citation29 In other words, the permeation rate of a drug from a microemulsion may be increased by using different internal phases and composition of a microemulsion, given that the affinity of the drug to the internal phase can be easily modified to favor partitioning into the external phase and then into the stratum corneum.Citation51
Another assumption for the enhancement of drug transfer relates to the transfer of ionized drugs through the ionic channels oriented in the lipid bilayers of the stratum corneum.Citation51 The higher transfer of piroxicam from formula F3 compared with formula F2 through the cellulose acetate membrane may also be related to the difference in permeability through the skin, given that drug release from the vehicle is essential for determination of drug permeability in skin.
Anti-inflammatory activity
NSAIDs administered topically penetrate the skin slowly and pass in small quantities into the systemic circulation. The bioavailability of NSAIDs after topical application is generally less than 5%–15% compared with equivalent oral administration. On the other hand, the ability of NSAIDs to reach deeply into the affected tissue to induce a topical local effect is still debatable. The multilayered composition of skin renders the extent of penetration of various drugs different at different skin levels. Generally, most drugs are applied with the purpose of enhanced topical delivery to target underlying tissues. For example, NSAIDs cannot accumulate sufficiently at the specified tissue because of absorption through the blood vessels in the dermis layer, preventing the drug from penetrating further into deeper tissues.Citation52
Correspondingly, it has been found that topical administration of NSAIDs leads to a relatively high concentration in the dermis as compared with the oral route.Citation53 The ability of piroxicam to achieve local enhanced delivery via topical application has been studied by McNeill et al.Citation52 A comparison was made between the muscle content of piroxicam beneath the area of topical application with that after an intravenous dose. It was found that topical application of piroxicam could achieve high local concentrations than the intravenous dose. The peak concentration of piroxicam in muscle was attained four hours after topical application and 12 hours after intravenous administration. Therefore, plasma drug concentrations achieved locally may not reflect drug bioavailability. In this situation, in vivo pharmacodynamic measurement (of anti-inflammatory and analgesic properties) may be of prime importance.Citation54
It can be seen in that formula F3 showed a significantly higher (P < 0.05) anti-inflammatory effect compared with the control, reference, and F2 formulae at two-, four-, and six-hourly intervals after injection of carrageenan. These results correlate well with in vitro results for aceclofenac reported previously by Shakeel et al.Citation55 Flux across the skin for formula F3 was nearly 3.3-fold higher than that obtained from the reference. The greater the flux across the skin, the greater the anti-inflammatory activity. Higher permeability indicates a greater amount of piroxicam available to generate an anti-inflammatory effect at the area of injection. Formula F2 also showed a significantly greater effect than that of the reference formulation ().
Figure 3 The effect of topical administration of different formulations of piroxicam on rat hind paw edema at 2, 4 and 6 hours after administration of carrageenan.
Notes: *** and ** indicate significant at P < 0.001 and P < 0.01, respectively. Mean ± S.D., N = 6.
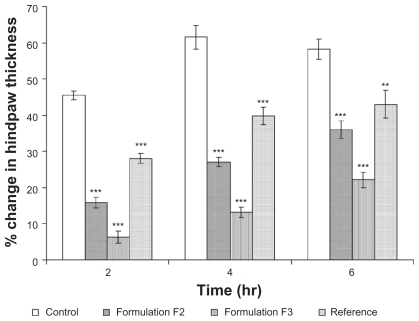
Both formulae F2 and F3 showed higher anti-inflammatory activity compared with the reference formulation. This rapid reaction may be attributed to the lower droplet size and the cumulative effect of the excipients used which would eventually increase the permeability of the drug. The anti-inflammatory effect of formula F3 was higher than that of F2. This may correspond to the difference in transport of piroxicam through the cellulose membrane and full thickness rat skin.
Analgesic activity
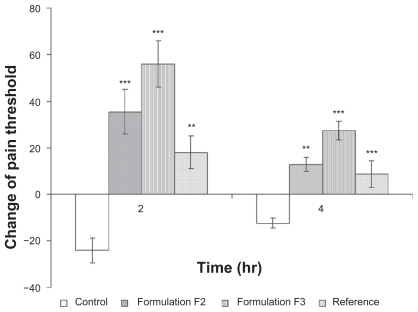
shows that F3 had significantly higher analgesic activity (P < 0.05) relative to that of the control, reference, and F2 formulae. Prostaglandins play an important role in promoting signs and symptoms of inflammation and in sensitizing terminal afferent C fibers in the periphery and enhancing the response of C fibers to algesic stimuli resulting in hyperalgesia.Citation56 Inhibition of the cyclo oxygenase enzyme and prostaglandin synthesis can rapidly reverse the hyperalgesia, pain, and inflammation.Citation57 Hence, a formula with faster and higher flux could produce a higher analgesic effect.
Figure 4 The effect of topical administration of different formulations of piroxicam on rat hind paw hyperalgesia at 2 and 4 hours after administration of carrageenan.
Notes: *** and ** indicate significant at P < 0.001 and P < 0.01, respectively. Mean ± S.D., N = 6.
Conclusion
Formula F3 had a high drug transfer rate through a cellulose acetate membrane compared with F2 and the reference gel during the initial hours of permeation. Both F3 and reference gel showed 100% drug release after eight hours of permeation. However, formulation F2 and F3 exhibited a higher skin permeation flux at steady state compared with the reference gel. This may be attributable to the reduced droplet size, along with the synergistic effect of the nanocream components which may enhance drug permeability. Formula F3 also demonstrated a higher flux compared with F2. This is perhaps due to a difference in partitioning of the drug from the internal oil phase to the external buffer phase of the emulsion system. The higher pH value of the external phase of formula F3 relative to that of F2 leads to higher availability of piroxicam in the external phase, probably leading to a higher release of the drug from formula F3. Furthermore, the study discovered that the prepared nanocream formulation of F3 and F2 had higher analgesic and anti-inflammatory activity compared with the currently marketed gel. The enhanced reaction may be due to the smaller droplet size along with increased permeability.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgments
The authors thank the Institute of Postgraduate Studies, Universiti Sains Malaysia, and the Biopharmacy Co-operative Center for providing financial assistance.
Notes
a Palm oil esters is the name of the oil used in our experiments and is not a mixture of esters.
References
- JinnoJOhDMCrisonJRAmidonGLDissolution of ionizable water-insoluble drugs: The combined effect of pH and surfactantInt J Pharm200089268274
- MihalicMPiroxicamAnalytical Profiles of Drug Substances19861523
- RobertsMSCrossSEPercutaneous absorption of topically applied NSAIDs and other compounds: Role of solute properties, skin physiology and delivery systemsInflammopharmacology1999733935017657437
- GoosenCPlessisJDMüllerDGJanse van RensburgLFCorrelation between physicochemical characteristics, pharmacokinetic properties and transdermal absorption of NSAIDsInt J Pharm1998163203209
- BeetgeEPlessisJDMüllerDGGoosenCvan RensburgFJThe influence of the physicochemical characteristics and pharmacokinetic properties of selected NSAIDs on their transdermal absorptionInt J Pharm200019326126410606790
- HadgraftJPlessisJDGoosenCThe selection of non-steroidal anti-inflammatory agents for dermal deliveryInt J Pharm2000207313711036227
- CheongHAChoiHKEnhanced percutaneous absorption of piroxicam via salt formation with ethanolaminesPharm Res2002191375138012403076
- HuangYBWuPCKoHMTsaiYHEffect of pretreatment by cardamom oil on in vivo percutaneous absorption of piroxicam gel: Pharmacokinetic analysis in rabbitsInt J Pharm1996134183191
- HsuLRTsaiYHHuangYBThe effect of pretreatment by penetration enhancers on the in vivo percutaneous absorption of piroxicam from its gel form in rabbitsInt J Pharm199171193200
- SantoyoSArellanoAYgartuaPMartinCPenetration enhancer effects on the in vitro percutaneous absorption of piroxicam through rat skinInt J Pharm1995117219224
- SantoyoSYgartuaPEffect of skin pretreatment with fatty acids on percutaneous absorption and skin retention of piroxicam after its topical applicationEur J Pharm Biopharm20005024525010962234
- PénzesTBlazsóGAignerZFalkayGErosITopical absorption of piroxicam from organogels – in vitro and in vivo correlationsInt J Pharm2005298475415893893
- ShinSCChoCWOhIJEnhanced eff icacy by percutaneous absorption of piroxicam from the poloxamer gel in ratsInt J Pharm200019321321810606784
- CurdyCKaliaYNNaikAGuyRHPiroxicam delivery into human stratum corneum in vivo: Iontophoresis versus passive diffusionJ Control Release200176737911532314
- KreilgaardMInfluence of microemulsions on cutaneous drug deliveryAdv Drug Deliv Rev200254S779812460717
- KengPSBasriaMZakariaMRSNewly synthesized palm esters for cosmetics industryInd Crops Prod2009293744
- AbdulkarimMFAbdullahGZChitneniMFormulation and characterization of palm oil esters based nano-cream for topical delivery of piroxicamInt J Drug Deliv20102287298
- AbdulkarimMFAbdullahGZChitneniMStability studies of nano-cream containing piroxicamInt J Drug Deliv20102333339
- DjordjevicJMichniakBUhrichKEAmphiphilic star-like macromolecules as novel carriers for topical delivery of non-steroidal anti-Inflammatory drugsAAPS PharmSciTech20035112
- NgSFRouseJSandersonDEcclestonGAComparative study of trans-membrane diffusion and permeation of ibuprofen across synthetic membranes using Franz diffusion cellsPharmaceutics20102209223
- ÖzgüneyISKarasuluHYKantarcGSözerSGüneriTErtanGTransdermal delivery of diclofenac sodium through rat skin from various formulationsAAPS PharmSciTech200674E1716584139
- SinghSGajraBRawatMMuthuMSEnhanced transdermal delivery of ketoprofen from bioadhesive gelsPak J Pharm Sci20092219319819339232
- WagnerHKostkaKHLehrCMSchaeferUFInterrelation of permeation and penetration parameters obtained from in vitro experiments with human skin and skin equivalentsJ Control Release20017528329511489316
- LarsonDLLombardinoJGThe topical anti-inflammatory effects of piroxicam in rodentsAgents Actions1980102462527405751
- YamMFLimVSalmanIMHPLC and anti-inflammatory studies of the flavonoid rich chloroform extract fraction of Orthosiphon stamineus leavesMolecules2010154452446620657453
- YamMFAngLFAmeerOZSalmanIMAzizHAAsmawiMZAnti-inflammatory and analgesic effects of Elephantopus tomentosus ethanolic extractJ Acupunct Meridian Stud2009228028720633503
- GuyRHHadgraftJOn the determination of drug release rates from topical dosage formsInt J Pharm199060R13
- ShahVPElkinsJLamSYSkellyJPDetermination of in vitro drug release from hydrocortisone creamsInt J Pharm1989535359
- DalmoraMEDalmoraSLOliveiraAGInclusion complex of piroxicam with beta-cyclodextrin and incorporation in cationic microemulsion. In vitro drug release and in vivo topical anti-inflammatory effectInt J Pharm2001222455511404031
- GuptaPKCannonJBLiuREmulsion and micro-emulsion for drug solubilization and deliveryWater-insoluble Drug FormulationNew York, NYCRC Press2000
- ChenHChangXDuDLiJXuHYangXMicroemulsion-based hydrogel formulation of ibuprofen for topical deliveryInt J Pharm2006315525816600540
- AbdullahGZAbdulkarimMFChitneniMPreparation and in vitro evaluation of mebeverine HCl colon-targeted drug delivery systemPharm Dev Technol4302010 [Epub ahead of print]
- ProniukSBlanchardJAnhydrous Carbopol® polymer gels for the topical delivery of oxygen/water sensitive compoundsPharm Dev Technol2002724925512066579
- WesterRCNoonanPKRelevance of animal models for percutaneous absorptionInt J Pharm1980799110
- NaikAKaliaYNGuyRHTransdermal drug delivery: Overcoming the skin’s barrier functionPharm Sci Technolo Today2000331832610996573
- WaltersKARobertsMSWaltersKAHadgraftJVeterinary applications of skin penetration enhancersPharmaceutical Skin Penetration EnhancementNew York, NYMarcel Dekker1993
- SantoyoSArellanoAYgartuaPMartinCIn vitro percutaneous absorption of piroxicam through synthetic membranes and abdominal rat skinPharm Acta Helv1996711411468810580
- TanojoHBouwstraJAJungingerHEBoddéHEIn vitro human skin barrier modulation by fatty acids: Skin permeation and thermal analysis studiesPharm Res19971442499034219
- ParkECuiYYunBJKoIJChiSCTransdermal delivery of piroxicam using microemulsionsArch Pharm Res20052824324815789759
- EndoMYamamotoTIjuinTEffect of nonionic surfactants on the percutaneous absorption of tenoxicamChem Pharm Bull1996448658678681419
- OkuyamaHIkedaYKasaiSImamoriKTakayamaKNagaiTInfluence of non-ionic surfactants, pH and propylene glycol on percutaneous absorption of piroxicam from cataplasmInt J Pharm199918614114810486432
- LópezALlinaresFCortellCHerráezAMComparative enhancer effects of Span® 20 with Tween® 20 and Azone® on the in vitro percutaneous penetration of compounds with different lipophilicitiesInt J Pharm200020213314010915936
- ShinSCChoCWOhIJEffects of non-ionic surfactants as permeation enhancers towards piroxicam from the poloxamer gel through rat skinsInt J Pharm200122219920311427350
- KreilgaardMPedersenEJJaroszewskiJWNMR characterisation and transdermal drug delivery potential of microemulsion systemsJ Control Release20006942143311102682
- TsaiYHHsuLRNaitoSIPercutaneous absorption of piroxicam from ointment bases in rabbitsInt J Pharm1985246178
- HadgraftJPlessisJDGoosenCThe selection of non-steroidal anti-inflammatory agents for dermal deliveryInt J Pharm2000207313711036227
- HadgraftJWaltersKAWottonPKFacilitated percutaneous absorption: A comparison and evaluation of two in vitro modelsInt J Pharm198632257263
- GreenPGHadgraftJFacilitated transfer of cationic drugs across a lipoidal membrane by oleic acid and lauric acidInt J Pharm198737251255
- CheongHAChoiHKEffect of ethanolamine salts and enhancers on the percutaneous absorption of piroxicam from a pressure sensitive adhesive matrixEur J Pharm Sci20031814915312594008
- MurthySNZhaoYLSenAHuiSWCyclodextrin enhanced transdermal delivery of piroxicam and carboxyfluorescein by electroporationJ Control Release20049939340215451597
- HillJRWertzPWMolecular models of the intercellular lipid lamellae from epidermal stratum corneumBiochim Biophys Acta2003161612112614561469
- McneillSCPottsROFrancoeurMLLocal enhanced topical delivery (LETD) of drugs: Does it truly existPharm Res19929142214271475228
- HeynemanCALawless-LidayCWallGCOral versus topical NSAIDs in rheumatic diseases: A comparisonDrugs20006055557511030467
- ShargelLWu-PingSYuABCApplied Biopharmaceutics and PharmacokineticsNew York, NYMcGraw-Hill1999
- ShakeelFBabootaSAhujaAAliJAqilMShafiqSNanoemulsions as vehicles for transdermal delivery of aceclofenacAAPS PharmSciTech20078E19
- VeigaAPDuarteIDAvilaMNde MottaPGTatsuoMAFrancischiJNPrevention by celecoxib of secondary hyperalgesia induced by formalin in ratsLife Sci2004752807281715464832
- ZhangYShafferAPortanovaJSeibertKIsaksonPCInhibition of cyclooxygenase-2 rapidly reverses inflammatory hyperalgesia and prostaglandin E2 productionJ Pharmacol Exp Ther1997283106910769399978