 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Purpose
To develop an in situ gel system comprising liposome-containing paclitaxel (PTX) dispersed within the thermoreversible gel (Pluronic® F127 gel) for controlled release and improved antitumor drug efficiency.
Methods
The dialysis membrane and membrane-less diffusion method were used to investigate the in vitro drug release behavior. Differential scanning calorimetry (DSC) thermal analysis was used to investigate the “micellization” and “sol/gel transition” process of in situ gel systems. In vitro cytotoxicity and drug uptake in KB cancer cells were determined by MTT, intercellular drug concentration, and fluorescence intensity assay.
Results
The in vitro release experiment performed with a dialysis membrane model showed that the liposomal gel exhibited the longest drug-release period compared with liposome, general gel, and commercial formulation Taxol®. This effect is presumably due to the increased viscosity of liposomal gel, which has the effect of creating a drug reservoir. Both drug and gel release from the in situ gel system operated under zero-order kinetics and showed a correlation of release of PTX with gel, indicating a predominating release mechanism of the erosion type. Dispersing liposomes into the gel replaced larger gel itself for achieving the same gel dissolution rate. Both the critical micelle temperature and the sol/gel temperature, detected by DSC thermal analysis, were shifted to lower temperatures by adding liposomes. The extent of the shifts depended on the amount of embedded liposomes. MTT assay and drug uptake studies showed that the treatment with PTX-loaded liposomal 18% Pluronic F127 yielded cytotoxicities, intercellular fluorescence intensity, and drug concentration in KB cells much higher than that of conventional liposome, while blank liposomal 18% Pluronic F127 gel was far less than the Cremophor EL® vehicle and empty liposomes.
Conclusions
A thermosensitive hydrogel with embedded liposome is a promising carrier for hydrophobic anticancer agents, to be used in parenteral formulations for treating local cancers.
Introduction
Paclitaxel (PTX) is a well known antineoplastic agent which acts through a number of mechanisms; that is, as a mitotic inhibitor (microtubule stabilization), an antiangiogenic agent, and a radiation sensitizer. It has been clinically used in the treatment of a wide variety of cancers, such as ovarian, skin, breast, and lung cancers.Citation1–Citation3 The anticancer activity of PTX is dependent both on its concentration and the duration of exposure.Citation4–Citation6 Although it is widely used in cancer chemotherapy, several problems associated with its clinical use remain unresolved. Its poor aqueous solubility necessitates intravenous delivery of the drug via lipophilic solvents such as the Cremophor/ethanol combination (Taxol®). This formulation has limited stability on dilution and is associated with significant vehicle-related toxicity in the clinical setting.Citation7–Citation9 Like most chemotherapeutic drugs, PTX has a narrow therapeutic window and short elimination half-life, thus requiring higher doses. However, the toxicity of solvent systems (eg, Cremophor EL®/ethanol) limits the maximum intravenous dose of PTX that can be safely used. New formulations that overcome these problems will have clear clinical advantages. In recent years, various groups of researchers have investigated the development of novel formulations for PTX, including liposomes,Citation10–Citation15 emulsions,Citation16–Citation19 cyclodextrins,Citation20–Citation22 microspheres,Citation23,Citation24 nanoparticles,Citation25–Citation31 and implants.Citation32–Citation35 In addition, injectable thermoreversible hydrogels are also being developed with reasonable success for PTX delivery.Citation36–Citation39 The unique advantages of this kind of drug delivery system, which include the provision of targeted cytotoxicity, and controlled release of a drug without systemic toxicities, have made it more and more attractive as a system with great potential for treating cancer.
Pluronic® F127 (F127) is one of the most important thermoreversible hydrogels, and it is commercially available. It is usually regarded as nontoxic and has been applied in localized drug delivery such as intramuscular, intraperitoneal, and subcutaneous injections.Citation40 A moderately concentrated solution of F127 forms a free-flowing solution at or below normal ambient temperature and is able to form a gel at body temperature, as the micelles form partially ordered cubic phases as the micellar cores dehydrate.Citation41 Therefore, it has been widely exploited as a topical drug delivery carrier of anticancer agents, and for the covering of burn wounds. It also has been used in rectal, ophthalmic, parenteral, and nasal administrations, as well as in subcutaneous administrations.Citation42,Citation43 For localized cancer therapy, intratumoral or peritumoreal injection of this gel will lead to the formation of a ‘depot’ at the site of administration that slowly and continuously releases the drug to the tumor and surrounding tissue. Using a topical or injectable gel for physical targeting has additional advantages over passive or other actively targeted therapies in that it can deliver a drug throughout the tumor regardless of vascular status, thus providing accurate dosing without systemic toxicity.Citation38
Liposomes are widely used in parenteral pharmaceutical dosage forms because they offer many advantages for delivery. In addition to drug targeting, they can increase the stability of various amphipathic molecules, improve the solubility of poorly water-soluble drugs, and can substitute for common but toxic nonaqueous cosolvents. PTX has poor aqueous solubility; however, at low concentrations, PTX completely dissolves in poloxamer gel while forming a suspension at higher concentrations.Citation44 Dhanikula et al also observed that at higher loadings, direct incorporation of paclitaxcel in gel leads to its precipitation.Citation45–Citation47 On the other hand, we believe that the incorporation of liposomal PTX into poloxamer gel should increase drug loading while, in all probability, yielding slower, more prolonged drug release compared with pure gel. Therefore, a system containing both F127 and liposome could be of great interest because they pair up the best of two worlds – the thermogelling properties of F127 and the carrier ability of the liposome.
In this study, F127 aqueous systems with different amounts of liposome were evaluated. In vitro release studies, thermal analysis investigation, and cancer cell cytotoxicity and cell drug uptake assays were made in order to outline the systems’ potential uses as parenteral formulations for localized cancer therapy. This work lays the foundation for developing a PTX delivery system joining the in situ thermogelling characteristic of F127 with the advantages offered by liposomes.
Materials and methods
Materials
PTX (molecular weight 853.4) was obtained gratis from Jiang Su AoSaiKang Co, Ltd (Jiangsu, China). F127 was obtained gratis from BASF (Hanover, Germany). Soybean phospholipids (lipids containing soya phosphatidylcholile [SPC]) were purchased from Shanghai Taiwei Co, Ltd (Shanghai, China). Nitro-benzoxadiazol-labeled phosphatidylethanolamine (NBD-PE) was purchased from Avanti Polar Lipids (Alabaster, AL). All solvents used were of analytical grade and were purchased from Sigma-Aldrich (St. Louis, MO). All the solutions were prepared using water from a Milli-Q® Gradient filtration system (Millipore, Billerica, MA).
Methods
Preparation of liposome
The solvent dispersion (ether injection) method was adopted for liposome preparation. Briefly, lipids containing SPC (250 mg) with or without PTX (20 mg) were dissolved in 3 mL ether as the organic phase and mixed thoroughly. A needle was used to draw up all the organic phase, which was then deposited just below the surface of the aqueous phase, a 20 mL phosphate-buffered saline (PBS) (pH 7.4), as it was being stirred rapidly. Stirring continued with a magnetic bar at a temperature of 55°C for 20 minutes. The suspension of lipids was then extruded six times through a 0.1 μm pore-size polycarbonate membrane (Nuclepore Track-Etch Membrane) using a LipexTM Extruder (Northern Lipids Inc., Vancouver, BC, Canada) driven by pressurized nitrogen to remove unincorporated drug aggregates. Liposomes thus obtained were stored at room temperature for further research.
For fluorescence microscopy, 1.0 mol% of NBD-PE relative to the total lipids was added to the liposome formulations.
Preparation of gel
The “cold” method was adopted for preparation of Pluronic gels as described by Soga et al.Citation48 Briefly, F127 (1.8 g) was added to 10 mL PBS (pH 7.4) aqueous solution in flat-bottomed screw-capped glass vials, and gently mixed with magnetic stirrers for 24 hours at 4°C until all of the Pluronic granules were completely dissolved and a clear solution was obtained. Excess PTX powder was added to the clear 18% F127 solution prepared above and gently mixed with the magnetic stirrers for 24 hours at 25°C. This mixture was filtered through a 0.22 μm filter (Millipore) to remove unincorporated drug aggregates, and then the obtained gels were stored at room temperature for further research.
Preparation of liposomal gel
The required amount of Pluronic granules was added to blank liposome or PTX–liposome solution in flat-bottomed screw-capped glass vials, and gently mixed with magnetic stirrers for 24 hours at 4°C until all of the Pluronic granules were completely dissolved and a clear solution was obtained. In the present experiment, the F127 concentration in F127 gel is always expressed as the weight percentage (wt%), unless specified otherwise.
In vitro release studies of PTX formulation
In vitro release of PTX using dialysis membrane method
Drug release profiles from liposomal PTX, PTX 18% F127, and liposomal–PTX 18% F127 formulation, with equivalent PTX drug concentration (300 μg/mL), were evaluated using the dialysis membrane method. For comparison, a commercial formulation (Taxol) was prepared in our laboratory according to a previous report.Citation48 Briefly, 12 mg of PTX was dissolved in 1.0 mL ethanol, and to this solution 1.0 mL Cremophor EL was added. This mixture was then sonicated for 30 minutes. The obtained Taxol formulation (PTX = 6 mg/mL) was diluted with Cremophor EL/ethanol (1:1, v:v) solvent (Taxol/Cremophor EL–ethanol), or PBS (pH 7.4) solution (Taxol/PBS) to make 300 μg/mL of PTX.
An aliquot of each PTX solution (0.5 mL) was placed in a pre-swollen dialysis bag with a 12,000–14,000 Da molecular weight cutoff (Spectra/Por® 2 dialysis membrane; Spectrum Laboratories, Auckland, NZ) and immersed into 6 mL of release medium, ie, PBS (pH 7.4) containing 0.1% (v/v) Tween 80 to maintain sink condition,Citation11 contained in a flat-bottomed glass vial (internal diameter about 20 mm), and then the vial was shaken in a thermostatic shaker at 37°C at 40 ± 10 rpm.Citation49 Samples were withdrawn at predetermined time intervals over a 24-hour period and refilled with the same amount of the fresh medium. Concentration of PTX was determined using an ultra performance liquid chromatography (UPLC) system after appropriate dilution with acetonitrile.
The UPLC system was equipped with a Waters 2487 Dual l Absorbance Detector, 717 plus Autosampler and 515 dual pumps. A reverse phase ACQUITY UPLC® BEH Shield C18 column (2.1 mm × 100 mm, 1.7 μm; Waters, Wexford, Ireland) was used at room temperature, and the detector wavelength was set at 227 nm. A mixture of acetonitrile: water (50:50, v/v) was used as the mobile phase at a flow rate of 1.0 mL/min.
In vitro release of PTX using a membrane-less diffusion method
A membrane-less diffusion system was used for studying drug release from the PTX 18% F127 gels or liposomal–PTX 18% F127 gels, and the poloxamer dissolution from Pluronic gels or liposomal Pluronic gels. A volume of 1 mL of the thermoreversible solution at room temperature was put into the bottom of a pre-weighed empty glass tube (12 mm × 75 mm) and then placed in a water bath (37°C) until a clear gel formed; the initial weight of each tube plus the gel was recorded. A volume of 2 mL of the release medium (PBS, pH 7.4, containing 0.1% [v/v] Tween 80) pre-equilibrated at 37°C was carefully layered over the surface of the gel. The tube was then shaken in a thermostatic shaker at 37°C at 40 ± 10 rpm. At predetermined timepoints, the release medium was totally replaced by fresh medium, and the weight of the tube plus the gel was recorded to calculate the weight of gel dissolved. The total PTX released into the medium was determined after appropriate dilution with acetonitrile and submitted to the UPLC system. The proportions of free PTX and PTX still encapsulated in the liposome or micelle released from the gel was determined after ultracentrifugation (10000 g, 15°C, 30 minutes) in centrifugal filter tubes (MWCO 10ku), and filtrate containing only free PTX was submitted to the UPLC system. The difference between total PTX released into the medium and free PTX allowed us to calculate the amount of PTX released from the gel but still encapsulated in the liposome or micelle.
Drug release kinetics study
To investigate the kinetics of drug release from gels in membrane- less diffusion systems, several mathematical models describing the kinetic behavior of drug release from gels were assessed.Citation50–Citation52 The drug release kinetics were determined by the best fit of the experimental data (amount of released drug versus time) to the kinetic models as described below:
where Mt/M∞ is the fraction of drug released at each timepoint (t), and Kh represents the Higuchi release kinetic constant.
where Mt/M∞ is the fraction of drug released at each timepoint (t), and K0 represents the zero-order release kinetic constant.
where Mt/M∞ is the fraction of drug released at each timepoint (t), and K1 represents the first-order release kinetic constant.
Drug release mechanism study
The drug release mechanism was studied using the semiempirical Power law model and Ritger–Peppas model. The Power law exponential expression has been extensively used by various pharmaceutical systems as follows:Citation53–Citation56
where Mt/M∞ is the fraction of drug released at each timepoint (t), K is a kinetic constant incorporating structural and geometric characteristics of the device, and n is the release exponent, indicative of the mechanism of drug release. As the release mechanism from polymeric systems is complex, this model is used when the exact mechanism is unknown or when more than one release mechanism is involved in drug release. EquationEquation (4)(4) has two distinct meanings, one in the special case of n = 0.45 (indicating diffusion-controlled drug release, also called Fickian diffusion kinetics), and a second in the special case of n = 0.89 (indicating erosion-controlled drug release, also called case II transport kinetics) for a cylindrical polymeric matrix. Values of n between 0.45 and 0.89 can be regarded as an indicator for the superposition of both phenomena (anomalous transport). When determining the n exponent, only the portions of the release curve where Mt/M∞ < 60% were used.
Best fit of all datapoints (up to 100% of release curves) was achieved by another interesting Ritger–Peppas model:
where Mt/M∞ is the fraction of drug released at each timepoint (t), and Ka, Kb, and m are constants. This equation was previously proposed to describe the release of molecules from polymeric matrices when diffusion is not the only determining factor for release, but other mechanisms, such as polymer relaxation parameters, are also important. The first term on the right-hand side represents the Fickian diffusional contribution, whereas the second term represents the case II relaxational contribution. The m value of 0.45 was taken in our study based on the aspect ratio of the matrices. The ratio of relaxational to Fickian contributions is given by:
The effect of liposome on F127 system
The sol-gelation temperature of the F127 gel (16%–27%) in PBS (pH 7.4) with and without liposome was roughly determined using the tube inversion method described previously. Briefly, each formulation was heated slowly from 10°C to 50°C at a heating rate of 1°C/min, until the solution totally lost fluidity; the temperature obtained was recorded as the gelation temperature.
Differential scanning calorimetry (DSC) was used to investigate the interaction of liposome and F127, and also used to analyze the self-assembling and thermogelation properties of F127 systems more accurately. The aqueous solutions of F127 in the absence and presence of liposome were analyzed. Samples (approximately 50 mg) were loaded in the calorimetric cells at 0°C, heated from −10°C to 60°C at 5°C/min, then cooled from 60°C to −10°C at 5°C/min by Jade DSC (PerkinElmer, Waltham, MA). A delay of 5 minutes was utilized between sequential scans to allow thermal equilibration. Data analysis was performed with the software preinstalled in the instrument (Pyris Data Analysis; PerkinElmer).
Cell culture
Human oral cancer KB cell line was obtained from American Type Culture Collection and grown in RPMI medium 1640 (Gibco/Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Gibco/Invitrogen) and 1% penicillin/streptomycin (Gibco/Invitrogen) in a humidified incubator under the conditions of 37°C, 5% CO2, and 90% relative humidity. After reaching confluence, the cells were prepared by washing in PBS and detached from the dish with trypsin-EDTA (Gibco/Invitrogen). The cells were then transferred to a centrifuge tube and centrifuged at a speed of 1000 rpm for 5 minutes, and the cells were resuspended to obtain a concentration of 5 × 104 cells/mL.
Cell cytotoxicity study
KB cells were seeded in a 96-well plate at a density of 5 × 103 cells per well, 24 hours before the in vitro cytotoxicity studies started. PTX formulation loaded with Taxol, liposome, or liposomal 18% F127 gel was prepared with PBS (pH 7.4) as described above (with equivalent PTX concentration of 300 μg/mL. Control formulations without PTX were also prepared using the same procedure. The three stock solutions were filtered through sterile 0.22 μm Millipore paper, and then further diluted with sterile PBS (pH 7.4) stepwisely to make PTX concentrations in the range of 0.0003–300 μg/mL. To evaluate their cytotoxic effects, 100 μL of the PTX formulations and 100 μL of the culture medium (RPMI Medium 1640 + 10% FBS) were added to the cells. As the reference, 100 μL of the culture medium was added to the cells as well as 100 μL of PBS (pH 7.4). The cells were incubated for 24, 48, and 72 hours at 37°C in a humidified atmosphere with 5% CO2. After incubation, the number of viable cells was determined by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay.
A volume of 20 μL of MTT solution (5 mg/mL) was added to each well. The plates were incubated for an additional 4 hours, and then the medium was discarded. A volume of 150 μL of DMSO (dimethyl sulfoxide) was added to each well, and the solution was vigorously mixed to dissolve the reacted dye. The absorbance (A) of each well was read on a microplate reader (BIO-RAD Benchmark Plus/microplate spectrophotometer, UK) at a test wavelength of 570 nm and reference wavelength of 650 nm. The c sample was tested in triplicate, and six wells with only culture medium served as blanks; the relative cell viability (%) was calculated as (Aof treated cells/Aof untreated cells) × 100%.
Measurement of intracellular drug concentration
KB cells were suspended in the culture medium (1 mL) at a density of 1 × 106 cells/well in 24-well flat-bottom tissue culture plates. After 24 hours, the medium was replaced with 1 mL of FBS-free culture media containing a formulation loaded with 500 ng/mL of PTX. Each cell in triplicate was incubated for 1, 2, 4, and 6 hours (37°C). The cells in each well were washed with cold PBS twice to remove nonuptaken drug, and 100 μL trypsin-EDTA solution (0.25%) was added. A volume of 0.5 mL acetonitrile was added to destroy the cell and precipitate the protein, and then after centrifugation at 10,000 rpm for 10 minutes, 2 μL of the supernatant was used for UPLC detection to obtain the intracellular drug concentration.
Cellular uptake and distribution of fluorescent blank liposome
For qualitative study, KB cells were seeded in a 96-well plate at a density of 5 × 104 cells per well in 100 μL of the culture medium and cultured for 24 hours before the experiment. For studying liposome uptake and distribution by fluorescent microscopy, 20 μL of NBD-labeled liposome or NBD-labeled liposomal 18% F127 formulation without PTX (liposome contained 1.0 mol% of NBD-PE relative to the phospholipids) and 100 μL culture medium was added to the cells. As a reference, 100 μL culture medium was added as well as 20 μL PBS (pH 7.4) and 20 μL PBS (pH 7.4). After 1-hour and 4-hour incubation, cells were washed twice with cold PBS (pH 7.4) and directly observed under a fluorescence microscope (Inverted Microscope Axiovert 200; Carl Zeiss, Wetzlar, Germany).
For quantitative study, 20 μL trypsin-EDTA solution (0.25%) was added to detach the cells after washing the cells with cold PBS. The cells were then harvested by adding 200 μL acetonitrile three times to obtain the cell lysate and the dissolved NBD-PE. Finally, the cell lysate was centrifuged at 10,000 rpm for 10 minutes, and the supernatant was used for fluorescence assay with a fluorometer (LS 55, Luminescence Spectrometer; PerkinElmer) (excitation, 460 nm; emission, 534 nm). The fluorescence intensity of cell lysate was normalized to that of the reference solution.
Statistical analysis
Data were expressed as the means of three separate experiments, and were compared by analysis of variance (ANOVA). A P-value < 0.05 (or <0.01) was considered statistically significant in all cases. All data analysis was performed with Microsoft Excel (Microsoft Corp, Redmond, WA).
Results and discussion
Comparison of the effect of different drug-loaded formulations on PTX release using dialysis membrane method
The drug-release behaviors of f ive PTX formulations were measured in vitro by the dialysis membrane method (). Taxol solution showed the fastest release rate. PTX formulations, especially the Taxol diluted with PBS, simulating clinical treatment form, exhibited a higher release rate than that diluted with Cremophor EL/ethanol (1:1, v:v) solvent. This is due to the lower viscosity of PBS solvent compared with Cremophor EL/ethanol (1:1, v:v) mixture solvent. A marked reduction of drug release was observed when drug was incorporated into liposomes. The release of PTX was rapid and almost complete from the Taxol formulation within 12 hours, while the incorporation of drug into liposome significantly retarded drug release to only 50% released at 12 hours, suggesting that most of the drug remained incorporated in liposomes under the study conditions. The maintenance of the sink condition in the in vitro drug release media signifies that the outer environment of the dialysis membrane has a strong solubilization capacity for the insoluble drug; thus, the gradual release of drug from the liposomes depends on the incorporating efficiency, and its diffusion to the external medium depends on the concentration gradient. Our results agree with the general conclusion that liposomal entrapment of drugs sustains their release. Moreover, further reduction in drug release was observed from drug-loaded gels which could be attributed to the much higher viscosity of gels compared to the PTX solutions. The latter represented a less permeable matrix with lower drug release; however, no significant difference was observed between liposomal-PTX 18% F127 gel and PTX 18% F127 gel under the conditions studied.
Figure 1 In vitro drug release kinetics from Taxol® (![]()
 ), liposomal-PTX solution (
), liposomal-PTX solution ( ), liposomal-PTX 18% F127 gel (
), liposomal-PTX 18% F127 gel ( ), and PTX 18% F127 gel (
), and PTX 18% F127 gel (Abbreviations: F127, Pluronic® F127; PBS, phosphate buffered saline; PTX, paclitaxel.
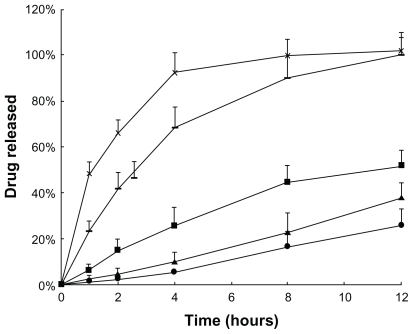
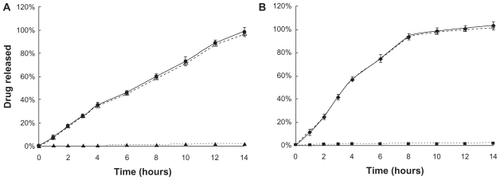
Figure 2 In vitro drug release kinetics from F127 gels as a function of time (hours), using the membrane-less model at 37°C. Data are expressed as mean ± standard deviation (n = 3). A) Liposomal-PTX 18% F127 gel:
 for liposomal drug. B) PTX 18% F127 gel:
for liposomal drug. B) PTX 18% F127 gel:  for total drug,
for total drug,  for micellar drug.
for micellar drug.Abbreviations: F127, Pluronic® F127; PTX, paclitaxel.
Comparison of PTX release from drug-loaded F127 gels performed by membrane-less method
When the PTX-loaded gels were separated from the release medium by a dialysis membrane, the dissolution of F127 gel was almost prevented, and diffusion dominated the drug release mechanism. In contrast, the membrane-less model, also used in our study, allowed the release medium solution to directly contact the gel surface and thereby dissolve the gel. As shown in , the dispersion of liposomes within the 18% F127 gel led to a much slower drug release than the simple micelle gel formulation without liposomes at each timepoint. The difference is more distinct compared with the dialysis membrane method. The form of PTX released from 18% F127 gels (free PTX or PTX still encapsulated in liposomes or micelles) was quantified. The results indicated that total PTX released was almost encapsulated in liposomes or micelles during the whole periods of release time from both gel formulations, which was due to the high PTX entrapment efficiency (~100%) of PTX liposomes or micelles determined by substracting free drug fraction, as described in the section ‘In vitro release of PTX using membrane-less diffusion method’ above.
One reason for the lower release of liposomal PTX from 18% F127 could be the ‘reservoir effect’; that is, the liposome carrier acts as a reservoir within the gel, lowering the diffusion rate from the inner gel to the outer medium. This phenomenon is also present in dialysis membrane experiments. However, besides the diffusion mechanism, there could be another important mechanism contributing to the drug-release behavior because the gradually decreasing volume of gel could be clearly observed in the membrane-less dissolution experiments, and most of the gel formulations had completely disappeared by the final timepoint.
The best fit between dissolution profiles and kinetic models was obtained with the Power law equation (R2 > 0.997 for both drug-loaded gels), and values of n higher than 0.89 (n = 0.951 for the liposomal PTX gel, n = 0.974 for the PTX gel) indicated that gel erosion was the predominant release mechanism ().
Table 1 Data from regression fitting between dissolution profiles (obtained from liposomal-PTX 18% F127 gel and PTX 18% F127 gel) and several kinetic models (Higuchi, zero-order, and first-order) and the Power law and Ritger-Peppas model (release mechanism)
Since only the first four or six datapoints (for the first 60% of release curves) could be used when analyzing release results with the Power law equation, the best correlations were further obtained when applying the two-parameter equation (EquationEquation (5)(5) ) (R2 > 0.99 for both drug-loaded gels) for the 100% of release curves. With this analysis, the kinetic constant Kb (relaxational contribution) was much higher than Ka (diffusion contribution) in both formulations (Kb = 0.0914 and Ka = 0.0073 for liposomal-PTX 18% F127 gel compared with Kb = 0.1118 and Ka = 0.0643 for PTX 18% F127 gel). This proves that the most significant processes for PTX release are erosion-type processes ().
Correlation between gel dissolution and drug release
To investigate whether the difference observed in PTX release between the liposome-in-gel system and the micelle-in-gel system is due to the extreme difference in the gel dissolution rate or erosion rate in each case, extra experiments were performed, in which the dissolution behavior of these two drug-loaded gel formulations was investigated by weighing them at the same timepoints (). The remaining weight% of gels also followed zero-order kinetics (R2 > 0.99), and the good linear correlation between the percentage of PTX released and the percentage of gel dissolved further indicated a distinct polymer dissolution-controlled release mechanism (R2 > 0.99) (). Similar zero-order release kinetics could be found in previous reports for recombinant hirudin,Citation57 interleukin-2,Citation58 insulin,Citation43 and ceftiofur.Citation59 However, first-order kinetics and Higuchi kinetics have also been reported.Citation60–Citation62 This discrepancy might be due to differences in experimental conditions, or in drug and gel properties. In our study, PTX was very poorly water soluble, which differs from those water soluble compounds previously reported. Thus, the molecular diffusion from the inner gel to the outside medium would be limited, as demonstrated in the dialysis membrane method, especially in PBS (pH 7.4) without Tween-80 solution, like the circumstance of gel inner-side. This implies that the release rates of lipophilic drugs are more dependent on the polymeric matrix dissolution rate than the diffusion rate.
Figure 3 In vitro drug release (%) from F127 gels (
 for liposomal-PTX 18% F127 gel,
for liposomal-PTX 18% F127 gel, Abbreviations: F127, Pluronic® F127; PTX, paclitaxel.
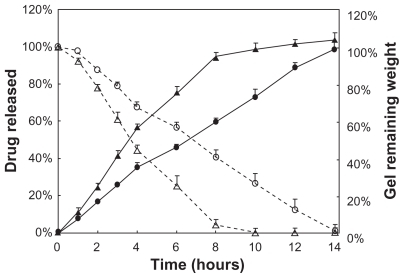
Figure 4 Correlation between PTX release and gel dissolution; data taken from . The line represents a linear regression fit. A) Liposomal-PTX 18% F127 gel. B) PTX 18% F127 gel.
Abbreviations: F127, Pluronic® F127; PTX, paclitaxel.
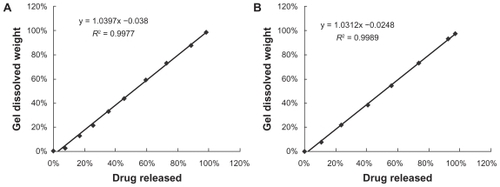
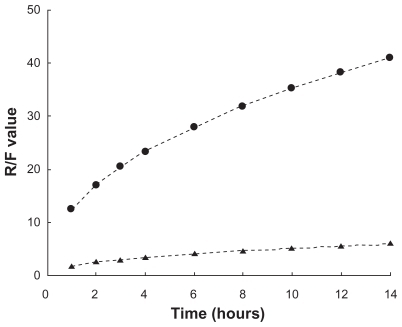
In fact, gel relaxation or erosion contributes almost 90% of drug release from liposome-in-gel systems in the whole dissolution process, whereas these processes contribute only 60%–80% of drug release from micelle-in-gel systems (). This phenomenon was demonstrated more clearly with liposomal gels than pure gels, because the ratio of relaxational over Fickian contributions (R/F value) is much higher for the liposome-in-gel system compared with the micelle-in-gel system at every timepoint (). This further proves that polymer dissolution is an important factor determining release of poorly water-soluble drugs from F127 gels. The liposome dispersed in the F127 gel could significantly reduce the erosion rate of formulation. So, it is important that the dissolution rate of the gel would be significantly affected by the formulation composition, especially when liposome is involved in the poloxamer gel.
Figure 5 Fickian diffusional contribution (%) (
for liposomal- PTX 18% F127 gel, Abbreviations: F127, Pluronic® F127; PTX, paclitaxel.
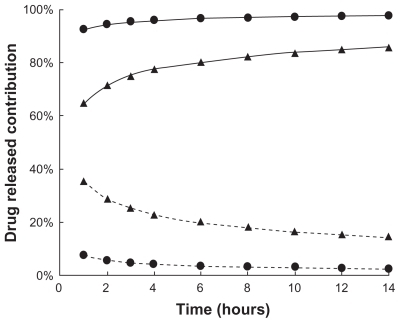
Figure 6 R/F value profile (
Abbreviations: F127, Pluronic® F127; PTX, paclitaxel.
Effect of different concentrations of poloxamer or liposome on gel dissolution
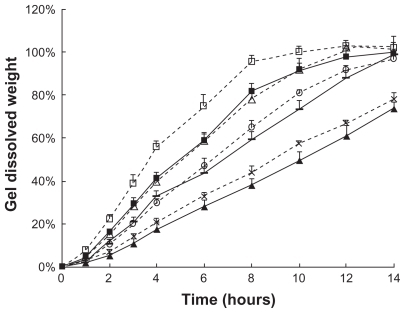
To investigate whether gel dissolution could be modified by adding different amounts of liposomes, the dissolution rate was evaluated in the case of drug-free liposomal F127 gel with low (0.21%, w/v), middle (0.42%, w/v), and high (1.25%, w/v) lipid content at the same polymer concentration of 18%. For comparison, the dissolution rate of drug-free F127 gels containing no liposome (at polymer concentrations of 18%, 21%, 24%, and 27%) were also investigated. It was seen that the rate of gel dissolution significantly decreased with the increase of F127 concentration (P < 0.05) and also with increase of lipid content in the gel (P < 0.05), and the addition of PTX did not significantly change the gel dissolution rate when compared with drug-loaded gel (P > 0.05) (). On the other hand, the dissolution profile of liposomal 18% F127 gel with low, medium, and high lipid content of liposome seemed practically similar to pure F127 gel with polymer concentrations of 21%, 24%, and 27%, respectively (). As shown in ,
Table 2 Data from regression fitting between gel dissolution profiles (obtained from F127 gels and liposome F127 gels) and zero-order model
Figure 7 In vitro gel dissolution (dissolved wt%) of F127 gel without drug (
 for 24% F127 gel,
for 24% F127 gel,  for 27% F127 gel, for liposome (with low lipid content) 18% F127 gel,
for 27% F127 gel, for liposome (with low lipid content) 18% F127 gel, Abbreviation: F127, Pluronic® F127.
In our study, an easier and safer way to reduce the drug release rate was found to meet long-term treatment needs for certain diseases, such as localized injection of certain antitumor drugs in cancer therapy. The adding of liposomal drug to thermoreversible hydrogel can not only avoid the potential toxicity of greater amounts of copolymer (usually reported for above 20% F127 gel formulation),Citation47,Citation57,Citation60,Citation63,Citation64 but also provide higher drug loading (about 1020 μg/mL, determined in our experiment) than that possible with copolymer micelle gel alone (about 430 μg/mL, determined in our experiment). Potentially, the drug dose could be flexibly adjusted according to therapeutic needs for different individual patients and different disease states, simply by dispersing different liposome concentrations in gels.
Moreover, liposomal PTX incorporated into an 18% F127 hydrogel vehicle could improve the stability of PTX liposome. The poor physical stability of PTX liposomes was apparent after 7 days from the date of preparation at room temperature by our visual observation, and was generally due to sedimentation and aggregation during storage. However, in the case of liposomal 18% F127 formulations, no perceptible change in appearance by microscope observation was observed when stored at room temperature for 30 days; meanwhile no significant changes were found in drug entrapment efficiency and drug content value (data not shown). This could be due to a higher viscosity of drug delivery system produced by adding F127, which contributes to favorable dispersion of liposomes.
Thermal analysis of thermo-sensitive F127 system
The inverted tube method showed that F127 PBS (pH 7.4) solution with F127 concentration equal to or higher than 18% exhibited reversal thermal gelation, transforming from liquid into a semisolid gel in 1–2 minutes at the characteristic sol-gel transition temperatures which were 29.2°C, 26.4°C, 23.1°C, and 20.9°C for 18%, 21%, 24%, and 27% F127 solutions without drug, respectively. Increasing the F127 concentration could decrease the sol-gel transforming temperature, which agreed with previous reports,Citation63,Citation65,Citation66 although the data were not identical since a different solvent was used and the experiment was performed under different conditions. Moreover, the gelation temperatures of 18% F127 containing low (0.21%, w/v), medium (0.42%, w/v), and high (1.25%, w/v) lipid concentrations of blank liposomes was found at 27.4°C, 25.8°C, and 23.1°C, respectively. In many published studies, the effect of smaller solutes, salts, cosolvents, or cell culture on the phase transform behavior of F127 has been well discussed,Citation41,Citation61,Citation63,Citation67–Citation69 but the influence of liposomes on the self-assembling and thermogelation property of the F127 system has not been examined. Therefore, a more easy and sensitive method (DSC thermal analysis) was considered for our study in order to get more useful information about both micellization and sol- gelation transition processes that are always accompanied by the thermoreversible behavior of F127.
It is well known that when F127 is put in water above a critical concentration, its structure changes from individual block copolymers (unimers) to self-assembling micelles. This process, involving aggregation, is defined as “ micellization”. At the same time, block copolymers at a given concentration are also characterized by a specific temperature at which they form micelles (critical micelle temperature, CMT). At elevated temperatures, these systems show phase transformation due to the complete dehydration of both PO (propylene oxide) and EO (ethylene oxide) blocks.Citation70,Citation71 Thus the systems are, in general, liquids at low temperature but give rise to a strong gel at a given higher temperature. This process involving phase transformation is defined as “sol-gelation transition”. The DSC technique was performed to outline the variation in the CMT and in the sol-gel transition temperature (SGT) of the systems containing different amounts of F127 and different amounts of liposomes. In order to better illustrate the sol-gel transition, the derivative heat flow (mW/min) is plotted against temperature (°C) in .
Figure 8 Comparison of the micellization and gelation processes of A) pure F127 formulations (16, 18, 21, 24, and 27 wt%) and B) 18% F127 gel containing pure liposome (low, medium, and high lipid content) solution, pure 18% F127 gel, and pure liposome (with high lipid content) by differential scanning calorimetry analysis at a scanning rate of 5°C/min. For better illustration of the sol-gel transition, the derivative heat flow was plotted against temperature.
Abbreviation: F127, Pluronic® F127.
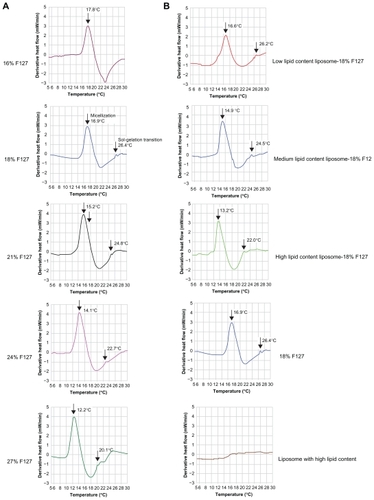
Under the experimental conditions of this study, there was a significant endothermic peak with increase in temperature for F127 gel samples, representing the CMT of F127 (cause of micellization), which could not be found in corresponding liposome solution samples in the absence of F127. In addition, a small shoulder peak representing SGT was found at a higher temperature, which appeared at the right-hand end of the CMT peak, corresponding to micelle rearrangement – a cause of thermogelation and consistent with previous reports.Citation41,Citation57,Citation71 However, thermogelation is only accompanied by a small enthalpy value; thus, it appears that DSC is less sensitive to the occurrence of the sol-gel transition, but more sensitive to the onset of rapid micelle formation.
As can be seen in , the CMT peak was shifted to a lower temperature with increasing F127 concentration for the samples without liposome, but the DSC curve pattern did not change. This result is in agreement with previously reported data.Citation57,Citation64,Citation71 The SGT as measured by DSC agreed with those measured with the inverted tube method. also shows an endothermic peak obtained for the pure 16% F127 solution. Although at this concentration the solution is known to be above the critical micellar concentration, it was not observed to undergo gelation, behaving as a liquid in the temperature range of 0°C–50°C, confirmed visually by the inverted tube method. Also, as can be seen in , no sol/gel transient weak endothermic peak was observed.
The phase behavior of this block copolymer is strongly affected by liposomes even when thermoreversibility is maintained. Both micellization and the micelle rearrangement phenomena are shifted to lower temperatures upon the addition of liposomes (). It is clear that the presence of liposomes with low lipid content allowed 18% F127 solutions to form thermoreversible gels similar to those of pure 21% F127 solution. Likewise, the 18% F127 solution with medium lipid content behaved similarly to those of pure 24% F127 solution, and 18% F127 solution with high lipid content was similar to those of the pure 27% F127 solution. The possible explanation for this phenomena is that some of the water molecules would have been taken into the hydrophilic inner core of the liposome bilayer structure, so the presence of liposomes probably caused dehydration of the micelles’ cores, which has been suggested as one of the major causes of gelation in these systems.Citation41,Citation66 The more liposome present in the F127 solution, the more water molecules would be located within the inner core of the liposomes, and consequently the greater the extent of dehydration.
This observation immediately highlights the benefits of using drug-loaded liposome carrier in F127 formulations. Localized cancer therapy is an important and promising application of F127 formulations,Citation36,Citation72–Citation74 and our results indicate that the mere presence of such liposome drug carrier could result in a 3–9-wt% decrease in the concentration of F127 required for a gel-based delivery formulation, while maintaining desirable themoreversibility and gel dissolution capabilities. We have proven this to be true in in vitro gel dissolution experiments. The benefits of such a system are many, including lower toxicity concerns regarding the copolymer, reduced costs for formulation production, and better control over the drug release rate.
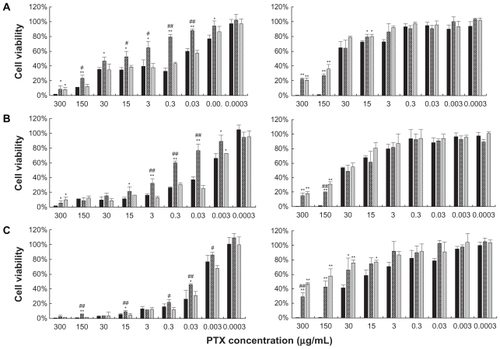
Cell cytotoxicity study
shows the in vitro viability of KB cancer cells after 24, 48, and 72 hours treatment with PTX formulated in liposome, liposomal 18% F127, and its current clinical dosage form Taxol at the same drug concentration range (0.0003–300 μg/mL). From , a few general conclusions can be immediately made. The Taxol vehicle showed greater cytotoxicity than any other blank formulation, especially at higher concentrations (300 and 150 μg/mL). The cellular viability of the Taxol vehicle was nearly 0%, while others were close to 20% (P < 0.01). Therefore, it could be expected that in vivo liposome and liposomal F127 gels would be far less toxic than the Taxol vehicle; if so, then the administration of PTX-loaded liposome and liposomal F127 gels at higher drug doses than PTX formulated in the Taxol vehicle would be possible. Secondly, in most cases, the liposomal-F127 gel demonstrated cytotoxicity greater than that of liposome with similar drug loading during the entire period. For example, after 24, 48, and 72 hours treatment, KB cell viability was decreased to 79.4%, 59.7%, and 22.0% by drug, and to 43.5%, 30.7%, and 11.6% by liposomal drug-F127 at 0.3 μg/mL, respectively (P < 0.01). Thus, therapeutic advantages might be provided after incorporating liposomal PTX into F127 gels. Cytotoxic activity was comparable in Taxol and liposomal drug-F127 formulation. Therefore, PTX remains biologically active as an anticancer agent even after incorporation into liposomal F127 gels. However, at higher drug concentrations, the cytotoxicity of Taxol was stronger than that of liposomes, which was apparently due to the Cremophor EL vehicle rather than PTX. In contrast, for drug-loaded liposome or liposomal F127 gel, the cytotoxicity is largely from the toxicity of the drug itself. Besides, the safety of blank liposomal F127 gel was higher than pure liposome, which was probably due to the capacity of increasing the cell viability of F127 alone below and at the CMC; nevertheless, cell viability declined steadily above the CMC.Citation75
Figure 9 Cytotoxicity of formulations on KB cells after 24 hours A), 48 hours B), and 72 hours C) incubation (■ for Taxol, 
Notes: *denotes P < 0.05 and **denotes P < 0.01 compared with Taxol. #denotes P < 0.05 and ##denotes P < 0.01 compared with liposomal 18% F127 formulation.
Abbreviations: F127, Pluronic® F127; PTX, paclitaxel.
Cellular uptake study
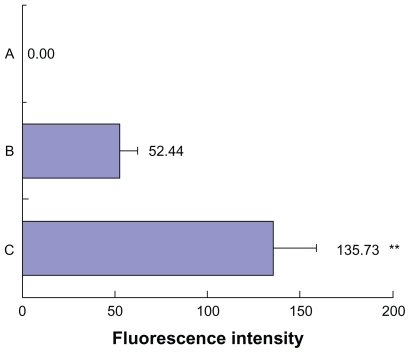
To clarify the relationship between cytotoxicity and drug concentration in the cells, the cellular uptake of drug was measured after incubating cells with drug-loaded formulation. As shown in , the highest intracellular drug concentration was obtained at 4 hours, and then decreased in most cases. The reduction of drug concentration might be caused by cellular drug metabolisms. A slightly lower PTX uptake of cells from the liposomes than that from Taxol appeared at 2 and 4 hours, which might be due to the time delay caused by endocytosis of transmembrane transport and sustained drug release from the liposomes caused by the “reservoir effect”. It is interesting to note that for the liposomal– F127 formulation, a significantly higher intracellular drug concentration was found than that for the liposome alone, when the cells were incubated for 2, 4, and 6 hours (P < 0.01). These results provide a good explanation for the stronger cytotoxicity of drug-loaded liposomal–F127 than that of drug-loaded liposomes alone observed in cell variability assay. Moreover, the cell uptake and distribution of fluorescent (NBD-PE)-labeled blank liposome in KB cells was observed by a fluorescence microscope for these two formulations after 1 hour and 4 hours incubation (), while the fluorescence uptake into the intracellular matrix was quantitatively estimated by a fluorometer after 4 hours incubation. From the images of cells, it could be seen that the KB cells with no treatment of formulation () showed a weak undetectable green fluorescence. In the case of cells exposed to liposome formulations, the intact liposome was found within the cellular components, and at the same time, some diffused fluorescence was clearly observed in the entire intracellular matrix due to the liposome degradation caused by the enzymes or environmental factors in cells during the incubation period, resulting in the loss of structural integrity and release of the fluorescent lipid molecule into the cytosol (). However, a more significant finding was that a notably higher fluorescence intensity was detected for liposomal–F127 formulation than pure liposome (P < 0.01) (). So far, no similar experimental results related to this phenomenon have been reported. For the moment, it is not clear what mechanism of the liposomes' uptake of cells may be enhanced by suspending in F127 gel. We speculate that it is probably related to surface activation to the cell membrane by an amphiphilic surfactant of F127. Therefore, further exploration needs to be carried out to reveal the nature of this phenomenon.
Figure 10 Intracellular PTX concentration against incubation time after the KB cells were incubated with the drug in different formulations (
for liposome, Notes: *denotes P < 0.05 and **denotes P < 0.01 compared with Taxol. #denotes P < 0.05 and ##denotes P < 0.01 compared with liposome.
Abbreviations: F127, Pluronic® F127; PTX, paclitaxel.
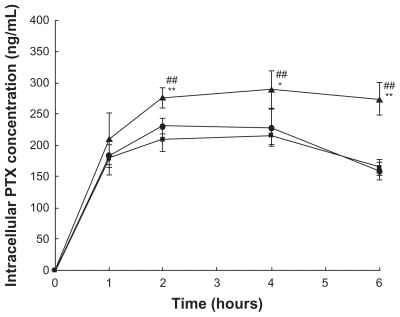
Figure 11 Fluorescence microscopic evaluation of NBD-labeled liposome and liposomal 18% F127 in KB cells after 1 and 4 hours treatment at 37°C. Left panels indicate cells visualized in the fluorescence mode; right panels indicate the same fields in the phase-contrast mode. A) Treated with no formulation. B) Treated with NBD-labeled liposome. C) Treated with NBD-labeled liposomal 18% F127 gel.
Abbreviations: F127, Pluronic® F127; NBD, nitro-benzoxadiazol.
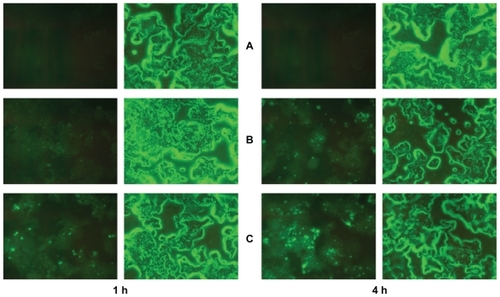
Figure 12 The cellular uptake intensity of fluorescent liposome in KB cells after 4 hours treatment. Data are expressed as mean ± standard deviation (n = 6). A) Treated with no formulation. B) Treated with NBD-labeled liposome. C) Treated with NBD-labeled liposomal 18% F127 gel.
Note: **P < 0.01 compared with liposome.
Conclusion
According to the results of this study, a thermoreversible hydrogel based on F127 containing liposomal PTX can be utilized for controlled drug delivery and enhanced drug uptake in cancer cells. Firstly, the desired drug release rate from this liposomal gel formulation could be obtained by incorporating an appropriate amount of liposome contained in the gels, instead of increasing the concentration of F127. Liposome may not only provide the means for substantially increased lipophilic drug loading in gels, but it may also act as a reservoir for sustained or delayed drug release. Our results indicate that the mere presence of liposome comprising 0.21%–1.25% SPC may substitute for 3%–9% F127 concentration required for a gel-based delivery formulation and still maintain similar thermoreversibility and gel dissolution behavior. This has significant positive implications for the toxicity and economic issues related to using F127 in drug delivery. Adjusting the amount of liposome in gels is an easy, flexible means of adjusting the drug dose required to meet different therapeutic needs of individual patients. Secondly, in vitro cytotoxicity and drug uptake in KB cancer cells of the PTX-loaded liposomal 18% F127 system was much higher than that of conventional liposomes, implying that F127 could be used as an intracellular penetration enhancer for liposomal drugs to improve their anticancer efficiency. At the same time, the cell cytotoxicity of corresponding blank liposomal 18% F127 gel was far less than that of the Cremophor EL vehicle and blank liposome. These results of our research collectively indicate that the utilization of liposomal thermoreversible gel as a non-Cremophor-based parenteral formulation for localized cancer therapy using hydrophobic anticancer agents such as PTX has a high potential for success.
Acknowledgments
The authors would like to thank Jiang Su AoSaiKang Co, Ltd (China) for the supply of PTX and BASF (Germany) for the supply of F127. The authors also would like to greatly thank Yuenfan Wong and Huihui Zhang for their excellent technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
- SchiffPBFantJHorwitzSBPromotion of microtubule assembly in vitro by TaxolNature1979277665667423966
- TishlerRBGeardCRHallEJSchiffPBTaxol sensitizes human astrocytoma cells to radiationCancer Res199252349534971350755
- MekhailTMMarkmanMPaclitaxel in cancer therapyExpert Opin Pharmacother2002375576612036415
- JangSHWientjesMGAuJLEnhancement of paclitaxel delivery to solid tumors by apoptosis-inducing pretreatment: effect of treatment scheduleJ Pharmacol Exp Ther20012961035104211181938
- AuJLLiDGanYPharmacodynamics of immediate and delayed effects of paclitaxel: role of slow apoptosis and intracellular drug retentionCancer Res199858214121489605758
- RaymondEHanauskeAFaivreSEffects of prolonged versus short-term exposure paclitaxel (Taxol) on human tumor colony-forming unitsAnticancer Drugs199783793859180392
- BeijnenJHBeijnen-BandhoeAUDubbelmanACvan GijnRUnderbergWJChemical and physical stability of etoposide and teniposide in commonly used infusion fluidsJ Parenter Sci Technol1991451081122051255
- FengSHuangGEffects of emulsifiers on the controlled release of paclitaxel (Taxol) from nanospheres of biodegradable polymersJ Control Release200171536911245908
- YehMKCoombesAGAJenkinsPGA novel emulsification-solvent extraction technique for production of protein loaded biodegradable microparticles for vaccine and drug deliveryJ Control Release199533437445
- YangTChoiMKCuiFDPreparation and evaluation of paclitaxel-loaded PEGylated immunoliposomeJ Control Release200712016917717586082
- YangTCuiFDChoiMKEnhanced solubility and stability of PEGylated liposomal paclitaxel: in vitro and in vivo evaluationInt J Pharm200733831732617368984
- WeiZHaoJYuanSPaclitaxel-loaded Pluronic P123/F127 mixed polymeric micelles: formulation, optimization and in vitro characterizationInt J Pharm200937617618519409463
- ShinHCAlaniAWRaoDARockichNCKwonGSMultidrug loaded polymeric micelles for simultaneous delivery of poorly soluble anticancer drugsJ Control Release200914029430019409432
- HuhKMMinHSLeeSCLeeHJKimSParkKA new hydrotropic block copolymer micelle system for aqueous solubilization of paclitaxelJ Control Release200812612212918155795
- SeowWYXueJMYangYYTargeted and intracellular delivery of paclitaxel using multi-functional polymeric micellesBiomaterials2007281730174017182095
- NornooAOChowDSCremophor-free intravenous microemulsions for paclitaxel: II. Stability, in vitro release and pharmacokineticsInt J Pharm200834911712317869458
- NornooAOZhengHLopesLBJohnson-RestrepoBKannanKReedROral microemulsions of paclitaxel: In situ and pharmacokinetic studiesEur J Pharm Biopharm20097131031718793723
- KanPChenZBLeeCJChuIMDevelopment of nonionic surfactant/phospholipid o/w emulsion as a paclitaxel delivery systemJ Control Release19995827127810099152
- LeeIHParkYTRohKChungHKwonICJeongSYStable paclitaxel formulations in oily contrast mediumJ Control Release200510241542515653161
- BouquetWBoterbergTCeelenWIn vitro cytotoxicity of paclitaxel/β-cyclodextrin complexes for HIPECInt J Pharm200936714815418938234
- HamadaHIshiharaKMasuokaNMikuniKNakajimaNEnhancement of water-solubility and bioactivity of paclitaxel using modified cyclodextrinsJ Biosci Bioeng200610236937117116587
- BouquetWCeelenWFritzingerBPaclitaxel/β-cyclodextrin complexes for hyperthermic peritoneal perfusion – formulation and stabilityEur J Pharm Biopharm20076639139717240125
- LiuJMeisnerDKwongEWuXYJohnstonMRA novel trans-lymphatic drug delivery system: implantable gelatin sponge impregnated with PLGA–paclitaxel microspheresBiomaterials2007283236324417434581
- JacksonJKHungTLetchfordKBurtHMThe characterization of paclitaxel-loaded microspheres manufactured from blends of poly(lactic-co-glycolic acid) (PLGA) and low molecular weight diblock copolymersInt J Pharm200734261717555895
- FangLJianingLXuejunWAnti-tumor activity of paclitaxel-loaded chitosan nanoparticles: an in vitro studyMater Sci Eng C Biomim Mater Sens Syst20092923922397
- DanhierFLecouturierNVromanBPaclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluationJ Control Release2009133111718950666
- FanLHongWHuiZAntitumor drug Paclitaxel-loaded pH-sensitive nanoparticles targeting tumor extracellular pHCarbohydr Polym200977773778
- LeeMKLimSJKimCKPreparation, characterization and in vitro cytotoxicity of paclitaxel-loaded sterically stabilized solid lipid nanoparticlesBiomaterials2007282137214617257668
- PulkkinenMPikkarainenJWirthTThree-step tumor targeting of paclitaxel using biotinylated PLA-PEG nanoparticles and avidin–biotin technology: formulation development and in vitro anticancer activityEur J Pharm Biophar2008706674
- LetchfordKLigginsRWasanKMBurtHIn vitro human plasma distribution of nanoparticulate paclitaxel is dependent on the physicochemical properties of poly(ethylene glycol)-block-poly(caprolactone) nanoparticlesEur J Pharm Biopharm20097119620618762253
- DongXMattinglyCATsengMChoMAdamsVRMumperRJDevelopment of new lipid-based paclitaxel nanoparticles using sequential simplex optimizationEur J Pharm Biopharm20097291719111929
- Lim SooPChoJGrantJHoEPiquette-MillerMAllenCDrug release mechanism of paclitaxel from a chitosan–lipid implant system: effect of swelling, degradation and morphologyEur J Pharm Biopharm20086914915718164931
- KraitzerAOfekLSchreiberRZilbermanMLong-term in vitro study of paclitaxel-eluting bioresorbable core/shell fiber structuresJ Control Release200812613914818201789
- ShikanovAShikanovSVaismanBGolenserJDombAJPaclitaxel tumor biodistribution and efficacy after intratumoral injection of a biodegradable extended release implantInt J Pharm200835811412018406086
- LaiYengLSudhirHRYilongFPaclitaxel release from microporous PLGA disksChem Eng Sci20096443414349
- GuoDDXuCXQuanJSSynergistic anti-tumor activity of paclitaxel-incorporated conjugated linoleic acid-coupled poloxamer thermosensitive hydrogel in vitro and in vivoBiomaterials2009304777478519524293
- ChunCLeeSMKimSYYangHKSongSCThermosensitive poly(organophosphazene)–paclitaxel conjugate gels for antitumor applicationsBiomaterials2009302349236019178941
- ElstadNLFowersKDOncoGel (ReGel/paclitaxel) – clinical applications for a novel paclitaxel delivery systemAdv Drug Deliv Rev20096178579419422870
- LiawJLinYEvaluation of poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (PEO–PPO–PEO) gels as a release vehicle for percutaneous fentanylJ Control Release20006827328210925135
- YangYWangJZhangXLuWZhangQA novel mixed micelle gel with thermo-sensitive property for the local delivery of docetaxelJ Control Release200913517518219331864
- SharmaPKReillyMJJonesDNRobinsonPMBhatiaSRThe effect of pharmaceuticals on the nanoscale structure of PEO–PPO–PEO micellesColloids Surf2008615360
- GuzmánMGarcíaFFMolpeceresJPolyoxyethylene- polyoxypropylene block copolymer gels as sustained release vehicles for subcutaneous drug administrationInt J Pharm199280119127
- BarichelloJMMorishitaMTakayamaKNagaiTAbsorption of insulin from Pluronic F-127 gels following subcutaneous administration in ratsInt J Pharm199918418919810387948
- AmijiMMLaiPKShenoyDBRaoMIntratumoral administration of paclitaxel in an in situ gelling poloxamer 407 formulationPharm Dev Techno20027195202
- DhanikulaABSinghDRPanchagnulaRIn vivo pharmacokinetic and tissue distribution studies in mice of alternative formulations for local and systemic delivery of paclitaxel: gel, film, prodrug, liposomes and micellesCurrent Drug Deliv200523544
- DhanikulaABPanchagnulaRPreparation and characterization of water-soluble prodrug, liposomes and micelles of paclitaxelCurrent Drug Deliv200527591
- DhanikulaRSDhanikulaABPanchagnulaRThermoreversible liposomal poloxamer gel for the delivery of paclitaxel: dose proportionality and hematological toxicity studiesPharmazie20086343944518604987
- SogaOvan NostrumCFFensMThermosensitive and biodegradable polymeric micelles for paclitaxel deliveryJ Control Release200510334135315763618
- YangYWangJZhangXLuWZhangQA novel mixed micelle gel with thermo-sensitive property for the local delivery of docetaxelJ Control Release200913517518219331864
- KorsemeyerRWGurneyRDoelkerEMechanisms of solute release from porous hydrophilic polymersInt J Pharm1983152535
- AhujaNKatareOPSinghBStudies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriersEur J Pharm Biopharm200765263816962750
- QuintenTde BeerTVervaetCRemonJPEvaluation of injection moulding as a pharmaceutical technology to produce matrix tabletsEur J Pharm Biopharm20097114515418511248
- PeppasNASahlinJJA simple equation for the description of solute release. III. Coupling of diffusion and relaxationInt J Pharm198957169172
- KimHFassihiRA new ternary polymeric matrix system for controlled drug delivery of highly soluble drugs: I. Diltiazem hydrochloridePharm Res199714141514219358555
- BettiniRColomboPGMassimoPLSwelling and drug release in hydrogel matrices: polymer viscosity and matrix porosity effectsEur J Pharm Sci19942213219
- SiepmannJPeppasNAModeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC)Adv Drug Deliv Rev20014813915711369079
- LiuYLuWLWangJCControlled delivery of recombinant hirudin based on thermo-sensitive Pluronic® F127 hydrogel for subcutaneous administration: in vitro and in vivo characterizationJ Control Release200711738739517207884
- JohnstonTPPunjabiMAFroelichCJSustained delivery of interleukin- 2 from a poloxamer 407 gel matrix following intraperitoneal injection in micePharm Res199294254341614979
- ZhangLParsonsDLNavarreCKompellaUBDevelopment and in vitro evaluation of sustained release poloxamer 407(P407) gel formulations of ceftiofurJ Control Release200285738112480313
- BochotAFattalEGulikACouarrazeGCouvreurPLiposomes dispersed within a thermosensitive gel: a new dosage form for ocular delivery of oligonucleotidesPharm Res199815136413699755886
- PanditNKWangDSalt effects on the diffusion and release rate of propranolol from poloxamer 407 gelsInt J Pharm1998167183189
- VeyriesMLCouarrazeGGeigerSControlled release of vancomycin from poloxamer 407 gelsInt J Pharm199919218319310567749
- MatthewJENazarioYLRobertsSCBhatiaSREffect of mammalian cell culture medium on the gelation properties of Pluronic F127Biomaterials2002234615461912322983
- ShishidoSMSeabraABLohWGanzarolli de OliveiraMThermal and photochemical nitric oxide release from S-nitrosothiols incorporated in Pluronic F127 gel: potential uses for local and controlled nitric oxide releaseBiomaterials2003243543355312809783
- CabanaAAït-KadiAJuhászJStudy of the gelation process of polyethylene oxidea-polypropylene oxideb-polyethylene oxidea copolymerJ Colloid Interface Sci19971903073129241171
- DesaiPRJainNJSharmaRKEffect of additives on the micellization of PEO/PPO/PEO block copolymer PF127 in aqueous solutionColloids Surf A Physicochem Eng Asp20011785769
- PanditNKKisakaJLoss of gelation ability of Pluronic® F127 in the presence of some saltsInt J Pharm1996145129136
- SharmaPKMatthewJEBhatiaSRStructure and assembly of PEO– PPO–PEO co-polymers in mammalian cell-culture mediaJ Biomater Sci Polym Ed2005161139115116231604
- WankaGHoffmannHUlbrichtWPhase diagrams and aggregation behavior of poly(oxyethylene)–poly(oxypropylene)–poly(oxyethylene) triblock copolymes in aqueous solutionsMacromolecules19942741454159
- GoldsteinREOn the theory of lower critical solution pointing hydrogen- bonded mixturesJ Chem Phys1984805340
- BonacucinaGSpinaMMisici-FalziMEffect of hydroxypropyl β-cyclodextrin on the self-assembling and thermogelation properties of Poloxamer 407Eur J Pharm Sci20073211512217656076
- LinJJChenJSHuangSJFolic acid–Pluronic F127 magnetic nanoparticle clusters for combined targeting, diagnosis, and therapy applicationsBiomaterials2009305114512419560199
- BatrakovaEVKabanovAVPluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiersJ Control Release20081309810618534704
- WellsJSenAHuiSWLocalized delivery to CT-26 tumors in mice using thermosensitive liposomesInt J Pharm200326110511412878399
- ExnerAAKrupkaTMScherrerKTeetsJMEnhancement of carboplatin toxicity by Pluronic block copolymersJ Control Release200510618819715951044