Abstract
The structure, physiology, and fate of living cells are all highly sensitive to mechanical forces in the cellular microenvironment, including stresses and strains that originate from encounters with the extracellular matrix (ECM), blood and other flowing materials, and neighbouring cells. This relationship between context and physiology bears tremendous implications for the design of cellular micro-or nanotechnologies, since any attempt to control cell behavior in a device must provide the appropriate physical microenvironment for the desired cell behavior. Cells sense, process, and respond to biophysical cues in their environment through a set of integrated, multi-scale structural complexes that span length scales from single molecules to tens of microns, including small clusters of force-sensing molecules at the cell surface, micron-sized cell-ECM focal adhesion complexes, and the cytoskeleton that permeates and defines the entire cell. This review focuses on several key technologies that have recently been developed or adapted for the study of the dynamics of structural micro-and nanosystems in living cells and how these systems contribute to spatially-and temporally-controlled changes in cellular structure and mechanics. We begin by discussing subcellular laser ablation, which permits the precise incision of nanoscale structural elements in living cells in order to discern their mechanical properties and contributions to cell structure. We then discuss fluorescence recovery after photobleaching and fluorescent speckle microscopy, two live-cell fluorescence imaging methods that enable quantitative measurement of the binding and transport properties of specific proteins in the cell. Finally, we discuss methods to manipulate cellular structural networks by engineering the extracellular environment, including microfabrication of ECM distributions of defined geometry and microdevices designed to measure cellular traction forces at micron-scale resolution. Together, these methods form a powerful arsenal that is already adding significantly to our understanding of the nanoscale architecture and mechanics of living cells and may contribute to the rational design of new cellular micro-and nanotechnologies.
Introduction
Normal tissue function depends on the coordinated action of a large number and diverse variety of cell types that must proliferate, function, and ultimately die at precisely defined times and places, and this intricate choreography relies heavily on a rich and continuous dialogue between individual cells and between cells and their environment. Cell biologists have long assumed that the words of this conversation are largely biochemical in nature, ie, that cells base their actions primarily on the concentrations of soluble signals to which they are exposed. However, one of the most exciting breakthroughs in cell biology in the past decade is the recognition that the physical microenvironment of a cell, including applied mechanical forces and the geometric arrangement, protein density, and mechanical compliance of the extracellular matrix (ECM), can regulate cell behavior in equally profound ways (CitationDischer et al 2005). For example, individual mesenchymal stem cells differentiate into adipocytes (fat cells) when they are prevented from completely spreading, even when they are saturated with growth factors that would normally induce them to form bone cells (CitationMcBeath et al 2004). Similarly, mammary epithelial cells assemble into normal tubular structures when cultured in relatively compliant (soft) ECMs; when they are cultured in ECMs that are slightly more stiff, these cells form abnormal structures, detach from their neighbors, and proliferate more rapidly – all hallmarks of a developing breast tumor (CitationPaszek and Weaver 2004). For this reason, any attempt to control or exploit cell behavior in a technological context must pay attention to how the physical microenvironment influences cell physiology. Viewed another way, physical crosstalk between the cell and its surroundings represents a design parameter that may be modulated to achieve a desired physiological outcome.
The crosstalk between cells and their surroundings is mediated by an intimately connected and interdependent set of structures that range in size from single molecules to networks that span the whole cell over tens of microns. The coordinated action of these nano-and microstructures lies at the heart of many of the characteristic processes of living cells, including adhesion, motility, and maintenance of shape. For example, cells adhere to specific extracellular matrix (ECM) components through transmembrane integrin proteins, which spatially cluster upon engagement and serve as nucleation sites for focal adhesion complexes between the cell and ECM. These micron-scale complexes contain literally dozens of distinct proteins that physically connect integrins to the cellular cytoskeleton, sense and biochemically transduce mechanical forces, and regulate the presentation and activity of various transmembrane receptors that initiate signaling (CitationIngber 2003; CitationVogel and Sheetz 2006). As cells spread and grip the ECM, they must assemble strong focal adhesion complexes that are capable of supporting increasing tractional forces generated by cellular contractile elements. Equally importantly, directional cell motility on a planar substrate depends on the ability to assemble focal adhesion complexes at the leading edge of the cell and disassemble them at the trailing edge (CitationLauffenburger and Horwitz 1996).
Thus, to dissect the molecular mechanisms through which cells sense and respond to their physical microenvironment, it is critical to understand how all elements in the cytoskeleton, focal adhesions, and the ECM come together in an organized fashion to allow the cell to generate and sense mechanical forces. This requires the ability to precisely and quantitatively measure a wide variety of biophysical properties, including binding and polymerization kinetics, dynamics, and mechanical properties. Traditionally, the vast majority of these measurements have been obtained in purified systems, ie, systems in which molecular components have been purified and removed from their cellular contexts. While these approaches enable precise, quantitative measurements that have contributed greatly to our understanding of cell structure and function, they cannot recapture the spatial and temporal architecture that is essential to transmission of mechanical information in living systems and enables such life-defining processes as locomotion. Over the past decade, however, a remarkable set of technologies has emerged that seeks to satisfy both goals by obtaining quantitative measurements in living cells. Some of these technologies represent increasingly sensitive ways to image and measure the dynamics and mechanics of structures on the molecular scale, and others represent platforms with which to directly manipulate micro-and nanoscale cell structural elements. Here, we review three of these technologies: subcellular laser ablation, advanced live-cell fluorescence imaging methods (including fluorescence recovery after photobleaching and fluorescent speckle microscopy), and cellular micropatterning. These technologies promise both to dramatically advance our ability to understand the conversation between cells and their physical environment and to build our own vocabulary of commands as we attempt to exploit this knowledge in the design of technologies that interface with living systems.
Subcellular laser ablation
Mechanical inputs from the ECM represent one of the most important and well-studied classes of cues in the cellular microenvironment. Because the cellular cytoskeleton confers shape and mechanical properties to the whole cell, it is absolutely critical to a cell’s ability to detect, process, and respond to these mechanical cues. More specifically, by setting its composition, three-dimensional architecture, and contractile state, the cytoskeleton serves as both a passive structural network through which environmental forces are distributed and an active generator of forces that deform the ECM and permit cell spreading and motility. While it is clear that the cytoskeleton is key to cell structure and mechanics in a global sense, considerably less is known about the loads borne by specific cytoskeletal filaments in living cells, or how individual filaments contribute to the structure and mechanics of the whole cell. Answering these questions requires methods capable of selectively probing and disrupting individual cytoskeletal structures in living cells. Ideally, these methods should be capable of interrogating living cells on the submicron length scale in a minimally invasive fashion without killing them in the process.
Subcellular laser ablation has emerged as a powerful tool to accomplish these goals. Pioneered in the late 1970’s by Michael Berns and colleagues (CitationStrahs and Berns 1979; CitationKoonce et al 1982; CitationBerns et al 1991; CitationBerns 1998; CitationBotvinick et al 2004), and then refined over the next two decades by the laboratories of Karsten Konig (CitationKonig et al 1999; CitationTirlapur et al 2001; CitationTirlapur and Konig 2002), Conly Rieder (CitationKhodjakov and Rieder 2001; CitationFaruki et al 2002; CitationMaiato et al 2005), and others, this technique utilizes a focused laser beam to selectively ablate nano-to microscale structures in living cells. While the first applications of this method used continuous-wave visible-range or ultraviolet lasers, more recently, pulsed lasers have become the norm. In particular, ultrashort laser pulses (eg, nano-to femtosecond) are focused through a high-numerical aperture objective lens onto an intracellular target that may be visualized by phase contrast or fluorescence. Material at the focus undergoes nonlinear multiphoton absorption of laser energy, leading to optical breakdown and photoablation. Ideally, the laser energy is delivered quickly enough to prevent energy dissipation by heat transfer, but at a sufficiently low energy and pulse frequency to limit the zone of damage. For this reason, pulse energy, pulse width, and repetition rate have all been shown to be important parameters in limiting the precision of this method. For example, in chemically fixed cells, delivery of femtosecond laser pulses at kilohertz repetition rates and at pulse energies ranging from 1.4 nJ to 2.3 nJ can produce zones of photodamage between 0 and 1 μm. Importantly, the spatial values were determined by transmission electron microscopy (TEM) imaging of cells that had been fixed and fluorescently stained; regions of cells irradiated at the lowest energies (∼1.4 nJ) were rendered nonfluorescent even though subsequent TEM failed to reveal damage, demonstrating that those regions had been photo-bleached rather than damaged (CitationHeisterkamp et al 2005). Thus, in interpreting these experiments, it is important to distinguish between photobleaching and photoablation.
Subcellular laser ablation has emerged as a powerful tool for the measurement of mechanical properties of load-bearing cytoskeletal elements in their living, intracellular context. Perhaps the most closely studied cytoskeletal structures by this method are actomyosin stress fiber bundles, which are contractile structures that connect to the cell-ECM interface and enable cells to exert tractional forces on their surroundings. In one of the first applications of laser ablation in living cells, Berns and coworkers visualized stress fibers by phase contrast microscopy, irradiated them with a laser microbeam, and observed their retraction and subsequent repair on a time scale of several hours (CitationStrahs and Berns 1979). In later work, they showed that the rate of stress fiber repair depended on the integrity of the microtubule and fine actin cytoskeleton and not, surprisingly, cellular protein synthesis (CitationKoonce et al 1982). In recent work, laser ablation has been used to study the mechanics of single stress fibers in living cells and their contributions to cell shape and ECM strain (). Here, the authors visualized the actin cytoskeleton in living endothelial cells using yellow fluorescent protein (YFP)-tagged actin and severed stress fibers at the cell base with femtosecond laser pulses (CitationKumar et al 2006). Following irradiation, the ends of the stress fibers retracted in a straight line (ie, in parallel with the axis of the fiber) with viscoelastic recoil dynamics with characteristic times of 15–20 sec; in some cases, it was possible to puncture, rather than incise, stress fibers, yielding holes with diameters as small as 300 nm that distended into ellipses as the weakened stress fiber retracted. Importantly, the degree to which incision of one stress fiber influenced the cytoskeletal architecture and shape of the rest of the cell depended strongly on the compliance of the ECM on which cells were cultured. For cells cultured on rigid substrates (eg, glass), incision of a single stress fiber, or even multiple parallel stress fibers, produced essentially no rearrangements in cell structure. Conversely, incision of even one stress fiber in cells cultured on compliant (∼4 kPa) polyacrylamide substrates produced cellular elongation and widespread rearrangements of cytoskeletal structures many microns from the site of incision. In the latter case, traction force microscopy measurements revealed that dissipation of tractional stresses into the ECM substrate were concentrated near sites of stress fiber insertion into the ECM; ie, focal adhesion complexes. In related studies, subcellular laser ablation was used to locally dissipate tension on focal adhesions by severing adjacent stress fibers, resulting in accelerated turnover of specific focal adhesion proteins. This result was consistent with results obtained upon global dissipation of cellular tension through the use of compliant culture substrates or through treating the cell with contractility-inhibiting drugs (CitationLele et al 2006a).
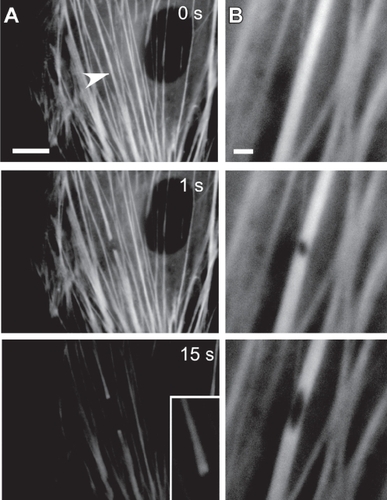
Figure 1 Subcellular laser ablation. In subcellular laser ablation (SLA), a series of high-intensity, ultrashort laser pulses are focused on an intracellular target, resulting in nanoscale destruction of material with minimal damage to surrounding structures and without killing the cell. Here, SLA is used to probe the micromechanics of yellow fluorescent protein-tagged actomyosin stress fiber bundles in an endothelial cell. Laser irradiation results in complete severing (A, arrow) or puncturing (B) of selected stress fibers, leading to profound remodeling on a time scale of 15 s; the ends of severed stress fibers retract (A) and splay apart (inset), and the irradiated region of punctured stress fibers deform into a progressively elongated ellipse (B) (Bar = 2 μm). Reproduced with permission from: CitationKumar S, et al. 2006. Viscoelastic retraction of single stress fibers and its impact on cell shape, cytoskeletal organization, and extracellular matrix mechanics. Biophysical Journal, 90:3762–73. Copyright © 2006, Biophysical Society, http://www.biophysj.org.
Subcellular laser ablation has also been used to sever cellular microtubules in various contexts. For example, laser ablation has been used to disrupt various parts of the mitotic spindle in dividing yeast in order to determine the distribution of tensile and compressive loads among astral and kinetochore microtubules (CitationKhodjakov et al 2004; CitationTolic-Norrelykke et al 2004). More recently, this method has been applied to cortical microtubules, which play mechanical roles in establishing and stabilizing cellular structure. Botvinick et al. (CitationBotvinick et al 2004) used a picosecond laser to irradiate and sever cytoplasmic microtubules tagged with various green fluorescent protein variants, including YFP and cyan fluorescent protein (CFP); as expected, following microtubule incision, one of the severed ends rapidly depolymerized, corresponding to the exposure of a bare minus end. Interestingly, based on subsequent TEM imaging of the ablated cells, the degree of damage differed significantly depending on whether CFP or YFP was used as a fluorescent tag, and both cases differed from green fluorescent protein (GFP). This strongly suggested that the mechanism of photodamage is different for each fluorophore, which in turn results from differences in their respective absorption spectra associated with differences in amino acid composition. Heisterkamp et al. (CitationHeisterkamp et al 2005) used a femtosecond laser to sever a curved GFP-tagged microtubule beneath the nucleus of living cells. Following laser severing, the bent microtubule rapidly straightened and then depolymerized, reflecting the release of stored elastic bending energy.
Fluorescence recovery after photobleaching
An important step in elucidating the influence of the physical microenvironment on cell function is to understand both the dynamics of structural elements in living cells, and how mechanical loads borne by these structures translate into specific biochemical changes at precise cellular times and locations. There are at least three challenges in obtaining these types of data. First, traditional biochemical assays rely on average measurements of populations of cells at fixed points in time, which, by definition, fail to capture information about events in specific portions of specific cells. Second, structures may appear relatively static macroscopically despite the presence of active and rapid molecular-scale remodeling events, such as subunit binding and unbinding; thus, even prolonged imaging of subcellular structures may fail to capture these subtle yet potentially important dynamics. Third, when fluorescently labeled, many cellular structural elements such as microtubules and intermediate filaments fluoresce along their entire length, thereby offering little to no negative contrast to track internal rearrangements in the body of the filament.
Fluorescence recovery after photobleaching (FRAP) has proved a powerful means to address all of these issues. In FRAP, regions of a fluorescently-tagged structure are selectively rendered nonfluorescent through irradiation with a pulse of high-intensity light at the excitation wavelength of the fluorophore, thereby photobleaching the population of fluorescent subunits in the region and rendering them optically invisible. Unlike subcellular laser ablation, FRAP does not destroy material or compromise protein function and only affects fluorescently tagged molecules whose excitation wavelength corresponds to the wavelength of irradiation. Because the loss of fluorescence in a bleached fluorophore is permanent on the time scale of the experiment, any recovery of fluorescence in the targeted structure must be due to a combination of the departure of the bleached subunits and the arrival of new, unbleached fluorophores into the bleached region. Both of these processes may result from either a combination of transport of material within the structure (eg, diffusion) or exchange of subunits between the material and the surrounding medium (ie, binding and unbinding). Thus, given a physical model that describes the dynamics of subunit binding and dynamics, FRAP data (fluorescence vs. time) may be fitted to the model to extract kinetic parameters that describe the binding and transport properties of the labeled molecule.
FRAP has been extensively applied to interrogate the dynamics of complex and heterogeneous cytoskeletal systems, particularly intermediate filaments and actin-based structures. In one study (CitationYoon et al 2001), FRAP was used to probe the network dynamics of keratin intermediate filaments within cultured epithelial cells. Here, cells were transfected with fluorescently-tagged keratin subunits which then incorporated into the tonofibril network (parallel bundles of keratin-rich cables) of cultured epithelial cells, thereby labeling the tonofibrils. When a line was bleached across many tonofibrils, the bleached spots recovered very slowly (half-times of 1–2 hours), thus yielding a relatively stable fiduciary marker to track internal tonofibril dynamics. Surprisingly, bleached zones on adjacent tonofibrils often moved in opposite directions and at different rates, thereby revealing for the first time the complexities of assembly of keratin transport and turnover in these cells. Similarly-inspired studies with the nuclear envelope proteins lamin A and lamin B1 provided insights into the molecular-scale dynamics of envelope assembly and disassembly during cell division (CitationMoir et al 2000). FRAP has also yielded tremendous insight into the internal dynamics of actin-based structures relevant to cell adhesion, mechanics, and motility, including actomyosin stress fiber bundles. Peterson et al (CitationPeterson et al 2004) expressed fluorescently-tagged α-actinin and myosin, two actin-binding proteins found in stress fibers, in cultured fibroblasts and used FRAP to investigate the binding kinetics of these proteins to various regions of the stress fibers. Interestingly, they found that both proteins exchange more rapidly at the center of stress fibers than near their peripheral attachments to focal adhesions; together with high-resolution imaging of myosin distributions during cell contraction, the FRAP data enabled the authors to demonstrate that stress fibers are capable of simultaneously stretching and contracting along their length. In another study, FRAP was used to examine the exchange kinetics of α-smooth-muscle-actin (SMA) in the presence of an SMA peptide fragment (CitationClement et al 2005). By showing that a peptide derived from the amino terminal sequence of SMA slowed recovery of SMA fluorescence and therefore retarded SMA exchange, the authors were able to develop a model in which the amino terminus of SMA facilitates incorporation of the protein into stress fibers.
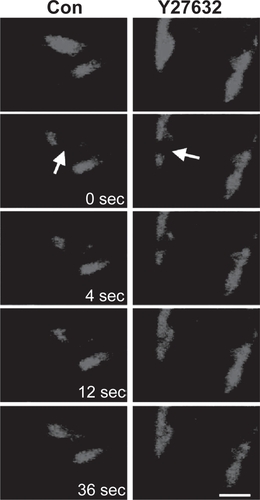
More recently, FRAP measurements have helped to elucidate the influence of the mechanical microenvironment, including mechanical force and ECM rigidity, on the unbinding kinetics of specific focal adhesion proteins at the cell-ECM interface. These studies were made possible by new innovations in the acquisition and analysis of FRAP data that permit separation of contributions from molecular binding and diffusion to the recovery curve (CitationLele et al 2004). In one study, the authors measured dissociation rates of zyxin and vinculin from focal adhesions before and after dissipating intracellular tension by a variety of methods, including adding contractility inhibitors, culturing the cells on compliant ECMs, and severing attached stress fibers by laser ablation (CitationLele et al 2006a). These studies revealed that the unbinding rate of zyxin, but not vinculin, increased when tension on the stress fiber was released. Thus, zyxin was postulated to serve as a molecular mechanosensor whose binding properties, and presumably conformational properties, are highly sensitive to the tensile state of the cell (). This approach has since been extended to the measurement of binding interactions between nuclear histone proteins and chromatin, thereby lending new insight into the biophysical basis of transcriptional control (CitationLele et al 2006b).
Figure 2 Fluorescence recovery after photobleaching. In fluorescence recovery after photobleaching (FRAP), a fluorescent intracellular structure is irradiated at high intensity and at the excitation wavelength, photobleaching the structure and rendering it optically invisible without destroying it. The kinetics with which the photobleached subunits are replaced by fluorescent subunits yields information about subunit transport and binding. Here, green fluorescent protein-tagged zyxin in focal adhesion complexes is photobleached (arrow) in the absence (left column) or presence (right column) of Y27632, a pharmacologic inhibitor of intracellular contractility. Inhibition of contractility dramatically accelerates the turnover of zyxin, demonstrating that the unbinding kinetics of this protein are sensitive to the mechanical properties of the cell. (Bar = 2 μm). Reproduced with permission from: Lele TP, et al. 2006. Mechanical forces alter zyxin unbinding kinetics within focal adhesions of living cells, Journal of Cellular Physiology, 207:187–94. Copyright © 2006 Wiley-Liss, Inc.
Fluorescent speckle microscopy
Fluorescent speckle microscopy (FSM) represents another powerful tool for the characterization of the microscale internal dynamics of cellular structural elements (CitationDanuser and Waterman-Storer 2006) (). In contrast to FRAP, which derives its ability to probe molecular dynamics from the selective inactivation of fluorophores within a continuously fluorescent structure, FSM follows the motions of fluorescent subunits within an otherwise nonfluorescent structure; this discontinuous fluorescence gives the structure a “speckled” appearance from which the name of the method is derived. Intracellular structures are typically made fluorescent in FSM experiments by either microinjecting a very low concentration of fluorescently-tagged proteins into a cell, or by expressing the protein as a GFP fusion construct at very low levels. While FSM may be performed with a wide-field epifluorescence microscope, it is increasingly being used in the context of total internal reflection fluorescence microscopy, which permits high-resolution imaging of structures at the cell-substrate interface such as the cortical cytoskeleton and focal adhesions. FSM has been used with great success to follow the internal dynamics of microtubules (CitationWaterman-Storer and Salmon 1998), actin filaments within cellular networks (CitationPonti et al 2004), and a wide variety of focal adhesion proteins, including vinculin, paxillin, and talin (CitationGeiger et al 2001). Whereas early FSM experiments were analyzed by manually tracking the motions of fluorescent particles, advanced automated tracking algorithms have emerged which allow rapid and highly quantitative interpretation of FSM data, leading to the direct estimation of local cellular viscoelastic properties and measurements of the degree of kinematic coupling between different cellular structural complexes (CitationWaterman-Storer and Salmon 1998).
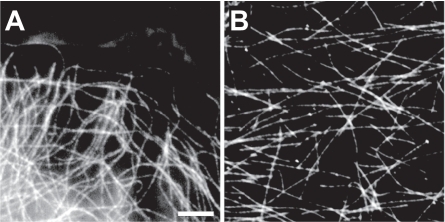
Figure 3 Fluorescent speckle microscopy. In fluorescent speckle microscopy (FSM), an intracellular structure is fluorescently labeled in a sparse fashion, such that the structure takes on a “speckled” appearance with discontinuous regions of fluorescence. The relative dynamics of these speckles reveals quantitative insight into the internal dynamics of the structure. Here, microtubules are labeled for an FSM study in a living epithelial cell (A) and in a purified preparation (B). (Bar = 5 μm). Reproduced with permission from: CitationWaterman-Storer CM, and Salmon ED. 1998. How microtubules get fluorescent speckles. Biophysical Journal, 75:2059–69. Copyright © 1998, Biophysical Society, http://www.biophysj.org.
FSM has uncovered particularly exciting details of the dynamics of the actin cytoskeleton in migrating cells. In a seminal FSM study (CitationPonti et al 2004), rhodamine-actin was microinjected into cultured epithelial cells, and the resulting actin speckles were utilized to track zones of actin polymerization and depolymerization near the leading edges of cells during migration. By carefully tracking the growth, disappearance, and trajectories of actin speckles, the authors constructed high-resolution spatial maps of actin flow which defined two distinct dynamic networks: the lamellipodium, a zone of fast, retrograde actin flow within 1–3 μm from the leading edge of the cell, and the lamella, an adjacent and much broader zone of slower retrograde flow. Subsequent immunofluorescence imaging revealed that the lamellipodium was enriched in the actin branch-promoting factors ADF/cofilin and Arp 2/3, and the lamella was enriched with the contractile proteins myosin II and tropomyosin. These observations, together with pharmacological inhibition data, supported a model in which the lamellipodium gives rise to protrusion of the leading edge, and the lamella drives advancement of the whole cell through actomyosin contraction. Thus, detailed analysis of a simple yet elegant FSM experiment provided rich and novel insight into the contributions of different portions of the actin cytoskeleton to coordinated cell movement.
All of the above methods represent ways of studying the molecular aspects of crosstalk between cells and their physical microenvironment by observing and manipulating structures inside living cells. We now turn to technologies that have been created to tap into this crosstalk by engineering the geometry of the extracellular matrix of cells. In some cases, these approaches have been used to study the role of cellular structure in driving cell function, and in others they have been used to measure physical interactions between cells and their surroundings at the micro-and nanoscale.
Microcontact printing approaches to controlling cell geometry
Direct patterning of extracellular matrix proteins by microcontact printing offers a powerful way to control the geometry of single cells and small groups of cells in two-dimensional culture. Analogous to conventional ink-stamp printing, microcontact printing is based on the controlled deposition of ECM proteins by apposition of a protein-coated stamp with a cell culture substrate (eg, glass) and subsequent passivation of the non-stamped regions of the substrate. By controlling the topographical relief pattern on the stamp, one may create micron-sized islands of defined location, size, and shape that in turn dictate the distribution and geometry of cells cultured on the substrate (CitationChen et al 1997; CitationLauer et al 2001; CitationBrock et al 2003; CitationMcBeath et al 2004). Typically, photolithography is used to create the inverse of the desired relief pattern on a silicon wafer that will serve as a “master.” A layer of photoresist (eg, SU-8) is spun onto the wafer at a controlled thickness. The photoresist layer is then selectively exposed to UV light via a photomask of the desired pattern. Finally, the resist is developed by application of a developer solution that, in the case of negative resist, removes the unexposed regions of the photoresist, leaving behind the desired microtopographical pattern on the wafer. An elastomer, frequently poly(dimethylsiloxane) (PDMS), is then cast and crosslinked atop the master; separation of the polymer and master yields a flexible stamp with the desired microtopographical pattern on its surface. Importantly, many PDMS stamps can be cast from the same master making this a highly cost-effective, reproducible, and high-throughput means of pattering proteins. Often, in order to facilitate adsorption of ECM proteins, the surface of the PDMS stamp is modified from a methyl-terminated surface to a hydrophilic hydroxyl-terminated surface with either air plasma or ultraviolet ozone (CitationYe et al 2006). Following passive adsorption of ECM protein, the coated surface is placed into conformal contact with a substrate, stamping the ECM protein into the desired pattern. Non-stamped regions of the substrate are often passivated with an amphiphilic block copolymer (eg, Pluronic) to inhibit protein adsorption and cell binding.
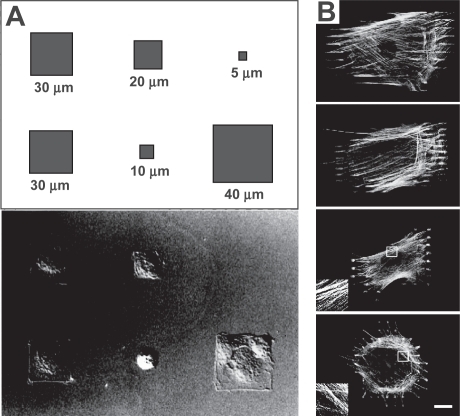
Ingber, Whitesides and coworkers have used microcontact printing to create two-dimensional fibronectin islands of defined geometry for the culture of single cells, a technique that has enabled them to study the effect of cell size on cell fate independent of ECM protein density, cell-cell contacts, or media conditions (CitationSinghvi et al 1994; CitationChen et al 1997) (). Endothelial cells cultured on the adhesive islands conformed to the size and geometry of the stamped ECM, and cell spreading could be promoted by patterning either a single, cell-sized ECM island (eg, 900 μm2) or multiple small focal adhesion-sized islands distributed over a cell-sized area. Importantly, spreading area strongly influenced whether cells proliferated or underwent apoptosis; specifically, as cells were allowed to spread to larger and larger areas, the frequency of proliferating cells increased whether the ECM was presented as a large, continuous island or multiple, discontinuous islands distributed over a large area (CitationChen et al 1997).
Figure 4 Control of cellular structure with micropatterning. Nomarski differential interference contrast image of endothelial cells cultured on square fibronectin islands (A). The cell shapes conform to the geometry of the adhesive islands as defined in the schematic. Reprinted with permission from: CitationChen CS, et al. 1997. Geometric control of cell life and death. Science, 276:1425–8. Copyright © 1997, AAAS. http://www.sciencemag.org. Microcontact printing of focal adhesion-sized islands of fibronectin onto glass (B). Islands of a variety of sizes (20, 10, 6, and 2 μm, from top panel to bottom panel) were used to support the culture of myofibro-blasts, which were then stained for F-actin. While all of the myofibroblasts cultured on the substrate developed actin-based stress fibers, stress fibers in cells cultured on islands larger than 6 μm preferentially incorporated α-smooth muscle actin from the cytosol, indicating higher contractility (not shown). Inset shows a 4 x magnification of F-actin stress fibers. (Bar = 20 μm). Reproduced with permission from: CitationGoffin JM, et al. 2006. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers, The Journal of Cell Biology, 172:259–68. Copyright © 2006, The Rockefeller University Press.
McBeath and coworkers (CitationMcBeath et al 2004) leveraged this approach to investigate the role of cell area in driving differentiation of human mesenchymal stem cells (hMSCs). Remarkably, they found that hMSCs confined to small (1024 μm2) islands appeared round and differentiated exclusively into adipocytes, whereas hMSCs cultured on large (10,000 μm2) islands spread and differentiated exclusively into osteoblasts. Cells cultured on islands of intermediate size (2025 μm2) yielded mixed populations of adipocytes and osteoblasts. The authors hypothesized that these cell area-dependent effects were due to changes in intracellular contractility; ie, cells allowed to spread to larger areas generated more contractile forces than those confined to small areas. To test this hypothesis directly, the authors manipulated hMSC contractility by controlling the activity of the small GTPase RhoA, which strongly promotes actomyosin activation, by transfecting hMSCs with either a constitutively active or a dominant negative version of RhoA. Cells made to express constitutively active RhoA predominantly differentiated into osteoblasts, while cells made to express dominant negative RhoA predominantly differentiated into adipocytes. Remarkably, this result held even when the hMSCs were cultured in media normally expected to promote proliferation or differentiation into the other cell type; eg, hMSCs with abrogated RhoA activity differentiated into adipocytes even when cultured in osteogenic media. Thus, the degree to which hMSCs can generate tensile forces against the ECM figures centrally in the fate decision between adipogenesis and osteogenesis. These studies, which provided fundamental insight into our understanding of hMSC physiology, would not have been possible without microcontact printing technology.
More recent studies have taken advantage of microcontact printing to control the geometry and distribution of individual focal adhesion complexes between the cell and ECM (CitationGoffin et al 2006) (). Here, arrays of fibronectin islets spaced several microns apart with a constant width (1.25 μm) and lengths ranging from 2–20 μm were printed on a rigid, planar substrate. Cells cultured on these substrates formed focal adhesion complexes that assumed the size and shape of the patterned islands. Varying the length of the focal adhesion sites influenced the biochemical composition of the associated intracellular stress fiber bundles; in particular, stress fibers associated with long (>8 μm) islands, and hence long focal adhesion complexes, preferentially incorporated α-smooth muscle actin (α-SMA), a marker of high contractility. The robustness of this system allowed for the simultaneous alteration of several mechanical cues including ECM elasticity and density as well as islet spacing and size. By systematically varying each parameter, the authors demonstrated that islet size dominantly influenced α-SMA localization to stress fibers.
Recent advances in soft lithography: Measuring cell-ECM traction forces
While the earliest applications of soft lithography to cell biology focused almost exclusively on the control of cell size and shape, more recent approaches have sought to measure functional properties, including the spatial distribution of cell-ECM traction forces. For example, Tan and coworkers (CitationTan et al 2003) developed a microfabricated post array detector (mPAD) system to measure cell traction forces and contractility. For this technology, the authors fabricated arrays of deformable, cell-adhesive microposts, schematically similar to a “bed of needles” (). As cells spread and exerted tractional forces on these microposts, they elastically deformed them; from the magnitude and direction of the deformation, the tractional force on a given micropost could be directly determined. The mechanical properties of each mPAD system, and therefore the range of measurable forces, could be tuned through the geometry of the constituent microposts (eg, 2–10 μm in diameter and 3–50 μm in height). Furthermore, by using modified microcontact printing methods to selectively stamp fibronectin islands onto each mPAD, the researchers added another dimension of control by controlling of area available for cell spreading. By varying the area of the stamped islands, the authors demonstrated that the total area of focal adhesions regulated the magnitude of traction force across the whole cell. Moreover, as spreading area increased, total tractional force increased; as expected, tractional force could be altered by manipulating intracellular RhoA activity.
Figure 5 Measurement of cellular traction forces with micropost systems. Scanning electron micrograph of a smooth muscle cell cultured atop an array of flexible PDMS pillars with fibronectin-coated tips (A). The degree to which each post deflects directly reveals the contractile force exerted by the cell at that position on the extracellular matrix. (Bar =10 μm). Reproduced with permission from: CitationTan JL, et al. 2003. Cells lying on a bed of microneedles: an approach to isolate mechanical force, Proceedings of the National Academy of Science of the United States of America, 100:1484–9. Copyright © 2003, PNAS. http://www.pnas.org. Scanning electron micrograph of an epithelial monolayer on an array of closely packed PDMS pillars (B). These measurements revealed that the highest contractile force is produced by the leading edge (inset) of the monolayer; moreover, cells within monolayers exerted greater tractional forces than isolated cells, suggesting cooperative mechanical behavior. (Bar = 20 μm). Reproduced with permission from: Citationdu Roure O, et al. 2005. Force mapping in epithelial cell migration, Proceedings of the National Academy of Science of the United States of America, 102:2390–5. Copyright © 2005, PNAS. http://www.pnas.org.
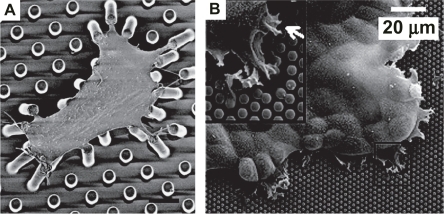
A similar technique was subsequently used to study traction forces exerted by an epithelial monolayer (du CitationRoure et al 2005) (). Here, the authors fabricated a substrate of narrow, densely packed PDMS micropillars (2 μm diameter, 1 μm post-post spacing) in an effort to improve the spatial resolution of the force measurements. In an important extension of the earlier technology, the authors examined cell-ECM tractional mechanics in a monolayer patch of epithelial cells, rather than only those of isolated cells. The authors found that the greatest force was exerted at the edge of the monolayer, and that tractional forces exerted by these cells exceeded those generated by isolated cells, suggesting cooperative mechanical behavior within the monolayer. This finding was later confirmed and significantly extended in a multicellular study with the mPAD technology (CitationNelson et al 2005).
Nanoscale patterning of cell adhesion receptors
Surface patterning can also be a useful tool on a scale unachievable with microcontact printing. The mechanics of cell adhesion have been controlled on the molecular level using nanopatterning. Arnold and colleagues (CitationArnold et al 2004) utilized a diblock copolymer micelle self-assembly system to pattern 8 nm diameter gold nanodots in a rigid hexagonal pattern with precise interparticle spacings (28, 53, 73, 85 nm). The dots were then functionalized with cyclic arginine-glycine-aspartate (RGD)-containing peptides that enabled ligation of transmembrane integrins. Importantly, the small size of each dot permitted conjugation of a single peptide, which in turn bound only a single integrin receptor. Thus, by controlling the spacing of the dots, the authors were able to precisely manipulate spacing between adjacent integrins. As expected, small spacing between dots permitted enhanced integrin clustering, which in turn promoted cell adhesion and spreading as well as formation of robust focal adhesion complexes and stress fiber bundles. Conversely, greater separation distances between the dots (≥73 nm) precluded integrin clustering and severely limited cell attachment and spreading.
Conclusions and future prospects
Traditional approaches to studying cell physiology have emphasized treating cultured cells with defined concentrations of soluble cues and characterizing their responses. While these studies have established the foundation for our modern understanding of receptor-mediated signal transduction, hormonal physiology, and other fundamental aspects of cell biology, it is becoming increasingly clear that these kinds of inputs do not tell the whole story. In particular, the physical microenvironment in which cells exist can provide signals that strongly modulate or even oppose the effects of these soluble cues, suggesting that elements of the physical microenvironment may provide an important but largely underappreciated role in regulating cell behavior and consequently tissue assembly, development, and pathology. This growing recognition is spawning an entirely new generation of cell biological experiments in which parameters such as ECM stiffness, geometry, and topography have been included as experimental variables, which has in turn created a need for entirely new platforms and tools to create the appropriate experimental conditions. Because much of the cellular machinery that senses, processes, and responds to these physical inputs range in size from single molecules to tens of microns, methods capable of probing nano-and microscale phenomena in living cells have proven tremendously useful in elucidating these signaling mechanisms and serving as a handle with which to engineer cell behavior. In this review, we have discussed three foundational technologies that are beginning to make particularly significant impact: subcellular laser ablation, advanced live-cell fluorescence methods including FRAP and FSM, and cellular micropatterning. The first two methods have enabled examination of subcellular mechanics and dynamics at unprecedented spatial and temporal resolution, and the third method has permitted both the precise engineering of cell structure and quantitative measurement of the mechanics of cell-ECM adhesion and traction.
Several challenges remain to be tackled in the future. In the case of subcellular laser ablation, efforts are ongoing to increase the spatial and temporal resolution of material destruction by using pulsed lasers that offer optimal combinations of pulse width, repetition rate, and energy (CitationVogel et al 2005). Moreover, because lasers may be used for capturing particles (eg, through an optical trap), cell stretching, and a variety of other processing modalities, efforts are beginning to emerge to incorporate laser ablation into multimodular laser processing systems, enabling microdissection and capturing capabilities in a single platform (CitationStuhrmann et al 2006). Similarly, because laser ablation is a multiphoton process that may be induced with the same sorts of near-infrared lasers that are typically used for multiphoton imaging, it has become attractive to develop combined systems for imaging and ablation. For example, the same femtosecond laser system was recently used to image Drosophila melanogaster embryos by both third harmonic generation and two-photon modalities as well as to ablate multicellular regions of the embryo (CitationSupatto et al 2005). In this way, the authors were able to correlate expression of a fluorescent, mechano-sensitive reporter gene with local disruptions of the multicellular force balance between cells in different portions of the embryo. Finally, one of the key limitations of subcellular laser ablation is the lack of intrinsic molecular specificity; in other words, all material at the laser focus is destroyed rather than specifically-tagged macromolecules. Here, a complementary technique known as chromophore assisted laser inactivation (CALI) is beginning to address this need; CALI is based on irradiation of a fluorophore-tagged intracellular target at the fluorophore’s excitation wavelength, but at a much higher intensity than would be required for imaging (CitationRajfur et al 2002; CitationTour et al 2003; CitationTanabe et al 2005; CitationTour 2005). The resulting laser-induced photochemical reaction leads to highly localized production of short-lived reactive oxygen species, which then inactivate the tagged protein.
Several new advances are also in progress for FRAP and FSM. First, beyond tracking the dynamics of single structural elements, FRAP and FSM are increasingly being used to study correlations between the dynamics of different structural complexes in relation to one another. For example, Danuser, Waterman-Storer and colleagues have begun to fluorescently label and simultaneously obtain FSM data for both actin and focal adhesion proteins in an effort to understand how focal adhesions mediate mechanical coupling between the cytoskeleton and ECM (CitationDanuser and Waterman-Storer 2006). Moreover, FRAP has been combined with various mechanical measurements or perturbations to gain insight into the connection between intracellular molecular dynamics and communication with the physical microenvironment. Earlier, we discussed the combination of FRAP with SLA and contractility-inhibiting drugs to study mechanical regulation of focal adhesion turnover (CitationLele et al 2006a). FRAP has also recently been combined with osmotic pressure manipulations to probe the effect of mechanical force on lipid mobility in outer hair cells (Citationde Monvel et al 2006), and FRAP has been used in parallel with optical trapping methods to correlate focal adhesion protein turnover with force-induced cytoskeletal remodeling (Citationvon Wichert et al 2003). With the expanding battery of methods to measure and deliver force to cells at ever-smaller length scales, these types of studies will increase and promise to revolutionize our understanding of how cells couple mechanics and biochemistry on the nanoscale.
Finally, there are several future opportunities for micropatterning approaches. One important challenge that remains is to develop patterning strategies that realistically capture the architecture of multicellular tissues. For example, Nelson and coworkers have cultured endothelial cells on micron-to-millimeter-sized ECM islands, thereby permitting adhesion of many cells in a confined area (CitationNelson et al 2005). Careful examination of proliferation rates in different portions of the island revealed that cells proliferate most rapidly at the edges, even when N-cadherin-mediated cell-cell contacts were broken. Finite-element calculations and mPAD force measurements demonstrated that cells at the edges of the islands also experienced the highest net mechanical forces, suggesting an intrinsic connection between mechanical force and cell fate determination. Micropatterning strategies are also increasingly being used to support co-culture of heterogeneous collections of cells, in an effort to work towards “bottom-up” engineering of functional tissues (CitationBhatia et al 1998; CitationBhatia et al 1999; CitationTourovskaia et al 2003). Perhaps the greatest unmet challenge in this area is to develop three-dimensional patterning strategies for single cells with precise control of ECM geometry and biochemistry. The creation of microwell-like structures that simulate a three-dimensional environment but still allow controlled ECM biochemistry while isolating cells appears to be one promising approach (CitationOstuni et al 2001); the culture of cells on substrates with topographically-defined three-dimensional structures is certainly another (CitationNorman and Desai 2005; CitationNorman and Desai 2006).
The intersection between cell biology and the micro-and nanosciences has never been greater. As cell biologists and bioengineers seek to understand the molecular-scale mechanisms that facilitate crosstalk between cells and their physical microenvironment with greater precision and sophistication, tools designed to interface with cells at mesoscopic length scales will become indispensable. While the technologies described in this review all began as highly specialized methods and in many cases were first developed by physicists and engineers, they are all rapidly becoming accessible to traditional cell biologists and are being closely integrated with the standard tools of cell and molecular biology. We are headed toward a future in which the well-rounded practice of cell biology will require comfort and facility with the micro-and nanotechnologies; if the nascent efforts described in this review are any indication, this will be a bright future indeed for our understanding of cellular structure and function.
Acknowledgements
We thank the members of the Kumar laboratory for critical reading of this manuscript. We also gratefully acknowledge the support of the University of California Cancer Research Coordinating Committee and the Arnold and Mabel Beckman Foundation Young Investigator Award.
References
- ArnoldMCavalcanti-AdamEAGlassR2004Activation of integrin function by nanopatterned adhesive interfacesChemphyschem5383815067875
- BernsMW1998Laser scissors, tweezersSci Am2786279532771
- BernsMWWrightWHWiegand SteubingR1991Laser microbeam as a tool in cell biologyInt Rev Cytol1291441917379
- BhatiaSNBalisUJYarmushML1998Microfabrication of hepatocyte/fibroblast co-cultures: Role of homotypic cell interactionsBiotechnol Prog14378879622518
- BhatiaSNBalisUJYarmushML1999Effect of cell-cell interactions in preservation of cellular phenotype: cocultivation of hepatocytes and nonparenchymal cellsFASEB J13188390010544172
- BotvinickELVenugopalanVShahJV2004Controlled ablation of microtubules using a picosecond laserBiophys J8742031215454403
- BrockAChangEHoCC2003Geometric determinants of directional cell motility revealed using microcontact printingLangmuir191611714674434
- ChenCSMrksichMHuangS1997Geometric control of cell life and deathScience276142589162012
- ClementSHinzBDuginaV2005The N-terminal Ac-EEED sequence plays a role in alpha-smooth-muscle actin incorporation into stress fibersJ Cell Sci118139540415769852
- DanuserGWaterman-StorerCM2006Quantitative fluorescent speckle microscopy of cytoskeleton dynamicsAnnu Rev Biophys Biomol Struct353618716689641
- de MonvelJBBrownellWEUlfendahlM2006Lateral diffusion anisotropy and membrane lipid/skeleton interaction in outer hair cellsBiophys J913648116603502
- DischerDEJanmeyPWangYL2005Tissue cells feel and respond to the stiffness of their substrateScience31011394316293750
- du RoureOSaezABuguinA2005Force mapping in epithelial cell migrationProc Natl Acad Sci USA1022390515695588
- FarukiSColeRWRiederCL2002Separating centrosomes interact in the absence of associated chromosomes during mitosis in cultured vertebrate cellsCell Motil Cytoskeleton521072112112153
- GeigerBBershadskyAPankovR2001Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalkNat Rev Mol Cell Biol279380511715046
- GoffinJMPittetPCsucsG2006Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibersJ Cell Biol1722596816401722
- HeisterkampAMaxwellIZMazurE2005Pulse energy dependence of subcellular dissection by femtosecond laser pulsesOpt Express133690616035172
- IngberDE2003Tensegrity I. Cell structure and hierarchical systems biologyJ Cell Sci11611577312615960
- KhodjakovALa TerraSChangF2004Laser microsurgery in fission yeast; role of the mitotic spindle midzone in anaphase BCurr Biol1413304015296749
- KhodjakovARiederCL2001Centrosomes enhance the fidelity of cytokinesis in vertebrates and are required for cell cycle progressionJ Cell Biol1532374211285289
- KonigKRiemannIFischerP1999Intracellular nanosurgery with near infrared femtosecond laser pulsesCell Mol Biol4519520110230728
- KoonceMPStrahsKRBernsMW1982Repair of laser-severed stress fibers in myocardial non-muscle cellsExp Cell Res141375846890458
- KumarSMaxwellIZHeisterkampA2006Viscoelastic retraction of single living stress fibers and its impact on cell shape, cytoskeletal organization, and extracellular matrix mechanicsBiophys J9037627316500961
- LauerLKleinCOffenhausserA2001Spot compliant neuronal networks by structure optimized micro-contact printingBiomaterials2219253211396899
- LauffenburgerDAHorwitzAF1996Cell migration: a physically integrated molecular processCell84359698608589
- LeleTOhPNickersonJA2004An improved mathematical approach for determination of molecular kinetics in living cells with FRAPMech Chem Biosyst11819016783931
- LeleTPPendseJKumarS2006aMechanical forces alter zyxin unbinding kinetics within focal adhesions of living cellsJ Cell Physiol2071879416288479
- LeleTPThodetiCKIngberDE2006bForce meets chemistry: analysis of mechanochemical conversion in focal adhesions using fluorescence recovery after photobleachingJ Cell Biochem9711758316408278
- MaiatoHKhodjakovARiederCL2005Drosophila CLASP is required for the incorporation of microtubule subunits into fluxing kinetochore fibresNat Cell Biol742715592460
- McBeathRPironeDMNelsonCM2004Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitmentDev Cell64839515068789
- MoirRDYoonMKhuonS2000Nuclear lamins A and B1: different pathways of assembly during nuclear envelope formation in living cellsJ Cell Biol15111556811121432
- NelsonCMJeanRPTanJL2005Emergent patterns of growth controlled by multicellular form and mechanicsProc Natl Acad Sci USA10211594916049098
- NormanJJDesaiTA2005Control of cellular organization in three dimensions using a microfabricated polydimethylsiloxane-collagen composite tissue scaffoldTissue Eng113788615871668
- NormanJJDesaiTA2006Methods for fabrication of nanoscale topography for tissue engineering scaffoldsAnn Biomed Eng348910116525765
- OstuniEChenCSIngberDE2001Selective Deposition of Proteins and Cells in Arrays of MicrowellsLangmuir172822834
- PaszekMJWeaverVM2004The tension mounts: mechanics meets morphogenesis and malignancyJ Mammary Gland Biol Neoplasia93254215838603
- PetersonLJRajfurZMaddoxAS2004Simultaneous stretching and contraction of stress fibers in vivoMol Biol Cell15349750815133124
- PontiAMachacekMGuptonSL2004Two distinct actin networks drive the protrusion of migrating cellsScience3051782615375270
- RajfurZRoyPOteyC2002Dissecting the link between stress fibres and focal adhesions by CALI with EGFP fusion proteinsNat Cell Biol42869311912490
- SinghviRKumarALopezGP1994Engineering Cell-Shape and FunctionScience264696988171320
- StrahsKRBernsMW1979Laser microirradiation of stress fibers and intermediate filaments in non-muscle cells from cultured rat heartExp Cell Res1193145570112
- StuhrmannBJahnkeHGSchmidtM2006Versatile optical manipulation system for inspection, laser processing, and isolation of individual living cellsRev Sci Instrum77063116
- SupattoWDebarreDMouliaB2005In vivo modulation of morphogenetic movements in Drosophila embryos with femtosecond laser pulsesProc Natl Acad Sci U S A10210475215657140
- TanJLTienJPironeDM2003Cells lying on a bed of microneedles: an approach to isolate mechanical forceProc Natl Acad Sci U S A1001484912552122
- TanabeTOyamadaMFujitaK2005Multiphoton excitation-evoked chromophore-assisted laser inactivation using green fluorescent proteinNat Methods2503515973419
- TirlapurUKKonigK2002Targeted transfection by femtosecond laserNature418290112124612
- TirlapurUKKonigKPeuckertC2001Femtosecond near-infrared laser pulses elicit generation of reactive oxygen species in mammalian cells leading to apoptosis-like deathExp Cell Res263889711161708
- Tolic-NorrelykkeIMSacconiLThonG2004Positioning and elongation of the fission yeast spindle by microtubule-based pushingCurr Biol141181615242615
- TourO2005EGFP as your targeted ‘hitman’Nat Methods2491215973416
- TourOMeijerRMZachariasDA2003Genetically targeted chromophore-assisted light inactivationNat Biotechnol211505814625562
- TourovskaiaABarberTWickesBT2003Micropatterns of Chemisorbed Cell Adhesion-Repellent Films Using Oxygen Plasma Etching and Elastomeric MasksLangmuir19475464
- VogelANoackJHüttmanG2005Mechanisms of femtosecond laser nanosurgery of cells and tissuesAppl Phys B81101547
- VogelVSheetzM2006Local force and geometry sensing regulate cell functionsNat Rev Mol Cell Biol72657516607289
- von WichertGHaimovichBFengGS2003Force-dependent integrin-cytoskeleton linkage formation requires downregulation of focal complex dynamics by Shp2EMBO J2250233514517241
- Waterman-StorerCMSalmonED1998How microtubules get fluorescent specklesBiophys J752059699746548
- YeHKGuZYGraciasDH2006Kinetics of ultraviolet and plasma surface modification of poly(dimethylsiloxane) probed by sum frequency vibrational spectroscopyLangmuir2218636816460119
- YoonKHYoonMMoirRD2001Insights into the dynamic properties of keratin intermediate filaments in living epithelial cellsJ Cell Biol1535031611331302