 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The aim of the present study was to prepare valdecoxib, a cyclo-oxygenase-2 enzyme inhibitor, as a loaded multiparticulate system to achieve site-specific drug delivery to colorectal tumors. Film coating was done with the pH-sensitive polymer Eudragit S100 and sodium alginate was used as mucoadhesive polymer in the core. The microspheres were characterized by X-ray diffraction, differential scanning calorimetry, and Fourier transform infrared spectroscopy and were evaluated for particle size, drug load, in vitro drug release, release kinetics, accelerated stability, and extent of mucoadhesion. The coated microspheres released the drug at pH 7.4, the putative parameter for colonic delivery. When applied to the mucosal surface of freshly excised goat colon, microspheres pretreated with phosphate buffer pH 7.4 for 30 minutes showed mucoadhesion. To ascertain the effect of valdecoxib on the viability of Caco-2 cells, the 3-(4,5-dimethylthiazol-2yl) 2,5-diphenyltetrazolium bromide) test was conducted using both valdecoxib and coated microspheres. In both cases, the percentage of dehydrogenase activity indicated a lack of toxicity against Caco-2 cells in the tested concentration range. Drug transport studies of the drug as well as the coated microspheres in buffers of pH 6 and 7.4 across Caco-2 cell monolayers were conducted. The microspheres were found to exhibit slower and delayed drug release and lower intracellular concentration of valdecoxib.
Introduction
Colorectal cancer is the third most common malignancy and the fourth most frequent cause of cancer deaths worldwide, with 945,000 estimated new cases and 492,000 deaths per year in industrialized countries, where the lifetime incidence of the disease is 5%.Citation1 Development of colorectal cancer is reported to be associated with multiple factors, which include, among others, acquired and inherited genetic susceptibility, environmental elements, and lifestyle choices.Citation2 Chronic ulcerative colitis, particularly when it involves the entire large intestine, predisposes individuals to colorectal cancer at a rate that is 4- to 20-fold greater than average.Citation3 There are three specific patterns in which colon cancer is generally observed: sporadic, inherited, and familial. Although the majority of cases of colon cancer are sporadic in nature, as many as 10% of cases are thought to be hereditary. The two most common forms of hereditary colon cancer are the familial adenomatous polyposis and the hereditary nonpolyposis colorectal cancer, both of which result from a specific germline mutation.Citation4
Studies have consistently demonstrated that regular use (at least two doses per week) of nonsteroidal anti-inflammatory drugs (NSAIDs) is associated with a reduced risk of colorectal cancer.Citation5 The conventional NSAIDs, such as aspirin, diclofenac, ibuprofen, and others, are reported to inhibit both the COX-1 and COX-2 forms of the enzyme cyclo-oxygenase (COX). The COX-1 enzyme is required for physiological processes such as maintenance of gastrointestinal mucosa and platelet aggregation. However, the potential mechanisms by which these NSAIDs exert their protective effects in colorectal cancer appear to be linked primarily to their inhibition of the enzyme COX-2 and free-radical formation. COX-2 overex-pression is seen in precancerous and cancerous lesions in the colon and is associated with a decrease in colon cancer cell apoptosis, as well as enhanced production of angiogenesis-promoting factors.Citation6 However, the dual COX inhibitory effect of the conventional NSAIDs results in gastrointestinal side effects, and hence these are not recommended to be used in the prevention of colorectal cancer. Specific inhibitors of the enzyme COX-2, such as valdecoxib and celecoxib, are quite selective in their action and hence are recommended for the treatment of colorectal cancer. However, some recent reports have focused on the cardiac toxicity of these COX-2 inhibitors, and the use of celecoxib, valdecoxib, and rofecoxib has been shown to be related to an increased risk of myocardial infarction.Citation7 On the other hand, in the APC (Adenoma Prevention with Celicoxib) trial, which was cosponsored by the National Cancer Institute and Pfizer Inc. of New York, it has been shown that cardiovascular problems associated with COX-2 inhibitors are more likely to occur with patients with previous cardiovascular problems, and their use is recommended for chemoprevention in patients without cardiovascular risks.Citation8,Citation9
Targeted delivery to the colon is an attempt to delay the release of the drug into the colon in order to achieve a high local concentration while reducing the dose and thus undue side effects of the drug. Researchers have reported alkaline pH (∼8.0) of colonic contents in patients suffering from colon cancer.Citation10–Citation12 These specific pH conditions of the colonic region may enhance the chances to successfully deliver the drug to the region by developing a suitable pH-dependent system.Citation13–Citation15 Moreover, dosage forms with mucoadhesive properties may prolong the residence time at the site of drug absorption. Mucoadhesive microparticulates coated with a pH-dependent polymer are proposed to initiate the release of the drug at the putative colonic pH 7–8.
Microspheres of valdecoxib (a COX-2 inhibitor) dispersed in a matrix of sodium alginate and subsequently coated with Eudragit S100 (copolymer of methacrylic acid and methyl methacrylate, a pH-sensitive polymer) were prepared. Characterization of the microspheres was carried out by Fourier transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD), differential scanning calorimetry (DSC), and scanning electron microscopy (SEM). The microspheres were evaluated for mucoadhesion, in vitro drug release in simulated gastrointestinal conditions, and stability of the formulations. Drug transport studies of valdecoxib and its coated microspheres were conducted to assess the permeability of the drug across the biological membranes using Caco-2 monolayers. Also, 3-(4,5-dimethylthiazol-2yl) 2,5-diphenyltetrazolium bromide test was performed to assess the toxicity of valdecoxib to Caco-2 cells in concentrations intended to be used in transport studies.
Materials and methods
Materials
Valdecoxib and Eudragit S100 were gifts from Aarti Drugs Ltd (Thane, India) and Evonik Industries (Mumbai, India), respectively. The following ingredients used in the studies were of analytical grade: sodium alginate (molecular weight: 198.11) and Span 80 (Loba Chemie Pvt. Ltd, Mumbai, India); light liquid paraffin, calcium chloride, isopropyl alcohol (IPA), potassium dihydrogen phosphate, disodium hydrogen phosphate, and sodium hydroxide (Merck Specialities Pvt. Ltd, Mumbai, India); petroleum ether (RFCL Ltd, New Delhi, India); ethanol (Changshu Chemical, China); methanol (SD Fine Chem Ltd, Mumbai, India); isobutanol and hydrochloric acid (Merck K GaA, Darmstadt, Germany); 3-(4,5-dimethylthiazol-2yl) 2,5-diphenyltetrazolium bromide) (MTT), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), bovine serum albumin (BSA), 2-(N-morpholino) ethanesulfonic acid (MES), and sodium dodecyl sufate (SDS) (Sigma-Aldrich Labochemikalien GmbH, Germany); and Hank’s buffered salt solution(HBSS) 10x (GIBCO batch 699997). Acetonitrile (Sigma-Aldrich Labochemikalien GmbH) was high-performance liquid chromatography (HPLC) grade.
Preparation of core valdecoxib microspheres
Core microspheres of valdecoxib in varying drug polymer ratios (VSA1–VSA5), as shown in , were prepared following previously published methods.Citation16–Citation18 In a typical experiment, a defined quantity of valdecoxib was dispersed in aqueous solutions of varying concentrations of sodium alginate in accordance with a drug polymer ratio as in . Subsequently, the dispersion was emulsified in light liquid paraffin containing 2% v/v Span 80, with the help of a mechanical stirrer (Remi Instruments Ltd, Mumbai, India) at 2000 rpm for 1 hour. A specified volume of the calcium chloride solution (5% w/v in IPA) (polymer CaCl2 ratio 1:1) was then added to the emulsion at a rate of 2 mL/min and the emulsion was stirred for 10 more minutes. Microspheres were collected by filtration and washed three times with petroleum ether to remove the residual liquid paraffin. Microspheres were kept in a deep freezer at −50°C for 10 hours and then kept in a vacuum desiccator for 12 hours.
Table 1 Various formulations of valdecoxib microspheres in sodium alginate polymer and their drug loading efficiency
Encapsulation of core microspheres
Selected formulations of core microspheres were coated with two different concentrations of Eudragit S100 viz. core coat ratio 1:2.5 and of 1:5 (VSAE-1 and VSAE-2, respectively). Thus, core microspheres were dispersed in Eudragit S100 solution (10% w/v) in methanol at room temperature followed by emulsification in light liquid paraffin containing 2% Span 80 in a beaker, with the help of a mechanical stirrer (1500–2000 rpm). Stirring was continued for 3 hours at room temperature in order to evaporate the solvent completely. Encapsulated microspheres were filtered and washed with petroleum ether to remove the liquid paraffin and dried in a vacuum desiccator for 24 hours.
Characterization of valdecoxib microsphere
FTIR spectroscopy
FTIR spectra of selected microspheres were recorded using a FTIR spectrophotometer (Perkin Elmer model 1600-FTIR) in the range of 4000–400 cm−1.
XRD
X-ray diffractograms of the selected microspheres were recorded using an X-ray diffractometer (X’Pert Pro, PW 3050/PW 3071; Lelyweg, the Netherlands) using nickel-filtered CuKα radiation (λ = 1.540598A°) generated at 40 kV and 30 mA and scanning rate 2°/min over a 2θ range of 10°–80°.
DSC
Thermal analysis of selected microspheres was performed using a DSC-TA system (Perkin Elmer). All samples were sealed in a crimped aluminum pan by application of the minimum possible pressure and heated at a rate of 10°C/min from 40°C to 260°C in a nitrogen atmosphere. An empty aluminum pan was utilized as the reference pan.
Surface morphology and particle size distribution
The shape and surface characteristics of selected microspheres were analyzed by SEM (ZEISS EVO Series Model EVO50 with a spatial resolution of 2.5 nm). Samples mounted on an aluminum stub were sputter coated with gold under reduced pressure and a 30–40 nm thick gold coat was applied using BIO-RAD POLARAN sputter coater. The sample assembly was placed in the microscope and vacuum was applied. The microspheres were observed under SEM at an accelerating voltage of 15KV.
The particle size distribution of the coated microspheres was determined using a Particle Size Analyzer (Brookhaven Instruments Corporation, Model 90Plus). The weighed microspheres (20 mg) were suspended in double-distilled water and the dispersion was examined to determine particle size distribution.
Drug loading and drug loading efficiency
Accurately weighed core microspheres equivalent to 20 mg of the valdecoxib were
dissolved in 100 mL ethanol and subjected to centrifugation at 3000 rpm for 10
minutes. The supernatant solution was withdrawn and diluted with ethanol to a
concentration ≈20 μg/mL. Absorbance of the resulting solution
was measured at 244 nm in an ultraviolet (UV) spectrophotometer (Shimadzu UV
Pharmaspec 1700), and the drug content in microspheres was determined. The same
procedure was followed for measuring drug content in the coated microspheres
using 100 mg of coated microspheres. The following formulae were employed for
the calculation of percentage drug loading and drug loading efficiency:
where DM is drug
contents in microspheres and WM is weight of
microspheres.
Drug release profile
Core microspheres
Accurately weighed core microspheres equivalent to 2 mg of valdecoxib were suspended in 20 mL of 7.4 pH phosphate buffer containing 1.5% w/v SDS. The mixture was stirred magnetically at 37°C at 50 rpm. Samples were withdrawn at specified time intervals with volume replacement. The withdrawn samples were centrifuged at 3000 rpm; supernatant was filtered through 0.45 μm membrane filter, diluted to 10 mL with phosphate buffer pH 7.4, and analyzed for drug content by measuring absorbance at 244 nm in a UV spectrophotometer. All the experiments were conducted in triplicate.
Coated microspheres
Accurately weighed coated microspheres equivalent to 2 mg of drug were placed in 20 mL 0.01 N hydrochloric acid (pH 2.0) and stirred magnetically at 50 rpm for 2 hours. The samples were centrifuged and supernatant filtered through 0.45 μm membrane filter and analyzed for the drug content as described previously.
In a similar experiment, coated microspheres equivalent to 2 mg of drug were placed in 20 mL of phosphate buffer containing 1.5% w/v SDS and stirred magnetically at 50 rpm. The initial pH of the buffer was maintained at 5.5 for 2 hours, which was increased by the addition of Na2HPO4 to 6.8 and maintained for 2 hours. Subsequently, the pH of the buffer was raised by further addition of Na2HPO4 to 7.4 and maintained until the completion of study. Hourly, 1 mL of the sample was withdrawn, and each withdrawn sample was replaced with fresh release medium. The samples were centrifuged and the supernatant was passed through a 0.45 μm filter and analyzed for drug content as described previously. All the experiments were run in triplicate.
Statistical analysis
The in vitro drug release data from microspheres were compared by statistical analysis using one-way analysis of variance (ANOVA). The Student’s t-test was performed to compare the significance of the difference between the means of two groups. P < 0.05 was considered significant.
In vitro evaluation of mucoadhesion
The in vitro wash-off test as reported by Lehr et alCitation19 was followed for the determination of mucoadhesion of microspheres. The proximal large intestine of freshly slaughtered goat was cut to expose the mucosal surface and washed with distilled water and phosphate buffer pH 7.4. The serosal side (2 cm × 2 cm) was fixed with the help of thread on a glass slide. Coated microspheres (5 mg) were spread on the exposed mucosal surface and rinsed with phosphate buffer pH 7.4 and the assembly was kept in a humidity chamber (Thermotech, India, Model TH-7004) at 37°C and 90% relative humidity (RH) for a period of 30 minutes. This pretreatment was performed to dissolve the Eudragit S100 coat and to expose the core alginate polymer. Subsequently, the complete assembly was mounted onto the tablet disintegration test apparatus (Veego, India, model VTD-AVP) with the help of a clamp and a thread. The apparatus was operated in such a manner that the tissue moved regularly up and down at a frequency of 28–32 cycles per minute while immersed in the phosphate buffer pH 7.4 contained in a 1000 mL beaker. The time for complete wash-off of microsphere from the tissue was considered the mucoadhesion time.
In vitro transport studies across Caco-2 monolayer
Protocols for culturing Caco-2 cells were as previously described.Citation20,Citation21 Caco-2 cells of passage 29 were seeded onto Transwell inserts (1.12 cm2, 0.4 μm pore size, 105 cells per insert), and all the experiments of transport studies were conducted at day 28 after seeding.
For the purpose of conducting the MTT test, approximately 4 × 104 suspended cells were seeded into each of 96 wells in enzyme-linked immunosorbent assay plates (MicroWells, Nunk, Denmark). The cells were then cultured under standard conditions for 20–24 hours before use.
MTT test
In order to assess the toxicity of valdecoxib and its coated microspheres to Caco-2 cells, an MTT test was conducted on the Caco-2 cell monolayers as described by Anderberg and Artursson.Citation22 The test is based on the principle that the enzyme dehydrogenase in the mitochondria of living cells converts the yellow MTT (3-[4,5-dimethylthiazol-2yl] 2,5-diphenyltetrazolium bromide) to a blue–purple formazan crystal.Citation23 If the cells are exposed to adverse treatment affecting their viability, the activity of the dehydrogenase enzyme is compromised.
Valdecoxib solutions and the coated microspheres were tested in eight concentrations (0.318–31.8 nM/mL). All the solutions were prepared a day before the experiment in HBSS. SDS (concentration ranging from 0.05 mM/mL to 5.0 mM/mL) was included as a positive control, and blank HBSS served as a negative control. HBSS was placed in cell-free wells as an additional background control.
On the day of the experiment, the cells were examined under microscope to ensure that they were attached to the bottom of the wells. The medium was discarded and the test solutions, positive, negative, and background controls, were transferred to the 96-well tray. All solutions were tested in triplicate. The tray was incubated at 37°C on a shaker table at 100 rpm (Edmund Bühler GmbH) for 4 hours. Next, the wells were emptied and 100 μL of HBSS was added to all the wells along with 20 μL MTT solution (5 mg/mL). The tray was wrapped in tin foil and incubated for 90 minutes at 37°C and 100 rpm (on the shaker table). At this time, insoluble blue–purple formazan crystals formed. Subsequently, 100 μL of solvent containing SDS (11%w/v), isobutanol (50%v/v), and 0.02 N hydrochloric acid (50%v/v) was added to all the wells, and the tray was again wrapped in tin foil and set aside on a shaker table at 37°C overnight, to allow dissolution of the blue–purple crystals. On the following day, the absorbance was measured at 595 nm using a plate reader (Labsystems Multiskan MS).
Drug transport studies across Caco-2 monolayer
The transport of valdecoxib across the Caco-2 cell monolayer was determined using HBSS as a medium supplemented with 1% BSA and 10 mM MES (pH 6.0; 2-(N-morpholino) ethanesulfonic acid) or 10 mM HEPES (pH 7.4; 4-2-hydroxyethyl-1-piperazineethanesulfonic acid). Before initiation of experiments and after the completion of the experiment, the barrier properties of the Caco-2 monolayers were assessed by transepithelial electrical resistance (TEER) measured at room temperature (25°C) in a tissue resistance measurement chamber (EndOhm) with a voltmeter (EVOM), both of which were from World Precision Instruments (Sarasota, FL). The cell monolayers were rinsed once in prewarmed HBSS, placed on a shaking plate, and heated to 37°C. The apical side was filled with 500 μL of drug solution (10 μg/mL or 31.81 nmol/ml) prepared in the aforementioned medium, and the basolateral side was filled with 1 mL of the medium. Samples (100 μL) from the apical solution were withdrawn at the start and end of the assays. Samples (100 μL) from the basolateral solution were withdrawn regularly at 30-minute intervals and replaced each time with the same amount of fresh buffer. Sampling was conducted for a period of 120 minutes for the valdecoxib drug solution and for a period of 240 minutes for the valdecoxib microspheres, in anticipation of the longer time required for drug release from them, as inferred from the in vitro drug release results. Transport of valdecoxib from solution and microspheres was investigated at both pH 6.0 and pH 7.4. All the experiments were performed in triplicate.
Estimation of the intracellular contents of the drug after the transport study was conducted by washing the cells along with supporting membrane twice with HBSS buffer and subsequent air-drying. Further, the individual supporting membrane, along with cells, was scraped out using a sharp scalpel and was kept in plastic centrifuge vials prefilled with 1 mL acetonitrile. All the centrifuge vials were swirled for 10 minutes and then centrifuged (Biofuge-15, Heraeus-Sepatech) at 4000 rpm for 10 minutes. The supernatant from all the vials was subjected to drug content analysis by HPLC.
Samples were transferred to HPLC vials and frozen until further analysis. The samples were analyzed using a method reported by Fronza et alCitation24 using a Merck-Hitachi HPLC system, with pump (L-7100), auto-sampler (L-7200), interface D-7000, UV detector L-7400, and column oven L-7350.
Stability studies
In order to assess long-term stability, three different batches of formulations for VSAE-2 were subjected to stability studies as per International Conference on Harmonisation (ICH) guidelines. Coated microspheres were wrapped in aluminum foil laminated on the inside with polyethylene. The samples were kept at 40°C ± 2°C/75% RH ± 5% in a stability chamber (Scope Enterprises, Delhi, India) for a period of 6 months. Samples were withdrawn after an interval of 15 days, 30 days, 90 days, and 180 days and were analyzed for drug content.
Results and discussion
Characterization of microspheres
FTIR spectroscopy
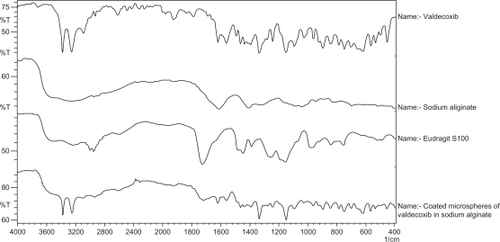
FTIR spectra of the drug valdecoxib, sodium alginate, Eudragit S100, and the coated microspheres are presented in . FTIR spectrum of the pure valdecoxib showed characteristic peaks at 3377 cm−1 and 3250 cm−1 due to N-H stretching of sulfonamide and at 1334 cm−1 and 1150 cm−1 due to S═O stretching vibrations of sulfonamide. The spectrum of Eudragit S100-coated microspheres of valdecoxib containing sodium alginate showed peaks at 3377 cm−1 and 3250 cm−1 due to valdecoxib, at 1728 cm−1 due to Eudragit S100, at 1620 cm−1 due to sodium alginate, and at 1334 cm−1 and 1155 cm−1 due to valdecoxib and Eudragit S100.
Figure 1 Fourier transform infrared spectra of valdecoxib, Eudragit S100, sodium alginate, and the coated microspheres (VSAE-2).
XRD
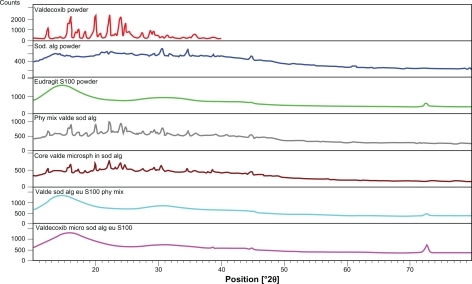
X-ray diffractograms () of valdecoxib indicated the presence of a crystalline material with principal peaks at 22.45° and 24.35° 2θ, whereas both the polymers sodium alginate and Eudragit S100 were found to be amorphous. Although the diffractogram of core microspheres of valdecoxib demonstrated the presence of crystalline drug embedded in the amorphous polymer, the diffractogram of coated microspheres showed an amorphous material devoid of any crystallinity. This could be attributed to a dilution effect by the amorphous polymers.
Figure 2 X-ray diffractograms of sodium alginate, Eudragit S100, their physical mixture, and core and coated microspheres (VSAE-2).
DSC
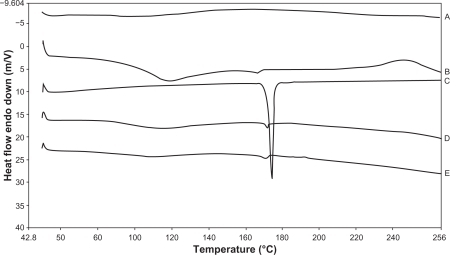
DSC thermograms of the drug, polymers, and microspheres are presented in . Pure valdecoxib exhibited a melting endotherm at 174.25°C. However, thermograms of the coated microspheres demonstrated a depressed, relatively broad endotherm at 168.38°C, which could be attributed to the dilution effect of the amorphous polymers.
Figure 3 Overlay showing differential scanning calorimetry thermograms of Eudragit S100 (A) sodium alginate (B) valdecoxib (C) physical mixture of valdecoxib, sodium alginate, and Eudragit S100 (D) and coated microspheres of valdecoxib in sodium alginate (E) (VSAE-2).
Surface morphology and particle size distribution
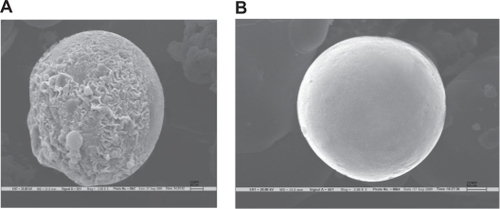
SEM of core valdecoxib microspheres in sodium alginate () revealed mostly spherical, rough-surfaced microspheres, the rough surface being indicative of the surface-associated drug crystals. On the other hand, SEM of the coated microspheres of valdecoxib in sodium alginate revealed mostly spherical and smooth-surfaced microspheres. The average diameter of the coated microspheres in sodium alginate was found to be 35.59 μm (22.67–46.51 μm).
Figure 4 Scanning electron microscopy photograph of formulation. A) VSA-1 uncoated microspheres, B) microspheres coated with Eudragit S100 (VSAE-2) (spatial resolution 2.5 nm).
Drug loading efficiency
The drug loading efficiency of various formulations is presented in . Retrofit analysis of the data reveals that increasing the drug polymer ratio from 1:5 to 1:15 resulted in an increased drug loading efficiency. However, the extent of the increase was not substantial beyond the drug polymer ratio 1:10. Hence, it was kept constant, and the quantity of drug and polymer were varied for compositions VSA-1, VSA-4, and VSA-5. Composition corresponding to composition VSA-1 was utilized for further studies incorporating coating with Eudragit S100 polymer in the core:coat ratio of 1:2.5 (VSAE-1) and 1:5 (VSAE-2). The drug loading efficiency in core micro-spheres of VSA-1 was 83.80% ± 1.62%, which is comparable with that of the core microspheres of chitosan, where it was 80.33% ± 2.24%, as reported in a previous publication.Citation18
In vitro release profile
Core microspheres
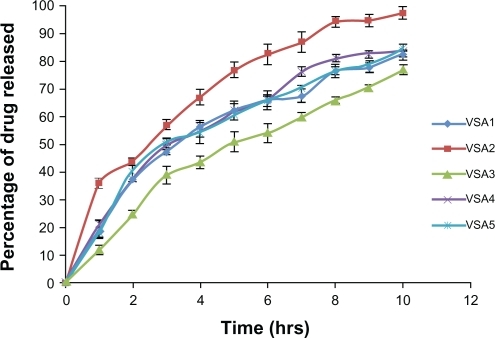
The in vitro release profiles of microspheres containing varying ratios of drug:sodium alginate as core, are presented in . ANOVA showed that there was no significant difference in the rate and extent of drug release from the formulations VSA1, VSA4, and VSA5 (P > 0.05), indicating that changing the quantity of polymer (sodium alginate) and drug, keeping their ratio constant, does not change the in vitro release profile of the microspheres. ANOVAs of VSA1, VSA2, and VSA3 indicated the significant difference in rate and extent of drug release (P < 0.05). The t-test indicated that rate and extent of drug release of VSA1 ≠ VSA2 ≠ VSA3. The difference may be attributed to the change in density of the polymer matrix and hence the diffusional path length.
Figure 5 In vitro release profile of sodium alginate core microspheres in phosphate buffer pH 7.4.
Coated microspheres
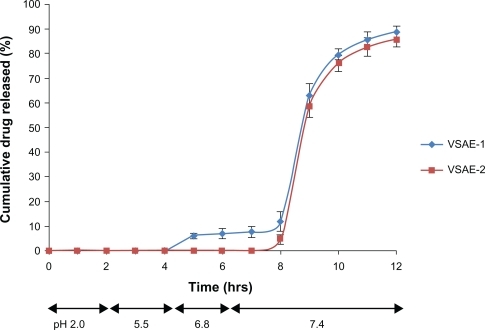
The in vitro release profile of the coated microspheres in release medium of pH 2 revealed the absence of drug release for the 2 hours tested (). In release studies conducted in phosphate buffer (), where the pH was gradually increased from 5.5 to 6.8, the drug release was negligible. However, as the pH of the release medium was raised beyond 7, the drug was released into the medium. This was expected, as Eudragit S100, an enteric copolymer made of methacrylic acid-methyl methacrylate, dissolves at a pH > 7. As the release medium pH was increased to 7.4, VSAE-1 microspheres showed around 12% drug release in a period of 2 hours against 4% drug release by VSAE-2. The amount of coating in VSAE-2 was larger, hence it took longer time to dissolve, as increased thickness of coating in tablets is known to affect oral bioavailability. However, 88% of the drug in VSAE-1 and 84% of that in VSAE-2 were found to be released at pH 7.4 in around 4.5–5 hours. The total drug release in the present case (88% and 84%) is better than that of coated microspheres of chitosan, where it was 82% and 79%, as reported earlier.Citation18
Figure 6 In vitro release profile of Eudragit S-100-coated microspheres in the release medium of different pH.
Drug release kinetics
The drug release data obtained from in vitro release experiments were subjected to various kinetics equations to evaluate the drug release mechanism and kinetics. The kinetic models used were zero order (as cumulative amount of drug released versus time), first order (as log cumulative percentage of drug remaining versus time),Citation25 and Higuchi model (as a cumulative percentage of drug released versus square root of time).Citation26 Moreover, Hixson–Crowell cube root lawCitation27 was used to evaluate the drug release with changes in surface area and diameter of the particles while the mechanism of drug release was also evaluated by plotting first 60% of drug release in Korsmeyer and Peppas (K–P) equation,Citation28 as log cumulative percentage of drug released versus log time, and the exponent “n” was calculated from the slope of the straight line. The release constants and regression coefficients (r2) for all the microsphere formulations using different kinetic equations are listed in .
Table 2 Fit for various drug and sodium alginate microspheres using different kinetic equations for describing release kinetics
A thorough analysis of the table revealed that in vitro release from the sodium alginate microspheres was best explained by the Higuchi equation, as the plots showed the highest linearity. This explains the reason for the slow diffusion rate as the distance for diffusion increases. The value of “n” as per the K–P model for all these microspheres was found to lie between 0.45 and 0.89, the range being indicative of anomalous behavior of drug release, where swelling, diffusion, and erosion play an important role.Citation29,Citation30 The coated microspheres, on the other hand, demonstrated the first-order release, ie, Fickian kinetics and the value of “n” (<0.45) as per the K–P model also complement the same. The dissolution data when plotted as per Hixson–Crowell cube root law indicated a change in surface area and the diameter of the microspheres with progressive dissolution of the matrix as a function of time.
In vitro mucoadhesive properties
Estimation of in vitro mucoadhesion revealed that the time required for complete removal of microspheres from the mucosal tissue was 95 minutes, indicating good mucoadhesive properties of alginate. Various polymer characteristics necessary for mucoadhesion include the presence of strong hydrogen bonding groups (– OH, – COOH), strong anionic charges, high molecular weight, sufficient chain flexibility, and surface free energy properties, favoring spreading onto mucus.Citation31 The interaction between mucoadhesive polymer and mucous membrane can be described by the two steps in the adhesion processCitation32 contact stage, where an intimate contact (wetting) occurs between mucoadhesive polymer and mucous membrane, and in the consolidation stage, where various physicochemical interactions occur to consolidate and strengthen the adhesive joint. Alginate is amongst the “first-generation” mucoadhesive polymer groups, a group that contains hydrophilic macromolecules with numerous hydrogen bond-forming groups.Citation33–Citation35 It is an anionic polymer with numerous carboxyl and hydroxyl groups. The mucous membrane secretes mucin that is rich in sialic acid and hexosamines. Mucin is also made up of sugars with a large number of hydroxyl groups. The concentrations of sialic acid and hexosamines are higher in mucin secreted by transitional mucosa that is immediately adjacent to colon tumor than the mucin secreted by normal mucosa.Citation36 At colonic pH, sialic acid will remain deprotonated and will carry a negative charge. The free carboxyl group of calcium alginate will also be deprotonated at the said pH and thereby repel the negatively charged sialic acid moieties. However, alginic acid also contains a large number of hydroxyl groups, which will form hydrogen bonds with hydroxyl groups of mucin, imparting mucoadhesion. Similar hydrogen bonding and mucoadhesion would also result from amino groups of hexosamines.
Higher concentrations of sialic acid with hydroxyl groups and hexosamines in mucin adjacent to colon tumor may lead to higher mucoadhesion and hence a long residence time of the alginate microspheres adjacent to the tumor. However, it has already been reported that the time required to completely wash-off the valdecoxib-containing chitosan microspheres was quite long (162 minutes)Citation18 compared with the similarly prepared microspheres of alginate (95 minutes). The better mucoadhesion with chitosan microspheres may be attributed to electrostatic attraction between positively charged chitosan and negatively charged mucin and also to the hydrogen bonding due to the presence of – OH and deprotonated –NH2 groups in chitosan.
Drug transport studies
MTT test
The color intensities of the wells were used as the measure of survival of cells after various treatments. The percentage dehydrogenase activity was calculated by the following formula and plotted against the concentration (): P = AT+/AT− × 100, where P is the percentage of dehydrogenase activity, AT+ is the absorbance of wells with test solution, and A T− is the absorbance of wells without test solution.
Figure 7 Percentage dehydrogenase enzyme activity in cell monolayers exposed to varying concentrations of valdecoxib and its coated microspheres.
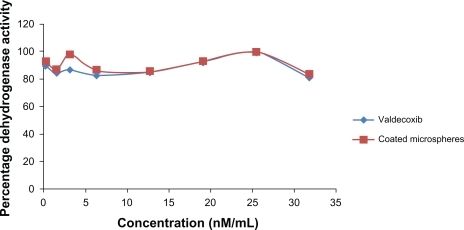
The graph shows that the dehydrogenase activity of the cells with all the samples varied from a minimum of 82% to a maximum of 99%, indicating the absence of toxicity of valdecoxib and its coated microspheres to Caco-2 cells in the concentration range used (0.318–31.8 nM/mL).
Transport studies
The major mechanism for drug uptake across the intestinal mucosa is passive diffusion where the driving force is concentration gradient. Penetration through Caco-2 cells is well established as an in vitro model for evaluating transcellular transport.
Accordingly, Caco-2 cell monolayer was used as a model for studying the cellular uptake of free valdecoxib as well as valdecoxib entrapped in microspheres, but cell uptake will only take place if the cells are alive. Thus, initially, the cytotoxicity of the free drug as well as drug entrapped in microspheres was evaluated in the Caco-2 cell layer (MTT test), and it was observed that neither the free drug nor the entrapped drug interferes with cell metabolism. In other words, the drug does not have any cytotoxicity. The lack of cytotoxicity in Caco-2 cell layer appears to be due to diminished expression of COX-2 by the Caco-2 cells.Citation37,Citation38 It will be appropriate to mention here that Schiffmann et alCitation39 did not observe any cytotoxicity of valdecoxib in HCA-7 cells with high COX-2 expression. Thus, it is quite obvious that valdecoxib will not show any cytotoxicity in Caco-2 cells with diminished COX-2 expression.
The TEER was measured before and after the experiment. A small decrease (8%) in the TEER was observed. TEER (mean ± standard deviation), before and after experiments, was 269 ± 15 Ω cm and 247 ± 34 Ω cm, respectively, indicating that membrane integrity was not compromised during the study.
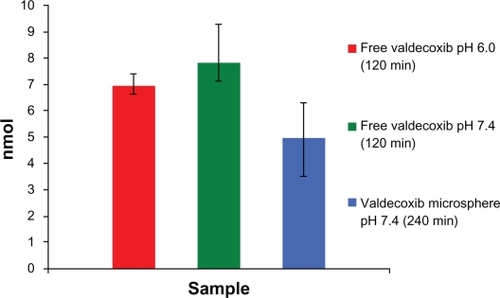
The transport of valdecoxib across Caco-2 cell monolayers was studied under four conditions: valdecoxib solutions at pH 6.0 and 7.4 and valdecoxib microspheres at pH 6.0 and 7.4. The results () suggest that there was no difference between the transport from the drug solutions at pH 6.0 and pH 7.4, indicating that the change in pH had no effect on the permeability of valdecoxib. This is supported by the fact that valdecoxib, a sulfonamide, with pKa of 9.8, would remain in a unionized state at either pH. At 120 minutes the total amount transported from the solutions was 27% and 31%, respectively. The high degree of transport of valdecoxib is supported by a predicted logP of 2.734 and logD of 2.82 at both pH values. The transport of valdecoxib from coated microspheres was studied at pH 6.0 and pH 7.4. Valdecoxib could not be detected in the basolateral chamber when the microspheres were suspended in buffer at pH 6.0. This is consistent with the in vitro release studies showing that no valdecoxib was released at pH < 7.
Figure 8 Amount of valdecoxib transported (cumulative).
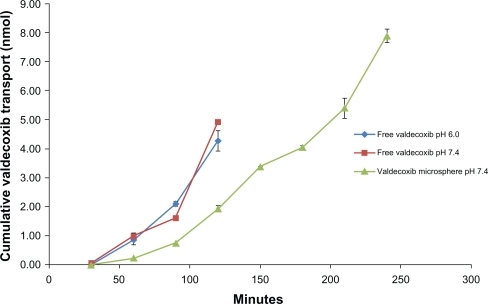
The transport across Caco-2 cell monolayers from valde-coxib microspheres at pH 7.4 was slower than from solutions, probably due to the processes involved in hydration and dissolution of the coating layer and diffusion of valdecoxib out of the sodium alginate microspheres. Thus, a prolonged and sustained release profile was achieved.
The intracellular contents of valdecoxib after transport studies are represented in . The intracellular contents of valdecoxib after transport studies for 120 minutes were significantly (P < 0.05) higher at pH 7.4 than after transport studies with microspheres for 240 minutes. Thus, the microspheres yield a slower release and a lower intracellular content of valdecoxib. This indicates that the local delivery of valdecoxib to colon cancer cells bypasses the systemic delivery of the drug, and hence reduced cardiac side effects are possible by the multiparticulate system studied.
Figure 9 Intracellular contents of valdecoxib after completion of drug transport studies across Caco-2 monolayer.
Stability studies
Selected formulation VSAE-2 was subjected to accelerated stability studies as per climatic zone IV condition for assessing long-term stability as per ICH protocol. After storage, the formulations were analyzed for the drug contents assay. No significant difference in drug contents was observed in the formulation. The degradation rate constant was found to be 6.91E-05 day−1 and t90% was found to be 1534 days, indicating that the said formulation may have a shelf-life of more than 3 years.
Conclusion
The present work was aimed at exploitation of pH-sensitive polymer Eudragit S100 for colon-specific delivery of valdecoxib, a COX-2 inhibitor, and, further, at achieving mucoadhesion of the core microspheres by use of mucoadhesive polymer sodium alginate. The results of the present study indicate that the microspheres prepared using sodium alginate as a core and subsequently coated with Eudragit S100 could be used for the colon targeting of drugs. The presence of sodium alginate in the core imparts mucoadhesion in the colon after the removal of Eudragit coat by the mildly alkaline pH of colonic contents, which might lead to increased residence time of the drug in the affected area. The concepts of colon targeting and mucoadhesion have been tried with mixed results, and one of the reasons for the lack of stellar results might be that the assumed difference in ileum/colon pH is not large enough. However, the chances for success might be improved if the higher pH reported around the tumors means that the pH difference is bigger in colorectal cancer. We believe that further research efforts are warranted to assess the potential of COX-2 inhibitors in mucoadhesive microspheres targeted to release around colorectal tumors and eventually to ascertain the efficacy of the formulation in real-life situations.
Disclosure
The authors report no conflicts of interest in this work.
References
- WeitzJKochMDebusJColorectal cancerLancet200536515316515639298
- DipiroRLTalbertRLYeeGCPharmacotherapy: A pathological ApproachNew YorkMcGraw-Hill2005
- MunkholmPThe incidence and prevalence of colorectal cancer in inflammatory bowlAlimentary Pharmacol Ther200318Suppl 215
- CalvertPMFruchtHThe genetics of colorectal cancerAnn Intern Med200313760361212353948
- SmalleyWRayWADaughertyJGriffinMRUse of nonsteroidal anti-inflammatory drugs and incidences of colorectal cancerArch Intern Med19991591611669927099
- ThunMJHenleyJPatronoCNonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issuesJ Nat Cancer Inst20029425226611854387
- FinckhAAronsonMDCardiovascular risks of cyclooxygenase-2 inhibitors: where we stand nowAnn Internal Med2005142212214
- NelsonNLCelecoxib shown effective in preventing colon polypJ Natl Cancer Inst20069866566716705118
- ArberNCyclooxygenase-2 inhibitors in colorectal cancer prevention: pointCancer Epidemiol Biomarkers Prev2008171852185718708371
- PietroiustiAGiulianoMVitaPFeacal pH and cancer of large bowelGastroenterology1983841273
- ThorntonJRHigh colonic pH promotes colorectal cancerLancet1981317108110836112450
- SamelsonSLNelsonRLNyhusLMProtective role of faecal pH in experimental colon carcinogenesisJ Royal Soc Med198578230233
- JainDPandaAKMajumdarDKEudragit S100 entrapped insulin microspheres for oral deliveryAAPS Pharm Sci Tech20056E100E107
- HoriMOnishiHMachidaYEvaluation of Eudragit-coated chitosan microparticles as an oral immune delivery systemInt J Pharm200529722323415885938
- JainDPandaAKMajumdarDKInsulin loaded Eudragit L100 microspheres for oral delivery: preliminary in vitro studiesJ Biomater Appl20062119521116443624
- CalisSAricaBKasHSHincalAA5-Fluorouracil-loaded alginate microspheres in chitosan gel for local therapy of breast cancerChitosan in Pharmacy and ChemistryMuzzarelliRAAMuzzarelliCGrottammare, ItalyAtec20026569
- RahmanZKohliKKharRKCharacerization of 5-fluorouracil microspheres for colonic deliveryAAPS Pharm Sci Tech20067E1E9
- ThakralNKRayARMajumdarDKEudragit S-100 entrapped chitosan microspheres of valdecoxib for colon cancerJ Mater Sci:Mater Med2010212691269920535630
- LehrCMBowstraJATukkerJJJungingerHEIntestinal transit of bioadhesive microspheres in an in situ loop in the ratJ Controlled Release1990135162
- NielsenCUAmstrupJSteffansenBEpidermal growth factor inhibits glycylsarcosine transport and hPepT1 expression in a human intestinal cell lineAm J Physiol Gastrointestinal Liver Physiol2001281G191G199
- LarsenMLarsenBBFrolundBNielsenCUTransport of amino acids and GABA analogues via the human proton-coupled amino acid transporter, hPAT1: characterization of conditions for affinity and transport experiments in Caco-2 cellsEur J Pharm Sci200835869518621125
- AnderbergEKArturssonPEpithelial transport of drugs in cell culture. VIII Effects of the pharmaceutical surfactant excipient sodium dodecyl sulfate on cell membrane and tight junctional permeability in human intestinal epithelial (Caco-2) cellsJ Pharm Sci1993823923988468683
- FreshneyRICulture of Animal Cells: A Manual of Basic TechniqueNew YorkWiley-Liss1987245256
- FronzaMBrumLWrasseMDevelopment and validation of a RP-HPLC method for the quantitation and dissolution studies of valdecoxibActa Farm Bonaerense200625117122
- WagnerJGInterpretation of percent dissolved-time plots derived from in vitro testing of conventional tablets and capsulesJ Pharm Sci196958125312575349114
- HiguchiTMechanism of sustained action medicationJ Pharm Sci1963521145114914088963
- HixsonAWCrowellJHDependence of reaction velocity upon surface and agitation. I Theoretical considerationsInd Eng Chem193123923931
- BourneDWPharmacokineticsModern PharmaceuticsBankerGSRhodesCTNew YorkMarcel Dekker20026792
- PeppasNAAnalysis of fickian and non-fickian drug release from polymersPharm Acta Helv1985601101114011621
- ChuehHRZiaHRhodesCPOptimization of sotalol floating and bioadhesive extended release tablet formulationsDrug Dev Ind Pharm19952117251747
- PeppasNABurimPASurface, interfacial and molecular aspects of polymer bioadhesion on soft tissuesJ Controlled Release19852257275
- WuSPolymer Interface and AdhesionNew YorkMarcel Dekker1982
- SmartJDKellawayIWWorthingtonHECAn in-vitro investigation of mucosa-adhesive materials for use in controlled drug deliveryJ Pharm Pharmacol1984362952996145763
- SmartJDDrug delivery using buccal adhesive systemAdv Drug Delivery Rev199311253270
- LeeJWParkJHRobinsonJRBioadhesive-based dosage forms: the next generationJ Pharm Sci20008985086610861586
- FilipeMICookeKBChanges in composition of mucin in the mucosa adjacent to carcinoma of the colon as compared with the normal: a biochemical investigationJ Clin Path1974273153184850957
- SmithMLHawcroftGHullMAThe effect of non-steroidal anti-inflammatory drugs on human colorectal cancer cells: evidence of different mechanisms of actionEur J Cancer20003666467410738133
- ShaharLLudmilaSDianaKCelecoxib and curcumin synergistically inhibit the growth of colorectal cancer cellsClin Cancer Res2005116738674416166455
- SchiffmannSMaierTJWobstIThe anti-proliferative potency of celecoxib is not a class effect of coxibsBiochem Pharmacol20087617918718547544