Abstract
Background
Recombinant human erythropoietin (rhEPO) is considered to be one of the most pivotal pharmaceutical drugs in the market because of its clinical application in the treatment of anemia-associated disorders worldwide. However, like other therapeutic proteins, it does not have suitable pharmacokinetic properties for it to be administrated at least two to three times per week. Chemoselective cysteine PEGylation, employing molecular dynamics and graphics in in silico studies, can be considered to overcome such a problem.
Methods
A special kind of EPO analog was elicited based on a literature review, homology modeling, molecular dynamic simulation, and factors affecting the PEGylation reaction. Then, cDNA of the selected analog was generated by site-directed mutagenesis and subsequently cloned into the expression vector. The construct was transfected to Chinese hamster ovary/dhfr− cells, and highly expressed clones were selected via methotrexate amplification. Ion-immobilized affinity and size exclusion (SE) chromatography techniques were used to purify the expressed analog. Thereafter, chemoselective PEGylation was performed and a nanosize PEGylated EPO was obtained through dialysis. The in vitro biologic assay and in vivo pharmacokinetic parameters were studied. Finally, E31C analog Fourier transform infrared, analytical SE-high-performance liquid chromatography, zeta potential, and size before and after PEGylation were characterized.
Results
The findings indicate that a novel nanosize EPO31-PEG has a five-fold longer terminal half-life in rats with similar biologic activity compared with unmodified rhEPO in proliferation cell assay. The results also show that EPO31-PEG size and charge versus unmodified protein was increased in a nanospectrum, and this may be one criterion of EPO biologic potency enhancement.
Discussion
This kind of novel engineered nanosize PEGylated EPO has remarkable advantages over rhEPO.
Introduction
Erythropoietin (EPO) is synthesized in the kidney and plays an important role in the production and differentiation of erythroid progenitors to mature erythrocytes in hypoxia conditions, which is a process called erythropoiesis.Citation1 A defect in the production of EPO in the body leads to a decrease in red blood cells (anemia), which results from chronic kidney disease, chemotherapy, and azidothymidine treatment of HIV.Citation2 The production of recombinant human EPO (rhEPO) is developed genetically by many biopharmaceutical industries, as it is widely used in anemia disorders. The available rhEPO is a highly glycosylated protein (40% of molecular weight) and comprises 165 amino acids with a ∼35 kDa molecular weight.Citation3 Due to post-translational modification, particularly in glycosylation pattern, and concerns about oncogenes in respect of human cell lines, it is expressed commercially in Chinese hamster ovary (CHO) and baby hamster kidney cells.Citation4,Citation5
Many scientists have studied the expression of EPO in systems such as Escherichia coli,Citation6 Saccharomyces cervisiae,Citation7 and bacullovirus,Citation2 but because of rapid clearance from the circulation due to differences in sialic acid content, expressed EPOs did not have efficient in vivo activity. Nevertheless, CHO-expressed rhEPO has a half-life of 4–13 hours. In order for it to have therapeutic effect, it should be administered two to three times per week, which, in addition to the high cost of treatment, means frequent intravenous administration for patients.Citation2 Therefore, there has been growing interest in the development of long-acting products to be administered less often than existing products.
One of the approved approaches for the improvement of pharmacokinetic properties of biologic products is PEGylation. PEGylation can lead to an increase in size, drug solubility, and bioavailability, while decreasing dose frequency as well as immunogenicity.Citation8,Citation9 The prominent marketing of some PEGylated proteins in the last decades has shifted the focus to the marketing of other PEGylated proteins.Citation10
Basic problems with the existing PEGylated proteins are inefficient conjugation yield, the formation of heterogeneous mixtures, and a decrease in biologic activity as a result of nonspecific PEGylation.Citation11 For example, the only marketed PEGylated form of rhEPO, CERA®, was generated by conjugation of 30 kDa methoxy-PEG via amide bonds to the N-terminal or the amino groups of lys-52 or lys-45.Citation12,Citation13 These drawbacks can be successfully compensated by chemoselective cysteine PEGylation, which is a strong tool for improvement of pharmacokinetic properties of therapeutic proteins. However, the addition of free cysteine residue to protein is not simple and occurs commonly in cysteine PEGylation because of the rare existence of free cysteine residue in the proteins. Introduced cysteine residue must not change the three-dimensional (3D) structure or stability of protein.Citation14 In addition, factors affecting PEGylation must also be considered because experimental analysis of all stable cysteine analogs is a very time-consuming and costly process. Therefore, rational selection of suitable sites for replacement with cysteine residue is a critical step for this method.Citation11,Citation14 It has already been shown that an increase or decrease in the protein nanosize or charge by PEGylation may significantly affect protein biologic properties, including half-life, potency, and stability.Citation15,Citation16
In the present study, a novel nanosize PEGylated rhEPO has been developed and evaluated based on a literature review, homology modeling, molecular dynamic (MD) simulations, factors affecting PEGylation, and biologic assays that have a bigger nanosize, more negative charge, and better pharmacokinetic properties with the same in vitro biologic potency compared with intact rhEPO.
Materials and methods
Selection of a position for replacement by cysteine residue using an in silico approach
A literature review was performed for finding nonstructural and nonfunctional residues. 3D structures of cysteine analogs were generated by homology modeling using MODELLER version 9.5 and a crystal structure of EPO (PDB code: 1EER, chain A, 1.9 Å) as a template (http://www.salilab.org).Citation17 For each cysteine analog or native EPO, 10,000 molecules were generated, and the molecule corresponding to the lowest value of probability density function was selected. The quality of models was checked using discrete optimized protein energy (DOPE) score profileCitation18 and PROCHECK (http://www.biochem.ucl.ac.uk).Citation11 The stability of modeled analogs was examined by MD simulation using a GROMACS 3.3 package (http://www.gromacs.org) and Gromos96 force field.Citation19 MD simulations were carried out at a constant temperature (300 K) and pressure (1 bar) for 5–15 ns. After calculating the average structures corresponding to the last 500–2000 ps of MD simulations, modeled cysteine analogs with a root mean square deviation (RMSD) of less than 4 Å from the initial structure were chosen for further analysis. Stable cysteine analogs were subjected to screening based on the RMSD from native EPO, surface area accessibility of engineered cysteine residues (cysteine-SAA), and pKa values of the thiol group. All-atom RMSDs between native EPO and the selected analogs were calculated by Qmol software (http://www.mbg.cornell.edu).Citation20 The pKa of thiol groups was determined by PROPKA 2.0 Web interface (http://propka.ki.ku.dk).Citation21 Cysteine-SAA in the modeled analogs was computed by GETAREA server (http://curie.utmb.edu).Citation22 Cysteine analogs with SSA more than 50% were considered to be solvent exposed. Finally, one cysteine analog was selected for experimental analysis.
Construction of an expression vector containing an engineered cysteine analog
The cDNA of EPO was obtained by the reverse transcriptase-polymerase chain reaction (RT-PCR) method from Hep G2 Cell line (ATCC No. HB-8065, USA) and cloned into the pTZ57R/T using first cDNA synthesis and a T/A cloning kit, respectively (Fermentas, Vilnius, Lithuania). A His-tag and kozak sequence were added to the C-terminal and N-terminal of EPO’s cDNA with a forward primer, 5′-ATT ATG GGG GTG CAC GAA TGT CCT G-3′, and a reverse primer, 5′-TCA ATG ATG ATG ATG ATG ATG GTC CCC TGT CCT GCA G-3′, respectively. Replacement of glutamic acid at position 31 with cysteine was performed by the site-directed mutagenesis method using a forward primer 31, 5′-GCT TGT CAC TGC AGC TTG AAT GAG AAT ATC AC-3′, and a reverse primer 31, 5′-CAT TCA AGC TGC AGT GAC AAG CAC AGC CCG TCG TG-3′. All PCRs were performed with Pfu DNA polymerase to avoid unexpected mutations (Fermentas). The mutated gene was cloned into the optiCHO vector (Topo T/A cloning kit, Invitrogen Inc, Carlsbad, CA). Finally, the accuracy of the construction was confirmed by restriction analysis and sequencing.
Expression of the engineered cysteine analog in the CHO cell line
CHO/dhfr− (1.5 × 105) cells (ATCC No. CRL-9096, USA) were cultured in Iscove’s modified Dulbecco’s medium supplemented with 10% (v/v) fetal bovine serum (GIBCO, Invitrogen Inc), penicillin (100 units/mL), streptomycin (100 μg/mL), hypoxanthine (0.1 mM) (Sigma-Aldrich, Munich, Germany), thymidine (0.016 mM) (Sigma-Aldrich), and methotrexate (0.002 mM) (Sigma-Aldrich) in a 24-well microplate. The final construct was linearized by SspI (Fermentas) and transfected using a lipofectamin 2000 kit (GIBCO, Invitrogen, Inc). Transfected cells were selected in dialyzed α-MEM medium containing 10% dialyzed FBS without any nucleosides (GIBCO, Invitrogen Inc). The resulting clones were subjected to gene amplification using increasing doses of methotrexate (Sigma-Aldrich). High-expressing clones were selected by EPO ELISA kit (Roche Diagnostics GmbH, Mannheim, Germany). RT-PCR and sequencing were carried out to confirm the existence of replaced cysteine codons at the mRNA level, first using a cDNA synthesis kit either by oligo-dT, random hexamer, or specific primers (Fermentas). Finally, secretion of cysteine analogs was analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in a 12% resolving gel at 200 V for ∼50 minutes using the Mini-PROTEIN® Tetra Cell system (Bio-Rad Laboratories, Hercules, CA) and visualized by the Coomassie brilliant blue staining method.Citation23 Electroblotting was performed using a Mini Trans-Blot® electrophoretic transfer cell (Bio-Rad Laboratories) to Whatman nitrocellulose membrane (Schleicher and Schuell, Riviera Beach, FL) for 1 hour. Membrane was blocked in 5% bovine serum albumin and 0.1% Tween 20 in phosphate buffered saline (PBS) at 4°C overnight. The identification of expressed E31C analog was performed by employing the mouse monoclonal antibody against rhEPO (GeneTex Inc, Irvine, CA) and goat antimouse immunoglobulin G (HRP) (GeneTex Inc) as a primary and secondary antibody for 1 hour at room temperature. The band was developed using a DAB chromogenic substrate (Sigma-Aldrich).
Purification of the expressed cysteine analog
Cell culture supernatant was concentrated 10-fold using a Labscale TFF system (Millipore, Billerica, MA) equipped with a PXC010C50 cassette (Pellicon XL, Millipore). Simultaneously, buffer exchange was carried out three times with PBS. An ion immobilized affinity chromatography (IMAC) was applied for purification of the cysteine analog using 5 mL Histrap columns (GE Healthcare Inc, Princeton, NJ). The column was pre-equilibrated with 0.02 M sodium phosphate 0.5 M NaCl and 20 mM imidazole adjusted to pH 7.4. After sample loading and washing of columns with equilibration buffer, anchored cysteine analog was eluted with 0.02 M sodium phosphate 0.5 M NaCl and 0.5 M imidazole (pH 7.4). An additional purification step was performed for elimination of imidazole and probable dimers by size exclusion chromatography (SEC) (HiLoad Superdex 75 prep grade, GE Healthcare). In this step, a solution containing 50 mM sodium phosphate and 150 mM sodium chloride (pH 7) was used as an equilibration and elution buffer. All fractions containing cysteine analog were pooled and adjusted to the desired concentration using Microcon® centrifugal filter devices (Millipore). Finally, purified analog was aliquoted and stored at −80°C for further analysis.
Chemoselective PEGylation of engineered cysteine analog
PEGylation reaction was performed at room temperature and under nitrogen gas. Briefly, a 30-fold molar excess of TCEP (Tris [2-carboxyethyl] phosphine- HCl) (Sigma-Aldrich) and a 30-fold molar excess of 30 kDa mPEG-maleimide (SunBio Inc, Anyang City, South Korea) were added to a solution of E31C analog (250 μg/mL) in 50 mM Tris buffer pH 8.0 and incubated for 3 hours. Then, the PEGylation reaction was terminated by adding 10-fold Tris buffer (20 mM, pH 8.0). Finally, the reaction was dialyzed against PBS, concentrated using Microcon® centrifugal filter devices (Millipore), and stored at −80°C for further analysis.
Characterization, size, and zeta potential of PEGylated and unmodified cysteine analog
SE-high-performance liquid chromatography (HPLC) (Shodex PROTEIN KW-802.5 SEC column, Shodex, Japan) was employed for analysis of the protein modification using 50 mM sodium phosphate containing 150 mM sodium chloride (pH 7) as an equilibration and elution buffer. 20 μL of each sample was injected separately and monitored at 214 nm. Also, the spectral changes of the E31C analog were analyzed by Fourier transform infrared (FTIR) spectroscopy (Thermo-Nicolet, NEXUS 870, Waltham, MA) before and after the conjugation. Through changes in IR spectra, we were able to show the surface conjugation and clarify which group was involved in the conjugation process. In addition, EPO31-PEG and E31C analog (0.02 mg/mL) were checked for any changes in the size and zeta potential distribution by DLS technique (Malvern, Zetasizer Nano ZS, Worcestershire, UK).
Biological assays and pharmacokinetic studies
In vitro assays of all samples were carried out based on proliferation of UT-7 cells as follows. UT-7 cells (ACC 137; DMSZ, Braunschweig, Germany) were grown in α-MEM supplemented with 20% FBS, 40 mg/mL gentamycin, 2 mM glutamine, and 5 ng/mL GM-CSF (Sigma-Aldrich). Five thousands cells were seeded in each well of a 96-plate. Twofold serial dilutions of the protein samples and standard were prepared in assay media. One hundred μL of the diluted protein samples was added in triplicate to each well of a 96-well microplate. Ten thousand cells were washed and seeded in each well and incubated at 37°C in humidified 5% CO2 for 3 days. PBS and 200 ng/mL rhEPO (R&D Systems, Minneapolis, MN) were added to the cultured cells as a negative and positive control, respectively. Proliferation of cells was measured by MTT assay, a technique based on the reduction of MTT to formazan by viable cells.Citation24 Finally dose-response curves were drawn for rhEPO, E31C analog, and EPO31-PEG. Animal experiments were performed with the approval of the Animal Ethics Committee of the Pasteur Institute of Iran. The five Sprague-Dawley rats (300 g ± 30 g weight) in each group received a single intravenous dose of 30 μg protein/kg.Citation12 Blood samples were collected from the lateral tail vein at 15 minutes, 1 hour, 8 hours, 24 hours, 48 hours, 96 hours, 120 hours, and 144 hours after the injection. The collected samples were centrifuged at 1500 g for 12 minutes, and separated plasma was analyzed using an EPO ELISA kit (Roche Diagnostics GmbH). The terminal half-life and the area under the curve were determined using linear regression of the last three plasma concentrations and a linear trapezoidal method with extrapolation to infinity, respectively. Protein concentrations were determined using a Bradford dye binding assay. Statistical analysis of the data was performed using Student’s t-test and one-way multivariate analysis of variance to calculate the level of significance value (P-value). Values of P < 0.05 were considered statistically significant.
Results
Selection of a position for replacement with cysteine residue using an in silico approach
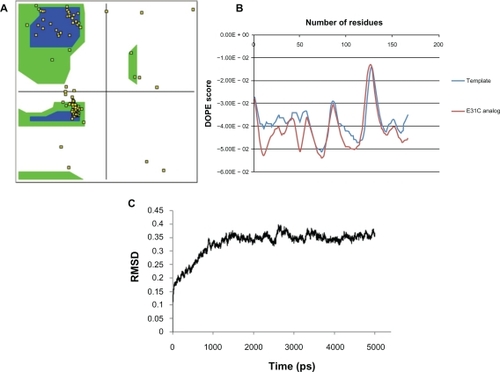
From 165 residues in the EPO, 115 positions were excluded based on a literature review (). The 3D structures of selected analogs were generated successfully from related sequences using homology modeling because of high homology (above 95%) between the template and cysteine analogs. The quality of modeled analogs was verified by Ramachandran plot and DOPE score profile. Over 98% of residues in the generated models are located in regions based on ψ and φ angels, as shown in for E31C analog. DOPE score profile of cysteine analogs did not show any energy differences compared with the template, as depicted in for E31C analog. MD analysis was performed for all analogs in 5–15 ns, and RMSD versus time was plotted for the evaluation of model stability and average structure calculations (). Stable cysteine analogs during MD simulations (40 analogs) were subjected to screening based on the SAA of replaced cysteine residue (), and 13 exposed cysteine analogs were selected. Then, pKa values of the thiol group and whole RMSD versus native EPO were determined (). Finally, based on having maximum cysteine-SSA and pKa first and then the minimum RMSD from native EPO in the final average structure, the E31C analog was chosen for experimental analysis.
Table 1 Exclusion of residues involved in receptor binding or structural stability of erythropoietin, based on a literature review
Table 2 Stability of modeled analogs after molecular dynamic simulations and surface area accessibility (SAA) of replaced cysteine
Table 3 All-atom root mean square deviation (RMSD) and pKa values of the thiol group for stable cysteine analogs that had solvent-exposed cysteine
Figure 1 Quality assessment of the modeled three-dimensional structure of the R31C analog as a sample. A) >98% of residues are in the allowed regions of the Ramachandran plot. B) Discrete optimized protein energy (DOPE) score profile of modeled E31C analog and template. C) Root mean square deviation (RMSD) plot for modeled E31C analog during molecular dynamic simulation. The fluctuations of the modeled structure reached plateau after ∼1500 ps of simulation.
Construction, expression, and biologic evaluation of E31C analog
The natural cDNA of EPO was obtained by the RT-PCR method and T/A cloned into pTZ57R/T. The addition of His-tag, kozak sequence, and replacing of glutamic acid by cysteine codon were performed simultaneously by overlap extension PCR using respective primers (). The mutated gene was cloned successfully into an optiCHO vector, and then the accuracy of the construct was confirmed by restriction analysis and sequencing (data not shown). The linearized construct was transfected into CHO/dhfr− cells, and expressing clones were obtained in selective mediums. The E31C-expressing clones were subjected to methotrexate amplification (), and a high-expressing clone was selected using the ELISA method. The existence of replaced cysteine codon in the mRNA was confirmed by RT-PCR and sequencing (data not shown). The identification of expressed cysteine analog was carried out using an anti-EPO antibody on nitrocellulose membrane (). The verified clone was cultured, and harvested supernatants were concentrated and immediately purified using IMAC and SE, as described in Materials and methods (). The biologic tests did not show any distinction in the activity of rhEPO, and the E31C analog EC50 (protein concentration achieving 50% of the maximum effect) for E31C analog and rhEPO were obtained, 0.53 ± 0.03 and 0.52 ± 0.04, respectively ().
Figure 2 Replacement of glutamic acid codon with cysteine codon using sitedirected mutagenesis. Lanes 1, 3, and 5: 100 bp markers (Fermentas, Lithuania); lane 2: first fragment of mutated cDNA containing kozak sequence and replaced cysteine codon (∼200 bp); lane 4: second fragment of mutated cDNA containing His-tag and replaced cysteine codon (∼400 bp); and lane 6: the complete cDNA of E31C analog (∼600 bp).
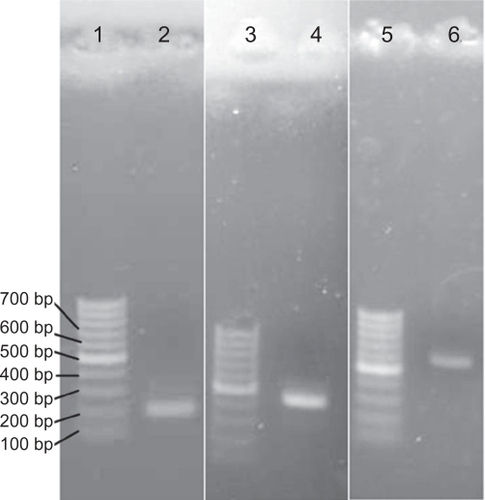
Figure 3 A) Methotrexate amplification of E31C analog (lanes 2, 3, 4, 5, and 6), visualized by Coomassie brilliant blue staining method. B) Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis of purified E31C analog (∼35 kDa) using immobilized affinity chromatography and size exclusion. C) Western blot analysis of E31C analog using monoclonal mouse antierythropoietin antibody.
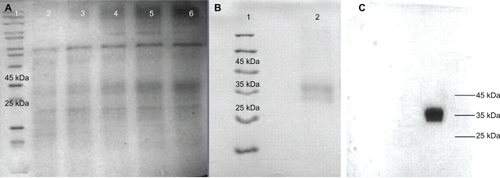
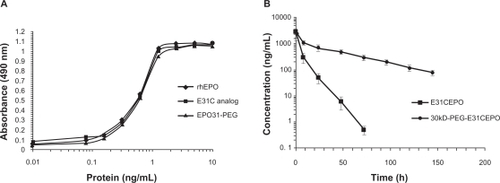
Figure 4 A) In vitro biologic assay based on proliferation of UT-7 cells. The EC50 (protein concentration achieving 50% of maximum effect) for E31C analog and EPO31-PEG were obtained (0.53 ± 0.03 and 0.54 ± 0.08, respectively), similar to recombinant human erythropoietin (rhEPO) (0.52 ± 0.04). B) Single intravenous injection of EPO31-PEG showed five-fold terminal half-life for unmodified protein in rats (36.47 hours ± 6.5 hours vs 7.22 hours ± 1.3 hours). Protein concentrations in all experiments were determined using Bradford dye binding assay.
Characterization of PEGylated and unmodified analog
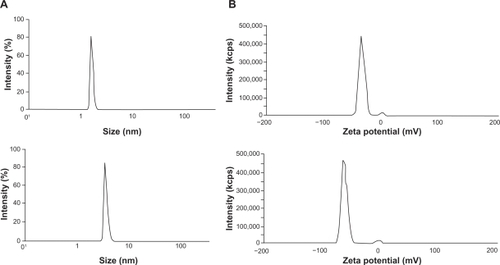
The purified E31C analog and the PEGylated form were monitored by injection of each sample to an analytical SE-HPLC. As revealed in the chromatogram, the peak PEGylated form was eluted from the column ∼6 minutes after the injection, which is earlier than the unmodified analog, as expected (). The unmodified protein peak appeared ∼20 minutes after the injection. Surprisingly, biologic activity of EPO31-PEG indicated no significant difference either by rhEPO or by E31C analog (). Also, in animal experiments, illustrated in , EPO31-PEG showed a five-fold terminal half-life compared with the E31C analog. In the FTIR spectra of engineered analog, a peak appeared at 2414 cm−1 (), which endorsed the substitution of free cysteine, whereas it was not observed in the rhEPO spectrum (data not shown). After PEGylation of the engineered analog with 30 kDa PEG-maleimide, a peak at 2414 cm−1 was missed and a peak raised at 2486 cm−1 (). This peak was related to C-H bound conjugated PEG polymer and confirmed that conjugation occurred in the free thiol group of replaced cysteine. The PEGylated analog showed an approximately two-fold increase in nanosize (from 3.1 nm ± 0.3 nm to 5.9 nm ± 0.2 nm) relative to the unmodified protein (). Zeta potential results also showed an increase in negative charge of PEGylated analog due to attachment of the PEG polymer (). Finally, animal studies showed that PEGylated analogs have a lower elimination rate than unmodified analogs, as well as other improved pharmacokinetic parameters (see ).
Table 4 Pharmacokinetic parameters for the E31C analog and its PEGylated form after injection of a single dose into the vein tail of rats. As shown, EPO31-PEG has a lower elimination rate for unmodified protein that leads to a five-fold terminal half-life in plasma
Figure 5 Size exclusion high-performance liquid chromatography chromatogram of PEGylated and unmodified analog. The peak at 6 minutes is related to EPO31-PEG, and the second peak is attributed to E31C analog eluted at 20 minutes after injection.
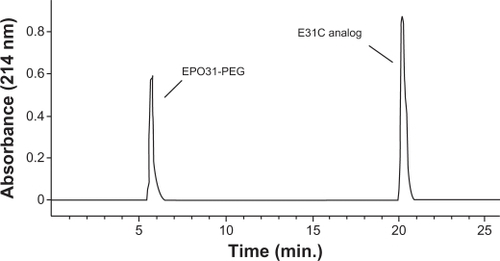
Figure 6 Characterization of the E31C analog and its PEGylated form. Fourier transform infrared spectra of the E31C analog before (A) and after (B) PEGylation reaction. A sharp peak at 2414 cm−1 is shown for the sulfhydryl group of free cysteine at position 31. Additionally, the omission of an observed peak at 2414 cm−1 is assumed for E31C analog–PEG conjugation. Also, the observation of a new apparent peak at 2486 cm−1 (CH-PEG) is a second confirmation of PEGylated conjugate synthesis.
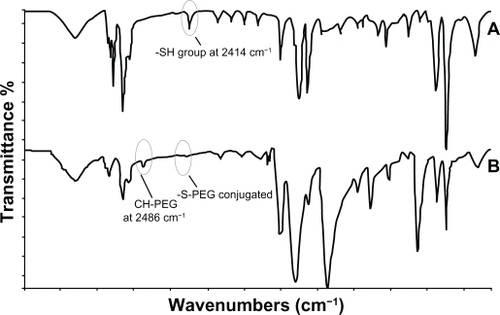
Figure 7 Changes in (a) size and (b) zeta potential of E31C analog before (up) and after (down) attaching to the PEG-30 kDa. As depicted, the size and negative charge of molecule increased due to the attaching of the polymer.
Discussion
Covalent attachment of PEG to proteins via cysteine residue, known as cysteine PEGylation, is considered an extremely efficient approach to circumventing product heterogeneity and loss of biologic activity characteristic of amine-PEGylation.Citation25 Several grants were allocated to Bolder BioTechnology Inc (www.bolderbio.com) by the National Institutes of Health for cysteine PEGylation of therapeutic proteins. This method has some limitations in practice. Functional or structural stability of cysteine analogs and factors affecting conjugation must be considered, because experimental analysis of all positions in the protein is a very time-consuming and costly process.Citation14
Desjarlais and ZalevskyCitation26 reported a computational method for rational cysteine PEGylation. The method determines the ratio of the ranges of motion of the PEG for protein alone versus protein complexed with its receptor. This ratio is related to activity when PEG is coupled at a specific site in the protein, but this method does not evaluate the effects of introduced free cysteine on the conformation and stability of the protein. Therefore, to the authors’ knowledge, there is no straightforward approach for rational site selection that comprises all aspects of cysteine PEGylation.
Our in silico approach consists of three steps. The first step is the selection of sites that do not involve protein function. This step can be carried out easily by a literature review, because almost all marketed recombinant proteins have been well studied. In this way, exclusion of residues involved in functional or structural stability of EPO was carried out successfully by literature review because of known EPO receptor binding sites (see ).
In the second step, structural stability of analogs due to cysteine substitution must be evaluated. For this purpose, 3D structures of cysteine analogs must be subjected to MD simulation.Citation27 As indicated in , the selected analog modeling and MD simulations revealed many possible sites (∼40 positions) in the EPO structure for cysteine residue replacement, which did not disturb EPO structure by RMSD measurement. These findings are consistent with previous studies that examined bioactivity of mutated EPOs by site-directed mutagenesis.Citation2,Citation28,Citation29
In the third step, PEGylation factors must be assessed on stable modeled analogs. Factors affecting protein PEGylation include SAA, inherent nucleophilicity, and pKa of residue. Other factors, such as time, temperature, pH, and protein/PEG ratio, depend on the PEGylation method and must be determined empirically.Citation30,Citation31 In the present study, PEGylation factors related to the protein were analyzed on modeled stable analogs of EPO. The thiol group of introduced cysteine residue must be accessible for a successful attack on PEG molecules.Citation30 Therefore, cysteine analogs with low thiol SAA must be excluded. Also, PEG attacks on the cysteine residue on the protein surface are dependent on the nucleophilicity of cysteine residue. PEG attacks will take place easier when the pH of PEGylation reaction is higher than the pKa of the thiol group. Therefore, cysteine residues have pKa values less than the pH of the PEGylation reaction and are preferred for cysteine PEGylation.Citation25 In combination with factors affecting PEGylation, only 13 positions remained for the further evaluations. As listed in , the selected positions were located structurally in the N-terminal, C-terminal, and AB, BC, and CD loops of EPO that are not involved in stability or activity. Among the remaining analogs, the E31C and E89C analogs showed suitable criteria, including high SAA and low conformational change relative to the EPO.
Our in silico findings are in agreement with another study, by Desjarlais and Zalevsky,Citation26 that measured the ratio of the ranges of PEG motion. It concluded that the best positions in the EPO for conjugation either by 2 kDa or 10 kDa PEG are Glu31, Ser85, and Ala125, which have optimal predicted conjugation efficiencies, stability, and functional activity. Substitution of Ser85 with cysteine disturbs N-glycosylation at Asn83, which is essential for in vivo activity of EPO. Long et alCitation2 also showed that S85C analog is glycosylated partially at position Asn83. The N85C analog was excluded in the first step of our approach. It must be noted that Desjarlais and ZalevskyCitation26 performed their simulations on the 1EER, which does not contain any glycosyl chain, but Ser126 is glycosylated in expressed EPO. Therefore, spatial restriction of the glycosyl chain at position 126 may influence the coupling efficiency of the polymer negatively, but, as indicated in , the pKa value of the thiol group in Ala125 is lower than the other selected analogs, which may compensate for lower thiol accessibility. The T27C analog has a similar condition as A125. The previously reported T26C analog lacks glycosylation at position 24, which affects in vivo bioactivity due to a decrease in sialic acid content and leads to rapid clearance from circulation.Citation2 Therefore, cysteine substitution at this position does not seem to be logical. Our data reveal that neighbor residue Thr27 is more suitable than Thr26 because cysteine substitution at this position does not affect N-glycosylation at position 24. As indicated in , the E89C analog has good criteria for cysteine PEGylation, high SAA and low RMSD, which is consistent with the previous study by Long et al.Citation2 Even being PEGylated with a high-molecular-weight (20 kDa) PEG-maleimide, the E89C analog shows the same activity.Citation2 L130C and T134C analogs are not suitable for PEGylation with a high-molecular-weight PEG because of their proximity to the R131, which is involved in receptor binding site 1 of EPO. PEGylation at these positions with 30 kDa PEG can reduce receptor binding affinity of PEGylated forms.Citation32 PEGylation of positions located in the N-terminal (R4 and L5) and C-terminal (R162, T163, and R166) in the EPO are not logical because of spatial restriction in interaction between EPO and its receptors. As shown by Long et al,Citation2 the attachment of 20 kDa PEG at positions A1, P3, and T163 decreases receptor binding affinity. In addition, it has been reported that terminal arginine of EPO is deleted from the CHO-expressed rhEPO. Therefore, remaining replaced cysteine at this position must be proved.Citation33 K116C and D123C analogs show higher RMSD than other analogs, but both analogs have reasonable cysteine accessibility with closer pKa value. As reported, PEGylation at neighbor position Q115 with a 20 kDa PEG-maleimide did not change receptor binding affinity.Citation2
Based on the in silico studies, E31C and A118C analogs were chosen as PEGylation-positive and -negative models. Further verification was obtained when E31C and A118C analogs showed the same biologic activity, which further proves that no structural or functional changes occurred (data not shown). The A118C analog showed a low PEGylation yield in the same conditions, as expected, because of low cysteine accessibility (see ). In the case of the E31C analog, FTIR spectra confirmed both the existence of replaced cysteine analog and chemoselective PEGylation. The data also showed an increase in nanosize of EPO and an increase in half-life. Extended terminal half-life due to attachment of 30 kDa PEG is in agreement with findings from previous studies.Citation12,Citation34 The attachment of a 30 kDa PEG to the E31C analog results in a five-fold increase in half-life, as reported by Agoram et al.Citation12 It has been shown that PEGylation influences protein clearance and plays a crucial role in their elimination from the circulation.Citation6 A 10-fold decrease in clearance of PEGylated E31C analog with 30 kDa PEG is comparable with another study in which a 30 kDa PEG attached to the NM385 variant.Citation12 In addition, it is reported that PEGylated proteins’ hydrodynamic volumes markedly increased as well.Citation35 The present data propound two additional important criteria for nanoEPO half-life or biologic activity enhancement observations: i) increasing the size by PEGylation, as indicated in ; and ii) the PEGylation was also found to be helpful in making the protein charge more negative rather than the intact EPO as well. On the other hand, an increase in EPO size (from 3 nm to 6 nm) or a negative charge resulted in a significant increase in biologic characteristics of new nanosize EPO. With this in mind, it has also been shown that the increase or decrease in the protein nanosize or charge by PEGylation may significantly affect protein biologic properties, including half-life, potency, and stability.Citation15,Citation16
Previous investigations have elucidated that longer-acting products can be created by amine-reactive PEG conjugation.Citation36–Citation40 However, as discussed, the PEGylated products consist of multiple PEGylated forms, and varying biologic activities have usually reduced activity due to the conjugation extent of the protein. Many studies have been carried out on PEG conjugation of EPO, but the only marketed form is CERA®, which is attained by conjugation of the PEG chain at the N-terminus and lysine residues predominantly at positions 45 or 52.Citation12,Citation13,Citation41 TGase-mediated PEGylation, provided by Centocor, allows the attaching of one to three PEGs to the protein, but the identity of the modified residue could not be ascertained completely.Citation42 Therefore, the major problem associated with PEGylation is due to nonselective conjugation of protein, which leads to heterogeneity or a significant decrease in receptor affinity. Our findings show that when a high-molecular-weight PEG has been conjugated to a protein chemoselectively, then the aforementioned problem does not arise.Citation16 As a result, our proposed in silico approach could be used as a promising methodology for rational PEGylation to remove all inconsistencies encountered.
Conclusion
The nanosize PEGylated E31C-EPO developed in this study shows better pharmacokinetic properties than rhEPO in rats, and further studies will be performed to prove its reliability in the management of anemia. Our results prove that using molecular modeling and MD simulations in rational site selection of cysteine-specific PEGylation is an emerging strategy in developing long-acting molecules. It is possible to use a similar approach to create long-acting forms of other recombinant proteins. Knowledge of factors affecting PEGylation in combination with computational techniques enable us to PEGylate proteins through cysteine residue rationally, without any of the problems that are common in other methods dealing with attaching a high-molecular-weight polymer to a protein.
Acknowledgements
This project was financially supported by the Pasteur Institute of Iran. The computational resource for running molecular dynamic simulations was provided by the National Institute of Genetic Engineering and Biotechnology (NIGEB). The authors wish to express their deep gratitude to all who provided support during the course of this research.
Disclosure
The authors report no conflicts of interest in this work.
References
- ElliottSPhamEMacdougallICErythropoietins: a common mechanism of actionExp Hematol200836121573158418922615
- LongDLDohertyDHEisenbergSPDesign of homogeneous, monopegylated erythropoietin analogs with preserved in vitro bioactivityExp Hematol200634669770416728273
- JelkmannWControl of erythropoietin gene expression and its use in medicineMethods Enzymol200743517919717998055
- WurmFMProduction of recombinant protein therapeutics in cultivated mammalian cellsNat Biotechnol200422111393139815529164
- TestaUErythropoietic stimulating agentsExpert Opin Emerg Drugs201015111913820158404
- WangY-JChenJHaoS-JEfficient preparation and PEGylation of recombinant human non-glycosylated erythropoietin expressed as inclusion body in E. coliInt J Pharm201038615616419932158
- ElliottSGiffinJSuggsSSecretion of glycosylated human erythropoietin from yeast directed by the [alpha]-factor leader regionGene19897911671802673934
- PasutGVeroneseFMPEGylation for improving the effectiveness of therapeutic biomoleculesDrugs Today200945968769519956810
- TillmannHCKuhnBKränzlinBEfficacy and immunogenicity of novel erythropoietic agents and conventional rhEPO in rats with renal insufficiencyKidney Int2006691606716374424
- JevševarSKunsteljMPorekarVGPEGylation of therapeutic proteinsBiotechnol J20105111312820069580
- FeeCJVan AlstineJAPEG-proteins: reaction engineering and separation issuesChem Eng Sci2006613924939
- AgoramBDoshiSGeggCInvestigation of the effects of altered receptor binding activity on the clearance of erythropoiesis-stimulating proteins: nonerythropoietin receptor-mediated pathways may play a major roleJ Pharm Sci20099862198221118837016
- MacdougallICEckardtKUNovel strategies for stimulating erythropoiesis and potential new treatments for anaemiaLancet2006368953994795316962885
- Gaberc-PorekarVZoreIPodobnikBMenartVObstacles and pitfalls in the PEGylation of therapeutic proteinsCurr Opin Drug Discov Devel2008112242250
- ChiuKAgoubiLLLeeIEffects of polymer molecular weight on the size, activity, and stability of PEG-functionalized trypsinBiomacromolecules201011123688369220979350
- MaldineyTRichardCSeguinJEffect of core diameter, surface coating, and PEG chain length on the biodistribution of persistent luminescence nanoparticles in miceACS Nano201133
- SaliABlundellTLComparative protein modelling by satisfaction of spatial restraintsJ Mol Biol199323437798158254673
- EramianDDevosDMeloFA composite score for predicting errors in protein structure modelsProtein Science2006151653166616751606
- LindahlEHessBvan der SpoelDGROMACS 3.0: a package for molecular simulation and trajectory analysisJ Mol Model200178306317
- GansJDShallowayDQmol: a program for molecular visualization on windows-based PCsJ Mol Graph Model200119655757960911552684
- DelphineCBDavidMRJanHJVery fast prediction and rationalization of pKa values for protein-ligand complexesProteins200873376578318498103
- FraczkiewiczRBraunWExact and efficient analytical calculation of the accessible surface areas and their gradients for macromoleculesJ Comp Chem199819319333
- MeyerTSLambertsBLUse of Coomassie brilliant blue R250 for the electrophoresis of microgram quantities of parotid saliva proteins on acrylamide-gel stripsBiochim Biophys Acta196510711441454159310
- AmanlouMSiadatSDAlaviAGd3+-DTPA-DG: novel nanoszied dual anticancer and molecular imaging agentInt J Nanomedicine2011674776321589643
- RobertsMJBentleyMDHarrisJMChemistry for peptide and protein PEGylationAdv Drug Deliv Rev200254445947612052709
- DesjarlaisJRZalevskyJMethods for rational PEGylation of proteinsUnited States Patent and Trademark Office Pre-Grant Publication2007
- IbragimovaGTWadeRCImportance of explicit salt ions for protein stability in molecular dynamics simulationBiophys J1998746290629119635744
- BoisselJPLeeWRPresnellSRErythropoietin structure-function relationships. Mutant proteins that test a model of tertiary structureJ Biol Chem19932682115983159938340419
- WenDBoisselJPShowersMErythropoietin structure-function relationships. Identification of functionally important domainsJ Biol Chem19942693622839228468077236
- WangYSYoungsterSGraceMStructural and biologic characterization of PEGylated recombinant interferon alpha-2b and its therapeutic implicationsAdv Drug Deliv Rev200254454757012052714
- HuaizhiDMinZYuZChunhuaYSynthesis and purification of mono-PEGylated insulinChem Biol Drug Des200769213213817381727
- SyedRSReidSWLiCEfficiency of signalling through cytokine receptors depends critically on receptor orientationNature199839567015115169774108
- RecenyMAScobleHAKimYStructural characterization of natural human urinary and recombinant DNA-derived erythropoietinJ Biol Chem19872623517156171633680293
- WangYJHaoSJLiuYDPEGylation markedly enhances the in vivo potency of recombinant human non-glycosylated erythropoietin: A comparison with glycosylated erythropoietinJ Control Release201014530631320427020
- FilpulaDZhaoHReleasable PEGylation of proteins with customized linkersAdv Drug Deliv Rev2008601294917884239
- ClarkROlsonKFuhGLong-acting growth hormones produced by conjugation with polyethylene glycolJ Biol Chem19962713621969219778703002
- BasuAYangKWangMStructure-function engineering of interfferon-β-1b for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective mono-PEGylationBioconjug Chem200617361863016704199
- BailonPPalleroniASchafferCARational design of a potent, long-lasting form of interferon: a 40 kDa branched polyethylene glycol-conjugated interferon α-2a for the treatment of hepatitis CBioconjug Chem200112219520211312680
- BakerDPLinEYLinKN-terminally PEGylated human interferon-β-1a with improved pharmacokinetic properties and in vivo efficacy in a melanoma angiogenesis modelBioconjug Chem200617117918816417267
- TsutsumiYKihiraTTsunodaSIPolyethylene glycol modification of interleukin-6 enhances its thrombopoietic activityJ Control Release1995333447450
- MacdougallICCERA (continuous erythropoietin receptor activator): a new erythropoiesis-stimulating agent for the treatment of anemiaCurr Hematol Rep20054643644016232379
- PoolC Inventor. Formation of novel erythropoietin conjugates using transglutaminase;2006
- TakeuchiMTakasakiSShimadaMKobataARole of sugar chains in the in vitro biologic activity of human erythropoietin produced in recombinant Chinese hamster ovary cellsJ Biol Chem19902652112127121302373681
- EisukeTGoseiKMasatsuguUThe role of carbohydrate in recombinant human erythropoietinEur J Biochem199018824054112156701
- WenDBoisselJPTracyTEErythropoietin structure-function relationships: high degree of sequence homology among mammalsBlood1993825150715168364201