Abstract
The treatment of brain disorders is one of the greatest challenges in drug delivery because of a variety of main barriers in effective drug transport and maintaining therapeutic concentrations in the brain for a prolonged period. The objective of this study was delivery of valproic acid (VPA) to the brain by intranasal route. For this purpose, nanostructured lipid carriers (NLCs) were prepared by solvent diffusion method followed by ultrasonication and characterized for size, zeta potential, drug-loading percentage, and release. Six groups of rats each containing six animals received drug-loaded NLCs intraperitoneally (IP) or intranasally. Brain responses were then examined by using maximal electroshock (MES). The hind limb tonic extension:flexion inhibition ratio was measured at 15-, 30-, 60-, 90-, and 120-minute intervals. The drug concentration was also measured in plasma and brain at the most protective point using gas chromatography method. The particle size of NLCs was 154 ± 16 nm with drug-loading percentage of 47% ± 0.8% and drug release of 75% ± 1.9% after 21 days. In vivo results showed that there was a significant difference between protective effects of NLCs of VPA and control group 15, 30, 60, and 90 minutes after treatment via intranasal route (P < 0.05). Similar protective effect was observed in rats treated with NLCs of VPA in intranasal route and positive control in IP route (P > 0.05). Results of drug determination in brain and plasma showed that brain:plasma concentration ratio was much higher after intranasal administration of NLCs of VPA than the positive control group (IP route). In conclusion, intranasal administration of NLCs of VPA provided a better protection against MES seizure.
Introduction
General methods that can enhance drug delivery to the brain are of great interest. A noninvasive technique for drug delivery to the brain is through intranasal administration. Intranasal drug administration was shown to present a safe and acceptable alternative to parenteral administration of various drugs that offers rapid absorption to the systemic blood, avoiding first-pass metabolism in the gut wall and the liver. Further, several studies have shown a direct route of transport from the olfactory region to the central nervous system (CNS) in animal models, without prior absorption to the circulating blood.Citation1 This neuronal connection constitutes a direct pathway to the brain.Citation2 However, it has been reported that the amount of drug which was delivered to the brain was very low indeed, with concentrations in the cerebrospinal fluid (CSF) and olfactory lobes from 0.01% to 0.1% bioavailability and dependent on characteristics of the molecule.Citation3 Although the clinical potential of this delivery route still remains controversial, there is considerable interest in exploring this route for the treatment of common intracerebral diseases.Citation4 A promising strategy for drug delivery without any modification to the drug molecule is using colloidal carriers such as nanoparticles. Lipid nanoparticles are interesting candidates for brain targeting, due to rapid uptake by the brain, bioacceptability, biodegradability, and less toxicity compared to the polymeric nanoparticles. Indeed, feasibility in scale-up and absence of burst effect make them promising carriers for drug delivery.Citation5,Citation6
Valproic acid (VPA) is widely used in the treatment of epilepsy, bipolar disorders, migraine, and cancer.Citation7 Administration of high doses of the drug is needed to provide clinical effects because of inefficient delivery of VPA to the brain. The variety of drug interactions and side effects of VPA are due to long-term use of drug and its rather high daily dosage requirements, particularly in adolescents and young women.Citation8 Based on the results found in a brain efflux index study, Scism et alCitation9 estimated that the efflux clearance of VPA at the blood–brain barrier (BBB) was 2.7-fold greater than influx clearance compared to control animals. Therefore, lowering the dosage of administration is important and one of the main aims of the current study. VPA distribution in brain is less than that of other anticonvulsants, such as phenytoin or phenobarbital via oral route. Possible mechanisms for this decreased distribution space in brain include a) increased plasma protein binding of valproate relative to the other anticonvulsants and b) asymmetric (BBB) transport of valproate such that the brain-to-blood flux exceeded the blood-to-brain flux.Citation10,Citation11
Three animal models which are commonly used in characterization of anticonvulsant drugs are the maximal electroshock (MES) seizure test, pentylenetetrazol (PTZ) seizure test, and kindling. The MES and PTZ tests are models of acute (reactive or provoked) seizures.Citation12 The MES test, which is a widely used animal model, identifies the drug activity against generalized tonic–clonic and partial seizures.Citation13,Citation14
The aim of this study is to develop intranasal nanostructured lipid carriers (NLCs) of VPA for lowering the dosage and providing prolonged action and evaluation of its efficacy against generalized tonic–clonic seizures by a simple study in rats.
Materials and methods
Materials
VPA was provided by Chemische Fabrik Berg GmbH (Bitterfeld, Germany). Cetyl palmitate (CP) (mp 46°C–53°C; Croda, Cowick Hall, UK), Poloxamer 188 (Sigma-Aldrich Chemie, Munich, Germany), Soy lecithin S100 (Lipoid GMBH, Ludwigshafen, Germany), and octyldodecanol (Sasol Germany, Hamburg, Germany) were used as received. Acetone, ethanol, high-pressure liquid chromatography (HPLC) grade acetonitrile, caproic acid, and chloroform were purchased from Merck Chemical Company (Darmstadt, Germany). All other chemicals and reagents were of analytical grade.
Preparation of VPA-loaded NLC
NLCs of VPA were prepared by an emulsion–solvent diffusion and evaporation method followed by ultrasonication as reported earlier.Citation15,Citation16 Briefly, the hot lipid phase at 60°C, containing 400 mg of CP, 100 mg of soy lecithin S100, 0.1 mL of octyldodecanol, and 400 mg of VPA, was dissolved in 10 mL of a mixture of three parts acetone and one part absolute ethanol in a water bath. The oily phase was then dispersed in 50 mL aqueous phase containing 1% Poloxamer 188 at 50°C and stirred for 1 minute at 2000 rpm. The resulting pre-emulsion was then ultrasonicated for 2 minutes using a probe sonicator (Bandelin Electronic, Berlin, Germany) by probe TT13 in 40% amplitude to produce oil in water (O/W) nanoemulsion. In the final step, the obtained nanoemulsion (O/W) was cooled down at room temperature while stirring at 600 rpm for about 1 hour. Optimization of the nanoparticle formulations was carried out using Taguchi screening design and Box–Behnken analysis as reported earlier by the authors.Citation15,Citation16
Characterization of NLCs of VPA
Photon correlation spectroscopy (Zetasizer 3000; Malvern Instruments, Malvern UK) was used to measure size and zeta potential of all drug-loaded NLC samples. All samples were diluted (1:10) with deionized water to get optimum 50–200 kilo counts per second for measurements. Z-average particle size, polydispersity index, and zeta potential were measured in triplicate.
Drug-loading percentage was determined by measuring the concentration of unentrapped free drug in aqueous medium.Citation17 To do this, approximately 1.5 mL of the NLC dispersion was placed in microtubes, acidified to pH 1.5 with HCl 1N, and centrifuged at 25,000 rpm for 30 minutes (Sigma 3K30; Osterode am Harz, Germany). The aqueous medium was separated, and the amount of VPA in the aqueous phase was determined by a reversed-phase HPLC (Waters, Milford, MA), with UV detector, according to the procedure described by Kishore et alCitation18 with modification. The analysis was performed at 210 nm using a Nova-pak, reversed-phase C18 column, 250 × 4.5 mm, employing a mobile phase of 40% acetonitrile and 60% phosphate-buffered solution (PBS) 0.02 M (pH 3), delivered at a flow rate of 1 mL/min.Citation18 The retention time of the drug was found to be 10.0 ± 0.1 min. Data analysis and processing was carried out using Millennium software (Version 32; Millenium, Milford, MA).
To determine the release rate of VPA from nanoparticles, 3 mL of aqueous dispersion of each formulation was added to the dialysis bags with molecular weight cutoff of 12,400 Da, and the sealed bags were placed in a glass test tube containing 100 mL PBS 0.1 M (pH 6) and 0.1% polysorbate 80 to provide sink conditions with agitation of 200 rpm. Samples of 1 mL were withdrawn at predetermined time intervals of 2, 24, 72, 96, 240, 336, and 504 hours (21 days) and replaced with fresh PBS and maintained at the same temperature. The content of VPA in the samples was determined by the described HPLC method.
Animals and study design
Male Wistar rats (body weight: 180–210 g) from animal house of the School of Pharmacy and Pharmaceutical Sciences of Isfahan University of Medical Sciences were used for the in vivo studies. The animals were housed in colony cages with free access to standard chow pellets and water, under uniform housing in the environmentally controlled conditions (22 ± 2°C, 12-hour light–dark cycle, and 55%–65% humidity), and placed in the laboratory for 4–6 days during the acclimatization period and during the course of the study. The animal study was approved by the guideline of the ethical committee of Isfahan University of Medical Sciences.
Experiments were performed between 11 am and 2 pm. The rats were divided into six experimental groups each comprising six animals. All animals underwent preliminary screening MES test, and only the responsive ones received drugs 48 hours after preliminary MES test. Electrical stimulation was performed via auricular electrodes (110 mA square wave with a 100 Hz current, 1 millisecond pulse duration, and 0.2 second shock duration) to produce tonic hind limb extension in more than 50% of rats.Citation19 Electrodes were wetted with 0.9% normal saline to induce stimulation. The duration of hind limb flexion and extension was recorded, and extension:flexion (E:F) ratio was taken as a measure of seizure severity. The animals with an increased E:F ratio, related to the treated ones, were considered to exhibit more severe seizures.Citation20 Intraperitoneal (IP) administration of phenytoin (90 mg/kg) was used as gold standard, and MES was carried out approximately 2 hours after injection. shows the details of preparations and routes of administration of the study groups.
Table 1 Preparations, dosage, routes, and times of administration of different preparations of VPA used in the different experimental groups
Drug administration
Prepared formulation of VPA was placed in dialysis bag with molecular weight cutoff of 12,400 Da in purified water with sink condition for about 24 hours in order to separate nanoparticles from free drugs adhered on their surface. Then, the amount of VPA loaded in the nanoparticles was determined to obtain the required dose for administration. The solution of VPA as positive control was prepared using its sodium salt equivalent to the corrected dose of VPA in purified water. Control suspension of NLCs was prepared using the same method of NLC preparation without VPA. For intranasal administration, 100 μL of drug solution was delivered in each nostril within a few minutes via 2 cm polyethylene tubing (outer diameter: 0.965 mm, inner diameter: 0.58 mm) attached to a Hamilton syringe, while the animal was held in a supine position under light ether anesthesia. The other groups were injected IP. Thereafter, rats were given shock in predetermined times (), and E:F ratio and the time that maximum protection obtained were recorded. Then, at the maximum protection time, the rats were sacrificed, and plasma and the whole hemisphere of brain tissue samples were prepared and processed according to the following methods and analyzed by gas chromatography (GC) (Shimadzu 17 A; Shimadzu Co., Kyoto, Japan).
Blood and plasma sample preparation
The blood samples of about 3 mL were taken by slaughtering the animal when it was under ether anesthesia and transferred into a heparinized glass tube at 60 minutes after dosing. Blood samples were centrifuged for 15 minutes at 3000 rpm, and the separated plasma was stored at −20°C until analysis. Following the completion of blood collection, the scull of the rat was opened after separation of the head, and the whole brain was collected. The brain samples were rinsed with normal saline and immediately stored at −70°C until analysis. In order to determine the concentration of VPA in the brain, one hemisphere was weighed and was homogenized in 0.5 mL of PBS.
Assay of VPA in plasma and brain tissue samples
VPA was assayed using a modified method of GC after extraction.Citation21–Citation23 Briefly, an exactly 200 μL sample of plasma was well mixed with 200 μL HCl (2 M) and 62 μg caproic acid (186 μg/mL in methanol) as internal standard using electrical vortex. The mixture was extracted for 2 minutes with 400 μL of chloroform and centrifuged at 3000 rpm for 20 minutes. A total of 1.0 μL of bottom layer of the sample was injected into the GC. To 0.5 mL of the brain homogenate, 0.5 mL of HCl 6N was added, and the mixture was extracted for 2 minutes with 600 μL chloroform containing 37 μg caproic acid (186 μg/mL in methanol) as internal standard and centrifuged at 3000 rpm for 20 minutes. Finally, 1.0 μL of bottom layer of the sample was injected into the GC (detector: FID; column: 30 m × 0.53 mm, a wide bore column 5 μm [TRB-G27 Teknokroma, Barcelona, Spain]). The column temperature was programmed as follows: 130°C at 10°C/min and isothermal at 240°C for 10 minutes. The detector temperature was 290°C, and injector temperature was 200°C. Helium was the carrier gas, air-hydrogen pressure for detection, column flow rate: 5.6 mL/min, split ratio: 5, linear velocity: 54, total flow: 35 mL/min. Retention times were 3.8 and 5.6 minutes for caproic acid and VPA, respectively. Calibration curves of VPA were prepared using plasma and brain tissue mixed with known amounts of the drug, utilizing its GC peak area ratios to the internal standard. Calibration curve of VPA was linear within the concentration range between 2 and 100 μg/mL (Y = 0.007X + 0.039, n = 5, r2 = 0.999). The mean extraction recovery of VPA from plasma and brain tissue homogenate was more than 95.4% ± 5.7% and 90.4% ± 7.2%, respectively.
Statistical analysis
The one-way analysis of variance and Tukey post hoc test were applied for determining the significance of differences in seizure protection effect between treatment, positive control, and control groups, which was shown using reduction ratio of E:F between groups in each time. Unpaired Student’s t-test was used to compare mean protective effect of NLCs of VPA in intranasal route and sodium VPA solution in IP route. All data are expressed as means ± SD. P ≤ 0.05 was considered statistically significant.
Results
The results of characterization of NLCs of VPA for size, polydispersity index, zeta potential, drug release, and drug loading are shown in .
Table 2 Composition and physical properties of the optimized NLCs of VPA
Table 3 Concentration of VPA in plasma and brain after administration of different formulations (n = 6)
Figure 1 Comparison between the effect of sodium valproate solution positive control (30 mg/kg), NLCs of VPA (4 mg/kg), and control NLCs (without VPA) in nasal route of administration on MES seizure protection which was assessed as the decrease of extension:flexion (E:F) ratio (n = 6).
Notes: *Statistically significant difference (P < 0.05) with respect to the control (empty) NLC group according to one-way ANOVA and Tukey test; ¥Statistically significant difference (P < 0.05) with respect to the positive control group according to 1-way ANOVA and Tukey test.
Abbreviations: E:F ratio, extension:flexion ratio; VPA, valproic acid; NLC, nanostructured lipid carrier; MES, maximal electroshock.
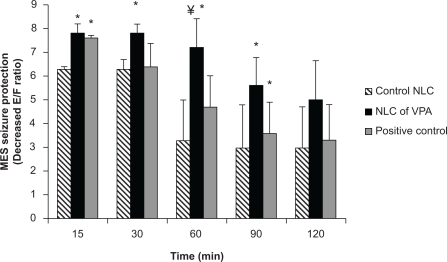
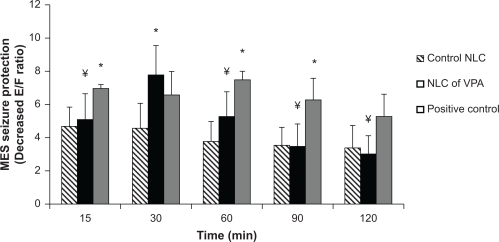
Figure 2 Comparison between the effect of sodium valproate solution positive control (150 mg/kg), NLCs of VPA (20 mg/kg), and control NLCs (without VPA) in IP route of administration on MES seizure protection which was assessed as the decrease of extension:flexion (E:F) ratio (n = 6).
Notes: *Statistically significant difference (P < 0.05) with respect to the control (empty) NLC group according to one-way ANOVA and Tukey test; ¥Statistically significant difference (P < 0.05) with respect to the positive control group according to 1-way ANOVA and Tukey test.
Abbreviations: E:F ratio, extension:flexion ratio; VPA, valproic acid; NLCs, nanostructured lipid carriers; MES, maximal electroshock; IP, intraperitoneal.
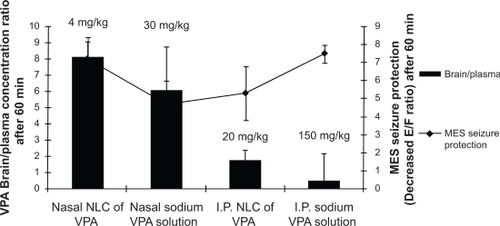
A t-test statistical analysis showed that there was no significant difference (P > 0.05) between the protective effect of NLCs of VPA in intranasal route and positive control in IP route, as a systemic route of administration, at any time after administration (). Results showed that protective effect of VPA nanostructures by intranasal route is similar to that of sodium valproate after systemic route of administration, while its dosage is significantly smaller than systemic route. Correlation between MES protective effect of different preparations and brain:plasma concentration ratio at 60 minutes (the most protective time) revealed that intranasal administration of VPA nanostructures provided the same protective effect as sodium valproate solution after systemic administration, with a dose of about 37 times less than that administered by the IP route (). This shows that brain/plasma concentration is higher in intranasal administration of NLCs of VPA.
Figure 3 Comparison between MES seizure protection effect of NLCs of VPA (4 mg/kg) in nasal route and sodium valproate solution (150 mg/kg) as positive control in IP route after different times of drug administration (P > 0.05) (n = 6). There is no statistical difference in any times according to independent t test.
Abbreviations: E:F ratio, extension:flexion ratio; VPA, valproic acid; NLCs, nanostructured lipid carriers; MES, maximal electroshock; IP, intraperitoneal.
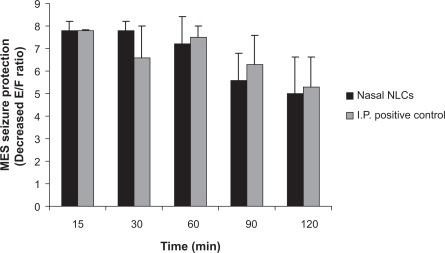
Figure 4 Correlation between MES seizure protection effect and brain:plasma concentration ratio of preparations in nasal and IP route of administration.
Abbreviations: E:F ratio, extension:flexion ratio; VPA, valproic acid; NLC, nanostructured lipid carrier; MES, maximal electroshock; IP, intraperitoneal.
Discussion
BBB is absent or very thin in diameter in the interphase of brain–olfactory nerve epithelium,Citation24 so intranasal route was chosen in the present study to bypass the BBB and promote drug distribution into the brain. The incorporation of drugs into nanoparticles might be a promising approach, since colloidal formulations were shown to protect drugs from degrading milieu in the intranasal cavity and facilitate their transport across the mucosal barriers.Citation25 The observed responses of size (154 ± 16 nm), polydispersity index (0.2 ± 0.1), and zeta potential (−10 ± 0.5 mV) in indicate that the nanoemulsions approached a monodisperse stable system and could deliver the drug effectively owing to large surface area. Intranasal is an efficacious route to deliver drugs (at small doses) rapidly and efficiently into the brain. Animal studies have shown that nanosized drug delivery systems can enhance nose-to-brain delivery of drugs compared to equivalent drug solution formulations. Protection of the drug from degradation and/or efflux back into the intranasal cavity and blood may be the reason for this effect of nanoparticles.Citation26 The nanoparticles and solid lipid nanoparticles (SLNs) with the size below 200 nm increased blood availability, eventually decreasing the time to pass the BBB and increasing uptake by the brain.Citation27 Higher concentration of clozapine SLNs, with 163 nm in size that were coated with Poloxamer 188, resulted in transport through the BBB by endocytosis.Citation28 Study on nanoparticle transit into olfactory epithelium in mice showed that intranasally administered chitosan-coated polystyrene or polysorbate-coated polystyrene nanoparticles of 100 nm can transport into olfactory epithelial cells.Citation29 The use of intranasal route via olfactory nerve was also reported for brain targeting of carbamazepin,Citation30 morphine,Citation31 zolmitriptan,Citation32 sumatriptan,Citation33 clonazepam, and nimodipine.Citation34,Citation35 All these reports showed promising transfer of adequate drug into the brain based on methods which are divided into four categories: nose–brain, nose–CSF, nose–brain/CSF, and pharmacodynamic studies.Citation24 There are a few studies monitoring pharmacologic effects as well as brain and/or blood levels of the active drug by using a large number of animals. It is evident that the majority of the studies in the area of nose–brain/CSF research do not meet the requirements as outlined in the theoretical study design. However, it is necessary to monitor drug levels within the blood or the brain to reveal the positive correlation between them.Citation36 For this purpose, in the present study, the VPA concentration is measured at the time of maximal protection against MES, leading to the minimal number of animals required. By this way, the number of animals to be killed was decreased significantly along with reaching the goal. As shows, there was a significant difference (P < 0.05) between protective effects of NLCs of VPA and control NLC in 15, 30, 60, and 90 minutes after administration. This revealed that the drug might be at a higher concentration in the brain. Blasi et al showed that the drug concentration of VPA in rats sacrificed 20 and 90 minutes after treatment with (14C) sodium valproate (150 mg/kg) was high in blood and moderate in liver, kidney, heart, and lung, but low in brain, fat, testis, and skeletal muscle.Citation37 shows that MES seizure protection 15, 60, and 90 minutes after IP injection of sodium valproate solution (150 mg/kg) was significantly different compared to the control NLC IP injection. Data in show that intranasal administration of NLCs of VPA and sodium valproate solution causes higher drug concentration in the brain than in plasma. As shows, maximum MES seizure protection happened 60 minutes after intranasal administration of NLCs of VPA when the higher concentration than plasma was measured. It was assumed that VPA was transported from the brain to the circulating blood across the BBB via a carrier-mediated efflux transport process.Citation38 Furthermore, the P-gp localized in the olfactory epithelium might prevent the drug to backflow through the membrane.Citation36 For this reason, in the present study, Poloxamer was added to our formulation to prevent the drug efflux from the membrane and to result in higher drug concentrations within the brain as it was shown in previous studies.Citation27 Friese et al used nanoparticles of a novel glutamate receptor antagonist drug coated with polysorbate 80 and increased the duration of anticonvulsive activity and the CNS availability of the drug.Citation39 As it is shown in and , the results of protective effect of NLCs of VPA in intranasal route are comparable with systemic route. The intranasal route is an alternative to systemic route for delivery of many drugs, and most of the research articles compared them with each other.Citation40 It was shown that olfactory bulb efferent projections may act as important sites of action for VPA because this projection might be involved in seizure generalization.Citation13 Furthermore, intranasal administration of VPA seems to accelerate binding of the VPA to the olfactory bulb and improving its efficacy even with smaller dosage of administration in intranasal route. Determination of effective brain concentrations is important for the interpretation of data on valproate. It should be noted that the effective brain concentration in rodents is much higher than respective doses in humans, because of the higher elimination rate of the drug. However, brain:plasma concentration ratio of valproate is almost equal to the respective ratio in humans.Citation13 The results of this study showed that brain:plasma concentration ratio increased to about 20 times after intranasal administration of NLCs of VPA compared to IP administration.
Conclusion
The present study clearly demonstrated that intranasal administration of nanostructures of VPA in rats is a suitable method to maintain the effect of VPA with much higher brain:plasma concentration ratio. The same protective effect as systemic administration was seen with much lower doses. Intranasal administration may represent a remarkable delivery route for targeting to the brain with an appropriate dosage form design. Further studies are required to evaluate whether this dosage form of VPA is clinically beneficial.
Acknowledgements
The authors thank the Vice Chancellery of Research of Isfahan University of Medical Sciences that supported this work. In addition, the authors appreciate financial support of Raha Pharmaceutical Company and technical assistance of Miss Moazen and Mr Sharifi.
Disclosure
The authors report no conflicts of interest in this work.
References
- Garcia-GarciaEAndrieuxKGilSCouvreurPColloidal carriers and blood–brain barrier (BBB) translocation: a way to deliver drugs to the brain?Int J Pharm2005298227429215896933
- IllumLTransport of drugs from the nasal cavity to the central nervous systemEur J Pharm Sci200011111810913748
- IllumLIs nose-to-brain transport of drugs in man a reality?J Pharm Pharmacol200456131714979996
- JonesNThe nose and paranasal sinuses physiology and anatomyAdv Drug Deliv Rev2001511–351911516776
- VinogradovSVBronichTKKabanovAVNanosized cationic hydrogels for drug delivery: preparation, properties and interactions with cellsAdv Drug Deliv Rev200254113514711755709
- KaurIPBhandariRBhandariSKakkarVPotential of solid lipid nanoparticles in brain targetingJ Control Release200812729710918313785
- StapletonSLThompsonPAOuCNPlasma and cerebrospinal fluid pharmacokinetics of valproic acid after oral administration in non-human primatesCancer Chemother Pharmacol200861464765217541590
- GibbsJPAdeyeyeMCYangZShenDDValproic acid uptake by bovine brain microvessel endothelial cells: role of active efflux transportEpilepsy Res2004581536615066675
- ScismJLPowersKMArtruAALewisLShenDDProbenecid-inhibitable efflux transport of valproic acid in the brain parenchymal cells of rabbits: a microdialysis studyBrain Res20008841–2778611082489
- BaltesSFedrowitzMTortósCLPotschkaHLöscherWValproic acid is not a substrate for P-glycoprotein or multidrug resistance proteins 1 and 2 in a number of in vitro and in vivo transport assaysJ Pharmacol Exp Ther2007320133134317043155
- CornfordEMDiepCPPardridgeWMBlood–brain barrier transport of valproic acidJ Neurochem1985445154115503921665
- LöscherWCurrent status and future directions in the pharmacotherapy of epilepsyTrends Pharmacol Sci200223311311811879677
- LöscherWValproate: a reappraisal of its pharmacodynamic properties and mechanisms of actionProg Neurobiol1999581315910321796
- LöscherWAnimal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsyEpilepsy Res2002501–210512312151122
- VarshosazJEskandariSTabbakhianMProduction and optimization of valproic acid nanostructured lipid carriers by the Taguchi designPharm Dev Technol2010151899619552542
- VarshosazJTabbakhianMMohammadiMYFormulation and optimization of solid lipid nanoparticles of buspirone HCl for enhancement of its oral bioavailabilityJ Liposome Res201020428629619958118
- VenkateswarluVManjunathKPreparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticlesJ Control Release200495362763815023472
- KishorePRajani KumarVSatyanarayanaVKrishnaDRHPLC determination of valproic acid in human serumPharmazie200358637838012856997
- EngelJPedleyTAAicardiJEpilepsy: A Comprehensive Textbook2nd edPhiladelphia, PALippincott Williams & Wilkins2007
- SwinyardEAElectrically induced convulsionsPurpuraDPPenryJKTowerDBExperimental Models of Epilepsy: A Manual for the Laboratory WorkerNew York, NYRaven Press1972434458
- WohlerASPoklisAA simple, rapid gas-liquid chromatographic procedure for the determination of valproic acid in serumJ Anal Toxicol19972143063099248950
- ShahdoustiPMohammadiAAlizadehNDetermination of valproic acid in human serum and pharmaceutical preparations by headspace liquid-phase microextraction gas chromatography-flame ionization detection without prior derivatizationJ Chromatogr B Analyt Technol Biomed Life Sci20078501–2128133
- DillsRLShenDDMethods to reduce background interferences in electron-capture gas chromatographic analysis of valproic acid and its unsaturated metabolites after derivatization with pentafluorobenzyl bromideJ Chromatogr B Biomed Sci Appl19976901–21391529106038
- BergMVIntranasal Drug Delivery: A Direct Approach to the Cerebrospinal Fluid? [doctoral thesis]. Leiden, The Netherlands: Leiden/Amsterdam Center for Drug Research, Faculty of Mathematics and Natural Sciences, Leiden University;2005
- AliJAliMBabootaSPotential of nanoparticulate drug delivery systems by intranasal administrationCurr Pharm Des201016141644165320210751
- MistryAGludSZKjemsJEffect of physicochemical properties on intranasal nanoparticle transit into murine olfactory epitheliumJ Drug Target200917754355219530905
- KumarMMisraABabbarAKMishraAKMishraPPathakKIntranasal nanoemulsion based brain targeting drug delivery system of risperidoneInt J Pharm20083581–228529118455333
- ManjunathKVenkateswarluVPharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administrationJ Control Release2005107221522816014318
- MistryAStolnikSIllumLNanoparticles for direct nose-to-brain delivery of drugsInt J Pharm2009379114615719555750
- BarakatNSOmarSAAhmedAACarbamazepine uptake into rat brain following intra-olfactory transportJ Pharm Pharmacol2006581637216393465
- WestinUEBoströmEGråsjöJHammarlund-UdenaesMBjörkEDirect nose-to-brain transfer of morphine after nasal administration to ratsPharm Res200623356557216489488
- VyasTKBabbarAKSharmaRKMisraAIntranasal mucoadhesive microemulsions of zolmitriptan: preliminary studies on brain-targetingJ Drug Target200513531732416199375
- VyasTKBabbarAKSharmaRKSinghSMisraAPreliminary brain-targeting studies on intranasal mucoadhesive microemulsions of sumatriptanAAPS PharmSciTech200671E816584167
- VyasTKBabbarAKSharmaRKSinghSMisraAIntranasal mucoadhesive microemulsions of clonazepam: preliminary studies on brain targetingJ Pharm Sci200695357058016419051
- ZhangQJiangXJiangWLuWSuLShiZPreparation of nimodipine-loaded microemulsion for intranasal delivery and evaluation on the targeting efficiency to the brainInt J Pharm20042751–2859615081140
- CostantinoHRIllumLBrandtGJohnsonPHQuaySCIntranasal delivery: physicochemical and therapeutic aspectsInt J Pharm20073371–212417475423
- BlasiPGiovagnoliSSchoubbenARicciMRossiCSolid lipid nanoparticles for targeted brain drug deliveryAdv Drug Deliv Rev200759645447717570559
- KakeeATakanagaHHosoyaKSugiyamaYTerasakiTIn vivo evidence for brain-to-blood efflux transport of valproic acid across the blood–brain barrierMicrovasc Res200263223323811866547
- FrieseASeillerEQuackGLorenzBKreuterJIncrease of the duration of the anticonvulsive activity of a novel NMDA receptor antagonist using poly(butylcyanoacrylate) nanoparticles as a parenteral controlled release systemEur J Pharm Biopharm200049210310910704892
- JadhavKRGambhireMNShaikhIMKadamVJPisalSSNasal drug delivery system-factors affecting and applicationsCurr Drug Ther2007212738