 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Scopolamine hydrobromide (SH)-loaded microparticles were prepared from a colloidal fluid containing ionotropic-gelated chitosan nanoparticles using a spray-drying method. The spray-dried microparticles were then formulated into orally disintegrating tablets (ODTs) using a wet granulation tablet formation process. A drug entrapment efficiency of about 90% (w/w) and loading capacity of 20% (w/w) were achieved for the microparticles, which ranged from 2 μm to 8 μm in diameter. Results of disintegration tests showed that the formulated ODTs could be completely dissolved within 45 seconds. Drug dissolution profiles suggested that SH is released more slowly from tablets made using the microencapsulation process compared with tablets containing SH that is free or in the form of nanoparticles. The time it took for 90% of the drug to be released increased significantly from 3 minutes for conventional ODTs to 90 minutes for ODTs with crosslinked microparticles. Compared with ODTs made with noncrosslinked microparticles, it was thus possible to achieve an even lower drug release rate using tablets with appropriate chitosan crosslinking. Results obtained indicate that the development of new ODTs designed with crosslinked microparticles might be a rational way to overcome the unwanted taste of conventional ODTs and the side effects related to SH’s intrinsic characteristics.
Introduction
Because of lifestyle changes, a demand has arisen for the development of pharmaceutical forms that can be readily handled and taken by many patients. Rapidly disintegrating or dissolving drug delivery systems, in which drugs are administrated orally for direct ingestion without water, are particularly useful for geriatric, pediatric, and traveling patients. These new forms of medicine are quite advantageous because they can be taken without any chewing or prior dispersion and/or dissolution processes.Citation1–Citation3 Some of them (sublingual or buccal tablets) display a spontaneous deaggregation and fast absorption in the mouth upon contact with saliva.Citation4 Therefore, numerous studies on various compositions and manufacturing methods of orally disintegrating or dissolving tablets (ODTs) have been performed.Citation5,Citation6 Unfortunately, the quickly disintegrating dosage forms usually have an unpleasant taste, local stimulation, and other side effects caused by short, intense exposure to high concentrations of the active agent.
Microencapsulation is a new technique that could be used to control the drug release rate, protect it from premature destruction, and avoid the unpleasant taste by entrapping the active drug into a microcarrier.Citation7 Spherically shaped and surface-distorted microparticles (embedded with the drug) could lessen the unwanted effects of ODTs by controlling the drug release rate. Recently, a novel nanoparticles-in-microparticles system (NiMS) has been developed using a double emulsion-like technique and evaluated for drug and gene delivery in specific regions of the gastrointestinal tract.Citation8,Citation9 This NiMS might give valuable hints for the development of new drug-loaded ODTs. However, as far as we know, few reports have made mention of this new formulation design.
Chitosan (CS), a biocompatible and biodegradable polysaccharide, is widely used in the development of drug delivery systems.Citation10 Our recent work has established a CS-based NiMS that displays high drug entrapment efficiency and controlled drug release.Citation11,Citation12 The obtained results are quite encouraging, in terms of the further application of NiMS for oral drug delivery.
Scopolamine, also known as levo-duboisine and hyoscine, is a tropane alkaloid drug that acts as a nonselective muscarinic antagonist and is used extensively in the clinic to treat motion sickness. It is obtained from plants of the Solanaceae (nightshades) family, such as henbane, weed, Angel’s Trumpets (Datura resp. Brugmansia spec.), and corkwood (Duboisia species).Citation13 Scopolamine can be administered by transdermal, oral, subcutaneous, ophthalmic, and intravenous routes. The oral, ophthalmic, and intravenous forms of the drug are usually scopolamine hydrobromide (SH). The commonly observed side effects are related to anticholinergic effects on postsynaptic receptors in the parasympathetic nervous system and include the following: dry mouth, throat, and nasal passages; in cases of overdose, impaired speech, thirst, blurred vision, and sensitivity to light; constipation; difficult urinating; and tachycardia. As for the fast disintegration behavior of conventional SH ODTs, the initial burst-released drug could be absorbed very quickly and result in high serum concentration. Then, an unpleasant taste and the aforementioned side effects of SH are inevitable. So it is desirable to design and develop a rational delivery system for SH using NiMS-based ODTs.
In the present study, to minimize the encountered side effects, a CS-based NiMS was prepared and used to encapsulate the model drug SH through an ionotropic gelation process followed by spray-drying. Orally disintegrating technology was then applied to construct orally disintegrating microparticle tablets (ODMTs) using a wet granule compression method. To compare these tablets with conventional ODTs, in vitro disintegration tests and drug release studies were also performed.
Materials and methods
Materials
CS that was over 95% deacetylated and had a molecular weight of 40–80 kDa was supplied by RuJi Biopharmaceutical Co. (Shanghai, People’s Republic of China). Tripolyphosphate pentasodium (TPP) and polyvinylpyrrolidone K30 were obtained from the Chemical Agent Station, Medicine Group of China (Shanghai). SH was obtained from HuanYe Pharmaceutical Co. Ltd (Guangzhou, People’s Republic of China). Xylitol (Danisco Sweeteners Co. Ltd, Anyang, People’s Republic of China), microcrystalline cellulose (USP/JRS, Shanghai), carboxymethylstarch sodium (USP/YongRi, Taiwan, People’s Republic of China), mannitol (LanJi Technological Co. Ltd, Shanghai), and gum arabic (USP/A250, Degussa, Germany) were also purchased for use as tablet adjuvants. All other chemicals were of reagent grade and used as provided by the manufacturer.
Preparation of SH-loaded CS nanoparticles
Ionotropic gelation for the formation of CS nanoparticles (CS-NPs) was performed as previously described.Citation11,Citation12,Citation14 CS solution (0.5%, w/v) was obtained by dissolving CS in dilute acetic acid (0.3%, v/v) at room temperature overnight and passed through a 0.22 μm filter to remove insolubles. SH had been previously dissolved into this CS solution at the CS/SH ratio of 4:1 (w/w) to obtain a homogenous mixture. Then, TPP was directly dissolved in distilled water at 2 mg/mL (w/v). Thereafter, the ionic gelification of CS in the aqueous medium was achieved by the addition of TPP solution (3 mL/min) corresponding to the CS solution and magnetically stirred (900 rpm) at room temperature. The ratio of CS/TPP was kept at 6:1 (w/w) for the construction of CS-NPs in both formulations. Thereafter, the resulting colloidal CS-NPs were continuously stirred for 30 minutes, and the product was stored at room temperature until the next microencapsulation step.
Preparation of SH-loaded CS microparticles by spray-drying
The obtained aqueous mixture of CS-NP colloids (including the NPs, drug not entrapped, and free CS), without centrifugation, was spray-dried (Büchi® Mini Spray Dryer, Type 290, Switzerland) at a feed rate of 6.0 mL/min. The spray-drying conditions were inlet temperature 128–132°C, outlet temperature 68–71°C, aspirator 90%, pump feed 20%, and Q flow 45. Finally, the product was kept under vacuum for 48 hours and stored in a desiccator at room temperature until use.
For use as a control, noncrosslinked CS microparticles (made without the addition of the TPP crosslinking agent) were prepared by spray-drying the aqueous sample made by mixing the drug directly with the CS solution.
Drug encapsulation efficiency and loading capacity
For the nanosuspension sample, the amount of drug entrapped was calculated as the difference between the total amount used and the amount presented in the centrifuged aqueous supernatant phase.Citation15 Then, the SH efficiency and loading capacity was determined directly in the spray-dried particles.Citation16 After extraction by sonication for 30 minutes with 0.3% (v/v) acetic acid solution, the drug encapsulation efficiency and the loading capacity of the microparticles were determined by analysis of the samples in suspension after centrifugation at 20,000 × g and 12°C for 30 minutes. The extracted SH in the supernatant after ultracentrifugation of the microparticle suspension was determined using the high-performance liquid chromatography (HPLC) method described here. Each sample was assayed in triplicate.
The HPLC analyses were carried out using a system (Shimadzu, LC-10A, Japan) that consisted of two LC-10AT pumps (Shimadzu), a Rheodyne 7125 manual injection valve with 20 μL sample loop, an SPD-10A UV detector (Shimadzu), and a C-18 reversed-phase column (4.6 mm ×200 mm, 5 μm, Hypersil ODS). The mobile phase was a mixture of methanol and 2 mmol/L ammonium acetate solution (40:60, v/v; pH 3.3). The flow rate was 1.0 mL/min at 25°C, and the wavelength was set at 210 nm.
The drug loading capacity (LC) of the microparticles and the encapsulation efficiency (EE) of the process were calculated according to the following equation:
where Ddetermined SH is the determined amount of SH extracted from microparticles, Wtotal SH is the total amount of SH added, and Wmp is the weight of the microparticles.
Scanning electron microscopy
The morphology and surface appearance of the spray-dried microparticles were studied by scanning electron microscopy (SEM, XL-30E, Philips, the Netherlands). The powdered sample was placed on double-sided tape that had previously been secured on aluminum stubs and then observed using SEM at an acceleration voltage of 15 kV after gold sputtering and under argon.
Drug release from the microparticles in vitro
The in vitro drug release profiles of SH-loaded microparticles were carried out as described here. About 30 mg of microparticles was placed into a dialysis membrane bag that was impermeable to molecules weighing more than 8 kDa. Then, the bag containing the SH microparticles was tied and put into 100 mL of phosphate-buffered saline (PBS, pH 6.8). The entire system was incubated at 37°C ± 1°C with stirring at 50 rpm. At designated time intervals, 0.5 mL of the release medium was removed and replaced with the same volume of fresh PBS. The amount of SH in the release medium was determined by HPLC under the described chromatographic conditions. All measurements were performed in triplicate.
Manufacture of SH-CS microparticle-based ODMTs
A wet granulation compression method was used to prepare the ODTs. First, the powdered drug-loaded NiMS and the various quickly disintegrating tablet adjuvants (with appropriate amounts of diluent, disaggregating agent, and lubricant at the specified ratios listed in ) were screened to produce a homogeneous mixture. The resulting mixtures were passed through a 30-mesh sieve with openings of less than 600 μm in diameter and then granulated in a mortar using 10% (w/v) polyvinylpyrrolidone K30 ethanol solution (95%, v/v) as an adhesive. The obtained granulates were dried in a 60°C oven for 1 hour. The final granules were then passed through a 14-mesh sieve and stored in a desiccator at room temperature until use.
Table 1 Percentage composition of the orally disintegrating tablet formulations investigated
The filtered dry granules were directly compressed using a single-punch tablet formation machine (Tianxiang Jiantai Machinery Drug Making Co., Shanghai) fitted with a 5 mm diameter normal biconcave punch and die set. Relatively constant tablet hardness was maintained at around 8 kg (measured on a 78X-2 multifunctional determinator for tablets, Huanghai, People’s Republic of China) for compression. All tablets generated using this method contained 0.2 mg of entrapped active drug. The complete compositions of these tablets are shown in .
Determination of disintegration time
The disaggregation tests for ODTs were performed at 37°C using a disintegration time determinator (LB-2D, Huanghai Drug Testing Instrument Co. Ltd, Shanghai) and purified water as a dispersion medium. The disintegration time is the time required to transform a tablet immersed in water into small fragments with vertical vibrations at a rate of 30 vibrations per minute. The disintegration time of each formulation was recorded (n = 6).
Dissolution testing of the prepared tablets
The dissolution tests of the prepared tablets were performed at 37°C ± 1°C using a stir method (50 rpm) with 10 mL phosphate buffer (pH 6.8) as a dissolution medium. At predetermined time intervals, 0.5 mL of the release medium was sampled and filtered. The filtrates were analyzed by the HPLC method described previously. It is worth noting that none of the additives used in the formulations interfered with the assay. The results are expressed as the percentage of released drug as a function of time.
Statistical analysis
Data were expressed as mean ±standard deviation. Results of the in vitro drug release studies were analyzed using F-test. Differences were considered to be significant at a level of P < 0.05.
Results and discussion
CS nanoparticles and the resulting microparticles constructed by direct spray-drying
TPP is a nontoxic and multivalent anion that can form cross-linkages involving ionic interactions between the positively charged amino groups of CS and multivalent, negatively charged TPP molecules.Citation17,Citation18 With the dropwise addition of TPP, the nanoencapsulation was processed with the electrostatic attraction. Meanwhile, with the incorporation of ion TPP to CS solution, the opalescence indicated the formation process of nanoparticles with a size range of 200–500 nm, a mean diameter of approximately 350 nm, and positive charge of 17 mV, analyzed with a Nicomp™ 380 ZLS Coulter (Santa Barbara, CA). As observed from the transmission electron microscopic (H-600 Hitachi, Japan) image in , the SH-loaded CS-NPs had a regular spherical shape with a relatively narrow size distribution. During the ionotropic gelation process, most of the available CS was formed into nanoparticles, and a particle containing more than 80% of the CS (as measured by a bicinchoninic acid protein assay) was generated. In this case, the TPP reacted with a large amount of the CS, resulting in the formation of gelated nanoparticles. TPP-crosslinked CS-NPs and their conversion into spray-dried microparticles are expected to improve the particles’ stability and applicability for controlled drug delivery. Following the ionotropic gelation process using TPP as a crosslinking agent, the spray-drying method was performed to reinforce the formation of drug-loaded CS microbeads. The spray-dried NiMS powder might as well have some merits for tablet formulation and granulation. Meanwhile, the disintegrated microparticles might be capable of modifying drug release rates as desired for masking the taste and proper absorption.
Figure 1 Transmission electron microscope photograph of scopolamine hydrobromide-loaded chitosan nanoparticles (×30,000) prepared by ionotropic gelation process.

As part of the microencapsulation technique, the nanoparticle colloids mixed with CS and free drug were directly spray-dried together prior to spray-drying the final samples composed of centrifuged CS-NPs. During the aqueous ionotropic gelation process, water-soluble properties of SH might lead to low drug encapsulation efficiency. The spray-drying was used to dramatically improve drug encapsulation efficiency for the resulting microparticles. Drug content typically denotes the percentage of drug that is entrapped, while the encapsulation efficiency is calculated as the ratio of the drug content to the actual drug added. According to our previous studies, the drug LC improved as the drug/material ratios increased.Citation16 Also, the drug content can be controlled using different weight ratios of SH to CS according to clinical needs. An adequate LC of 23.4% ± 2.6% was obtained for the selected SH:CS ratio of 1:4 (w/w). The production yield of spray-dried microparticles was about 65%, with satisfactory encapsulation efficiencies of more than 90%.
Morphology of the CS microparticles produced by the spray-drying method
The shape and surface morphology of CS microparticles prepared by spray-drying were observed using SEM (). As shown in , the spray-dried microparticles were almost spherical, with a regular shape and a particle diameter range of 1–5 μm. The observed small solid particles () could have been formed from the spraying of a single minor droplet containing one or two CS nanoparticles. A few relatively rough-surfaced particles were found in the noncrosslinked CS microparticle sample (). Most of the TPP-crosslinked CS microparticles were found to be spherical with smooth surfaces (). These might have been some of the pre-existing nanoparticles that remained unreacted after ionotropic gelation with aqueous TPP.
Figure 2 Scanning electron microscopy microphotographs of chitosan (CS) microparticles prepared by the spray-drying method: A) without tripolyphosphate pentasodium (TPP) and B) crosslinked with TPP (CS/TPP weight ratio of 6:1, w/w).
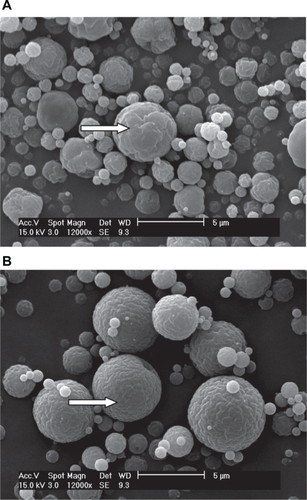
Generally, the size of the noncrosslinked CS microparticles was smaller than that of the crosslinked microparticles (). This effect is somewhat similar to that observed by Anal et al,Citation18 who showed a slight influence of TPP on the particle diameter. In the presence of sufficient amounts of TPP, fusion of smaller nanoparticles occurs, giving rise to CS microparticles of considerably larger size. The multifunctional TPP also might act here to facilitate intermicroparticle binding of the CS.
In vitro drug release behaviors of the spray-dried microparticles
The in vitro SH release profiles were obtained by relating the amount of SH released from microparticles to the amount of SH encapsulated. displays the drug release profiles of spray-dried CS microparticles and the effect of the crosslinker (TPP) on release rate. As shown in , the release rate of noncrosslinked microparticles was significantly faster (P < 0.05) than that of crosslinked microparticles during the first 1.5 hours. The cumulative release percentage from the noncrosslinked microparticles exceeded 74% within 30 minutes. In contrast, only about 55% of the drug was released from microparticles crosslinked with TPP in 30 minutes (). Statistical analysis of SH release values before 1.5 hours shows that there is a significant effect (P < 0.05) of the TPP. The addition of TPP is probably effective to occupy CS amino groups. The crosslinking of CS microparticles with TPP could retard the release rate because of the decreased water solubility of CS in this formulation, which could be important for maintaining further sustained drug release. Therefore, the degradation and enzymatic digestion of CS-NPs would be required for accomplishing the release process.Citation19 The delayed drug release from nanoparticle-entrapped microparticles might be valuable in order to control the unpleasant taste and local stimulation of the formulated tablets in the mouth.
Figure 3 In vitro drug release profiles for noncrosslinked and crosslinked chitosan microparticles (n = 3).
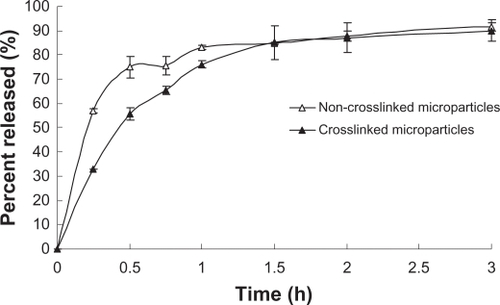
It was apparent that drug release showed a rapid initial burst effect in vitro. As shown in , about 85% of drug was released from the spray-dried microparticles within 1.5 hours. This initial burst release occurred because the drug was dispersed close to the surface of the particles. This portion of absorbed drug can be easily desorbed from the outer layer and will then diffuse out into solution. After 1.5 hours, similar drug release behavior for the two formulated microparticles (P > 0.05) could be found in . Generally, noncrosslinked CS microparticles cannot be kept suspended in water because of their swellability and dissolvability. In order to prepare highly stabilized CS microparticles, other crosslinking agents (glutaraldehyde or formaldehyde) are usually needed to solidify the microparticles.Citation20
Disintegration time of ODTs
According to the description of Kuno et al,Citation21 the preliminary test in volunteers was carried out previously. The oral disintegration time of an ODMT put into the mouth of a healthy male adult volunteer without water was recorded as the time until the volunteer felt that the tablet had disappeared in his mouth. The recorded oral disintegration times were close to those resulting in the procedure used in the present study. The dynamic state of 30 vibrations/minute was taken to mimic the condition in vivo.
The disintegration of a tablet was defined as the disappearance of the bead-like structure as observed in the baskets of the disintegration time determinator. The time needed for complete disintegration of various ODTs with drugs in different states is shown in . All the prepared tablets disintegrated completely within 45 seconds. The developed formulations were therefore demonstrated to be “quickly dispersible” ODTs.
Table 2 Disintegration times of orally disintegrating tablets with different drug dispersion states (n = 6)
The disintegrating agent swells when it comes into contact with water in a very predictable manner. It absorbs water rapidly, resulting in swelling that leads to the rapid disintegration of the tablets and granules. The disintegrating agents employed in the formulations were composed of 35% weight percentage microcrystalline cellulose and 10% weight percentage carboxymethylstarch sodium (). These formulations appeared to be the main factor responsible for the variations in disintegrating times shown in .
Disintegration time, which might also be affected by the hardness of the tablets, is related to the nature of the disintegrating agent, which allows the tablet to break up into smaller fragments upon contact with physiologic fluids. According to the results of Kuno et al,Citation21 tablets containing 1% xylitol showed a suitable hardness and oral disintegration time. Therefore, 1% xylitol was also used in the ODMT formulation () to ensure that the tablet hardness and disintegration properties were maintained.
Dissolution characteristics
ODTs disaggregate in the oral cavity, usually releasing the active agent and allowing it to dissolve remarkably quickly in the saliva. In this work, NiMS-based ODMTs were designed to overcome the local irritation to oral mucosa caused by the direct drug exposure from conventional ODTs. shows the in vitro drug release profiles of conventional ODTs, NiMS-based ODMTs, and noncrosslinked ODMTs. For the conventional ODTs, 94.6% of the drug was released within only 3 minutes. In contrast, 70% or less of the SH was released from the two prepared ODMTs in 30 minutes, and the time for 90% of drug release to occur was more than 90 minutes (). The statistical analysis shows that the drug released very quickly from conventional ODTs (P < 0.01) compared with that from the two ODMT formulations. Formulation of a microencapsulated drug into ODTs, as in the present case, might result in this controlled, sustained release.
Figure 4 In vitro drug release profiles for orally disintegrating tablets (ODTs) formulated with different drug dispersion states (n = 3).
Abbreviation: ODMT, orally disintegrating microparticle tablet.
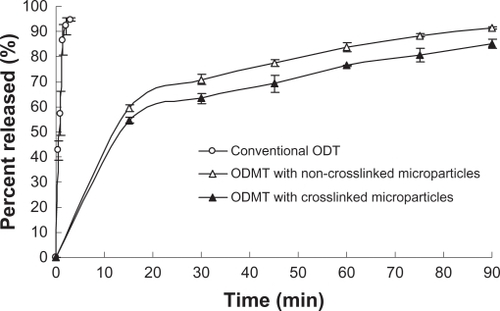
shows that the drug release rate of ODMTs formulated with crosslinked microparticles (NiMS-based ODMTs, 63% in 30 minutes and 85% in 90 minutes) was slower (P < 0.05) than that of ODMTs made with noncrosslinked microparticles (noncrosslinked ODMTs, 70% in 30 minutes and 91% in 90 minutes). This behavior might be due to the retardant effect of TPP, in accordance with the behavior of the microparticles described previously (). The controlled drug release could alleviate problems such as premature destruction of the tablet, unpleasant taste, or intense exposure to high concentrations of the active agent. Finally, it is important to note that the SH-NiMS, including novel ODMTs, could potentially improve compliance with drug administration regimens and promote a remarkably steady rate of drug absorption.
Conclusion
The SH-entrapped NiMS was successfully prepared using an ionotropic gelation process combined with a spray-drying method. The developed ODMT formulation has advantageous characteristics that distinguish it from commonly used ODTs. The presence of disaggregating agents makes it possible to produce a hard tablet that can still disaggregate within seconds and be considered “quickly dispersible”. The ODMTs can be formulated not only for oral dispersibility but also for delayed drug release that takes place far from the buccal region. Therefore, this novel ODMT prepared by means of a conventional tablet formation technique would be desirable to mask the drug taste in the oral cavity and lessen the common side effects related to the intrinsic characteristics of SH. Formulations containing NiMS represent an example of quickly dispersible/slowly releasing tablets that are an alternative to traditional ODTs.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Project 30973661) and Shanghai Nanotechnology Promotion Center (Project 0852nm04300).
Disclosure
The authors report no conflicts of interest in this work.
References
- ChangRKGuoXBurnsideBCouchRFast-dissolving tabletsPharm Technol20002415258
- HabibWKhankariRHontzJFast-dissolving drug delivery systemCrit Rev Ther Drug Carrier Syst2000171617210755211
- FiniABergamanteVCeschelGCFast dispersible/slow releasing ibuprofen tabletsEur J Pharm Biopharm200869133534118182280
- HisakadzuSYunxiaBPreparation, evaluation and optimization of rapidly disintegrating tabletsPowder Technol20021221188198
- MizumotoTMasudaYYamamotoTFormulation design of a novel fast-disintegrating tabletInt J Pharm20053061–2839016257154
- FukamiJYonemochiEYoshihashiYTeradaKEvaluation of rapidly disintegrating tablets containing glycine and carboxymethylcelluloseInt J Pharm20063101–210110916434157
- WattsPJDaviesMCMeliaCDMicroencapsulation using emulsification/solvent evaporation: an overview of techniques and applicationsCrit Rev Ther Drug Carrier Syst1990732352592073688
- BhavsarMDTiwariSBAmijiMMFormulation optimization for the nanoparticles-in-microsphere hybrid oral delivery system using factorial designJ Control Release2006110242243016338017
- BhavsarMDAmijiMMGastrointestinal distribution and in vivo gene transfection studies with nanoparticles-in-microsphere oral system (NiMOS)J Control Release2007119333934817475358
- IllumLChitosan and its use as a pharmaceutical excipientPharm Res1998159132613319755881
- LiFQFeiYBChenXAnchoring of ulex europaeus agglutinin to chitosan nanoparticles-in-microparticles and their in vitro binding activity to bovine submaxillary gland mucinChem Pharm Bull200957101045104919801856
- LiFQJiRRChenXCetirizine dihydrochloride loaded microparticles design using ionotropic cross-linked chitosan nanoparticles by spray-drying methodArch Pharm Res201033121967197321191762
- PutchaLCintrónNMTsuiJPharmacokinetics and oral bioavailability of scopolamine in normal subjectsPharm Res1989664814852762223
- FeiYBLiFQHuJHPreparation of chitosan nanoparticles loaded with HB vaccine by ionotropic gelation-homogenization processPharm Care Res200882119122
- LiFQSuHWangJPreparation and characterization of sodium ferulate entrapped bovine serum albumin nanoparticles for liver targetingInt J Pharm20083491–227428217870261
- LiFQHuJHLuBCiprofloxacin-loaded bovine serum albumin microspheres: preparation and drug-release in vitroJ Microencapsul200118682582911695645
- ShuXZZhuKJA novel approach to prepare tripolyphosphate/chitosan complex beads for controlled release drug deliveryInt J Pharm20002011515810867264
- AnalAKStevensWFLópezCRIonotropic cross-linked chitosan microspheres for controlled release of ampicillinInt J Pharm20063121–216617316497452
- JanesKAFresneauMPMarazuelaAChitosan nanoparticles as delivery systems for doxorubicinJ Control Release2001732–325526711516503
- JameelaSRJayakrishnanAGlutaraldehyde cross-linked chitosan microspheres as a long acting biodegradable drug delivery vehicle: studies on the in vitro release of mitoxantrone and in vivo degradation of microspheres in rat muscleBiomaterials199516107697757492707
- KunoYKojimaMAndoSNakagamHEffect of preparation method on properties of orally disintegrating tablets made by phase transitionInt J Pharm20083551–2879218182258