Abstract
Nanostructured agglomerated vesicles encapsulating ciprofloxacin were evaluated for modulated delivery from the lungs in a healthy rabbit model. An aliphatic disulfide crosslinker, cleavable by cysteine was used to form cross-links between nanosized liposomes to form the agglomerates. The blood levels of drug after pulmonary instillation of free ciprofloxacin, liposomal ciprofloxacin, and the agglomerated liposomes encapsulating ciprofloxacin were evaluated. The liposomes and agglomerated vesicles showed extended release of drug into the blood over 24 hours, while the free ciprofloxacin did not. The agglomerates also allowed modulation of the drug release rate upon the introduction of cysteine into the lungs post-drug instillation; the cysteine-cleavable agglomerates accelerated their drug release rate, indicated by an increased level of drug in the blood. This technology holds promise for the post-administration modulation of antibiotic release, for the prevention and treatment of pulmonary and systemic infections.
Introduction
Agglomerated vesicles for pulmonary delivery
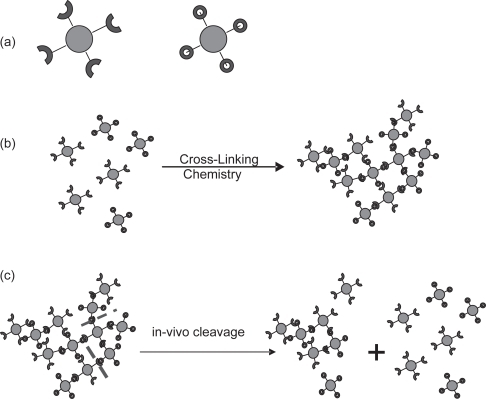
In previous work (CitationBhavane et al 2003) the concept of drug-containing nanoparticles that are chemically cross-linked to each other, to create porous agglomerates (AVT or Agglomerated Vesicle Technology), was introduced and demonstrated in vitro. The core particles for making the agglomerate could be polymeric nanoparticles with drug incorporated into them, or even drug nanoparticles to which the spacer arms have been conjugated. A particularly versatile choice of core nanoparticle is a liposome, that can conveniently be fabricated to present a variety of ligands at the distal ends of spacer PEG chains (CitationZalipsky 1993). The ligands at the distal ends of the spacer arms are then used to cross-link the nanoparticles into larger clusters or agglomerates, by using a suitable chemical linker. The preparation, size, aerodynamic characteristics, and drug release characteristics of such agglomerates in vitro were also demonstrated (CitationBhavane et al 2003). It was shown that by making the cross-links cleavable, post-administration modulation of the drug release rate could be achieved by selective cleavage of the cross links. A description of the agglomeration of liposomes with a cross-linker and the subsequent cleavage of the linkages between the liposomes is illustrated in . We have termed this technology as Agglomerated Vesicle Technology (AVT).
Figure 1 Schematic of core particles bearing ligands (a); agglomeration of core particles by linkers to form agglomerates (b); based on the linker chemistry some can be cleaved in-vivo by components of lung fluid (c).
Progressive cleavage in vitro showed increased release, which even exceeded that from the unagglomerated particles (CitationBhavane et al 2003). A possible reason for this unexpected increase in release could be attributed to the disruption in the structure of agglomerate. Particles in the lung, or in stirred sample in vivo, experience shear forces due to the motion of the suspending fluid. This in turn could lead to the disruption of the liposome bilayers due to shear forces acting over the entire length scale of the agglomerate, tearing out the lipid anchors from the vesicular bilayer and causing gross leakage from the disrupted liposomes.
One of the major shortcomings of these agglomerated vesicles made with the crosslinker DTBP was the need for aggressive cleaving agents to cleave the cross links. In these first-generation agglomerates, DTT (dithiothreitol) was the cleaving agent used. In subsequent work therefore (CitationKarathanasis et al 2005) a dithiobenzyl linkage cleavable by a biologically acceptable agent (cysteine) was developed. The release of ciprofloxacin from such agglomerates was demonstrated. In a departure from the behavior of the DTT cleavable particles, the cysteine-cleavable particles were shown to release drug continuously, possibly due to ongoing cleavage of the linkages by thiols present in the release experiment medium (pulmonary surfactant replacement, Survanta®). When administered in vivo, these particles faciliated post-administration-modulation of drug release. Thus, insulin release with the possibility of controlled post-adminstration changes in the release rate was demonstrated for the first time (CitationKarathanasis et al 2006). However, continuing concerns about the cleavage of this linkage by endogenous cysteine remained. This therefore led us to seek alternative linkages that would not cleave in the presence of endogenous thiols, but would cleave upon the administration of exogenous cysteine, at levels higher than that present normally in the body. The availability of such a linkage would facilitate more complete control over the release of drug from the nanostructured AVT particles.
In this work therefore, an aliphatic disulfide DTSSP was used to evaluate drug release upon cysteine introduction in vivo. In our previous studies of AVT agglomerates (CitationKarathanasis et al 2005) with the dithiobenzyl linkage, the aliphatic disulfide DTSSP was used as a cross-linker. When the cleavage of the particles by cysteine was attempted by the addition of exogenous cysteine, cleavage was indeed observed, but it was not clear whether the cleavage was occurring at the dithiobenzyl site or at the aliphatic disulfide of the DTSSP. In this work therefore, the cleavage of the aliphatic disulfide – DTSSP was studied in vivo, as a candidate for facilitating selective in vivo cleavability of AVT particles.
In previous studies done in our labs (CitationBhavane et al 2003), it has been shown that AVT particles with large geometrical sizes (1–100 + μm) exhibited aerodynamic sizes in the respiratory size range (1–5 μm). It was also shown that the nebulization of these large agglomerates produced minimal damage to the agglomerates on the basis of size and encapsulation of the drug. Hence, we have a high degree of confidence that the AVT particles can be used in inhalation therapy. These particles were instilled in animals in this study since nebulization into the lungs of these animals is difficult.
Liposomal ciprofloxacin vs free ciprofloxacin
Ciprofloxacin, a fluoroquinolone, was chosen for this study because of its potential for inhaled pulmonary delivery. Ciprofloxacin is a potent broad-spectrum antibiotic having good antibacterial activity against most bacteria including Pseudomonas aeruginosa, Klebsiella pneumoniae, Staphylococcus aureus, Streptococci, Mycobacterium tuberculosis, and Mycobacterium avium complex (CitationGay et al 1984; CitationFenlon and Cynamon 1986; CitationSanders et al 1987). While ciprofloxacin has better tissue penetration, phagocytic accumulation and low pH activity than most other antibiotics, it is still not sufficiently active against intracellular pathogens in a single dose. Hence, repeated doses of the drug are prescribed every 8 to 12 hours as an oral or intravenous treatment.
Liposomal encapsulation of ciprofloxacin followed by pulmonary delivery however, greatly improves its efficacy. It has been shown that ciprofloxacin encapsulated in liposomes when administered directly to the lungs as a single dose, is a potent treatment and prophylactic agent against pulmonary pathogens including intracellular ones (CitationWebb et al 1998; CitationWong et al 2003). Wong et al also showed that the survival of mice infected with Francisella tularensis was improved when ciprofloxacin encapsulated in liposomes was aerosolized into the lungs. Indeed, the liposomal drug was active both in “prophylactic” mode (administration pre-infection) and in “treatment” mode (administration post infection). Further, the work showed that the lung-retention of liposomal ciprofloxacin was higher than the free drug, leading to a better prophylactic efficacy in comparison to free ciprofloxacin. Pretreatment of mice up to 24 hours prior to bacterial challenge with liposome-encapsulated ciprofloxacin thus resulted in complete survival.
We anticipated that AVT particles being larger than liposomes would have lower susceptibility to macrophage uptake and therefore improve lung residence time and systemic bioavailability. AVT formulations, liposomal formulations and an unencapsulated ciprofloxacin formulation were therefore evaluated for release of drug from the lungs after instillation in healthy rabbits. Further, to measure lipid elimination as a function of particle size and correlate it to drug release, fluorescently tagged liposomes and agglomerates were instilled in the lungs of rats and the residual fluorescence in lung tissue was assayed over a 48 hour period.
Materials and methods
Materials
1, 2-Dipalmitoyl-sn-Glycero-3-Phosphatidylcholine (DPPC) and methoxypoly (ethylene glycol) – distearoylphosphatidylethanolamine (MPEG-DSPE) were purchased from Genzyme Pharmaceuticals (Cambridge, MA, USA). Cholesterol (CHOL) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Distearoyl phosphoethanolamine [Amino (polyethylene glycol)] (DSPE-PEG-NH2) conjugate was purchased from Avanti Polar Lipids (Alabaster, AL, USA). Fluorescently tagged lipid, N-(7-nitrobenz-2-oxa-1, 3-diazol- 4-yl)-1, 2-dihexadecanoyl-sn- glycero-3-phosphoethanolamine (NBD-PE) was purchased from Invitrogen Corp. (Carlsbad, CA, USA). The cross-linkers dimethyl 3, 3′-dithiobispropionimidate • 2HCl (DTBP) and 3, 3′-Dithiobis[sulfosuccinimidylpropionate] (DTSSP) were purchased from Pierce (Rockford, IL, USA). Ciprofloxacin (Bayer Pharmaceutical Corp., West Haven, CT, USA) was purchased from a local pharmacy as Cipro-IV solution and purified as follows: The pH of Cipro-IV solution was raised from 2.1 to 7 with sodium hydroxide to precipitate the ciprofloxacin. The suspension was then centrifuged at 1500 × g (4000 rpm) for 10 minutes, and the supernatant discarded. The powder was then washed with DI water and centrifuged 4 times to remove lactic acid (an inert excipient in the iv. solution). A final wash and centrifugation with ethanol yielded a wet powder which was collected and dried in a vacuum desiccator. Ciprofloxacin yield was 90%.
All other reagents were purchased from Fisher Scientific (Hampton, NH, USA).
Animals
New Zealand White male rabbits (2.9 ± 0.5 kg) were purchased from Myrtle Rabbitry (Thompson Station, TN, USA). Male Sprague Dawley rats (330–350 gm) were purchased from Harlan (Indianapolis, IN, USA). The use of animals for this study was approved by the Center for Laboratory Animal Medicine and Care (CLAMC) of UTHSC-Houston. Care and handling of the animals was in accordance with all policies of the United States Department of Agriculture (USDA) and U.S. Public Health Service (PHS).
Preparation of ciprofloxacin-loaded liposomes and agglomerates for pharmacokinetic studies
Fabrication and characterization of parent liposomes
Two separate sets of liposomes (~120 mM lipid content) were prepared. A lipid composition of DPPC:CHOL: DSPE-MPEG (57:40:3, mol%) (MPEG terminated, for Stealth® liposomes), and DPPC:CHOL:DSPE-PEG- NH2 (57:40:3, mol%) (Amino terminated, for making AVT) was used. The liposomes were extruded through a 400 nm polycarbonate membrane by the extrusion process described in previous work (CitationBhavane et al 2003). The liposomes were characterized by Dynamic Light Scattering (DLS) using a Brookhaven Instruments BI-9000AT Digital Autocorrelator, a BI-200SM goniometer and a Hamamatsu photomultiplier (Brookhaven Instruments Corp., Holtsville, NY, USA). The light source was a 532 nm, Ti-sapphire, frequency doubled laser. For the DLS measurement, the liposomal suspension was appropriately diluted in saline (150 mM).
Loading of ciprofloxacin into liposomes
Ciprofloxacin was loaded into the liposomes by the ammonium sulfate gradient method (CitationLasic et al 1995). Blank liposomes were first prepared in a 400 mM ammonium sulfate solution. Liposomes were diafiltered for 1–2 hours using 50 nm cutoff tubings with the external phase of the liposomes being replaced with saline (150 mM) at pH of ~5.3 in order to remove ethanol and non-encapsulated ammonium sulfate. For the remote loading procedure, Ciprofloxacin was dissolved in saline (60 mg/ml) at pH 4.2 and 60 ºC, and was gradually added (0.5 ml added every 3–5 minutes) to the liposomal suspension with the temperature maintained at 60 ºC. The loading was terminated after 1 hour by rapidly dropping the temperature using an ice bath. The drop in temperature reduces the permeability of the liposome bilayer thus preventing the transfer of species across the bilayer and entrapping the loaded drug. Finally, the suspension was separated from unencapsulated Ciprofloxacin by overnight dialysis against saline solution (100 times volume of liposomes) at pH 5.3. The final lipid content of the ciprofloxacin-loaded Stealth® liposomes used in the study was approximately 110 mM, and of the liposomes that were later agglomerated was ~50 mM.
Agglomeration of liposomes
Liposomes containing 3% PEG-NH2 conjugate in the lipid bilayer were agglomerated using DTBP or DTSSP as the linker. The amount of DTBP used was a 50 fold molar excess over the number of PEG-NH2 groups on the outer leaflet of the liposomes. The pH of the liposomes was raised from 5.3 to 8.5 (the optimal pH for DTBP activity is in the 8–9 range) with NaOH solution, and the linker was added to the liposomes with constant stirring for 1.5 hours after which the pH was dropped to 5.3 to arrest the cross-linking reaction. DTSSP used for the agglomeration was a 12 fold molar excess over the PEG-NH2 groups on the outer leaflet of the liposomes. The pH was raised from 5.3 to 7.4 (optimal for DTSSP crosslinking activity) and the linker was added to the liposomes and constantly stirred for 1.5 hours.
The size distribution of the agglomerates was determined using Fraunhofer diffraction technique on a Malvern Mastersizer with 100 mm lens (Malvern Instruments Inc., Southborough, MA, USA). The morphology of the agglomerates of DTSSP preparations was examined by negative staining electron microscopy (EM). Copper grids coated with colloidion-carbon and freshly glow discharged were used for sample adsorption. Each grid was floated on a drop of sample for 5 min. Excess fluid was removed by blotting with a filter paper, and the grid was washed for 2 seconds on a drop of water, floated on a drop of 1% uranyl acetate for 15 seconds, and air dried for 2–5 minutes. All electron micrographs were taken with a JEOL JEM1230 electron microscope operating at 80 kV and 56 μA beam current.
Determination of ciprofloxacin
The concentration of ciprofloxacin was determined by HPLC. The HPLC system consisted of a Shimadzu SCL-10Avp liquid chromatograph, SPD-10Avp UV-Vis detector (278 nm) and SIL-10Advp auto-injector. Chromatography was carried out with a Waters Symmetry 5 μm C18 column (150 × 4.6 mm) at room temperature and a mobile phase flow rate of 1.0 mL/minute; The mobile phase was a mixture of 15% (v/v) of acetonitrile and 85% (v/v) of 25 mM sodium phosphate (pH ~ 2.3) buffer; The sample injection volume was 20 μL. Under these assay conditions, ciprofloxacin eluted within 10 minutes.
To determine the amount of ciprofloxacin entrapped within liposomes, the liposomes were disrupted by methanol (30% of total volume) before assay. The resulting solution was then assayed as above.
Preparation of fluorescent tagged liposomes and agglomerates
Fabrication and characterization of fluorescent liposomes
A lipid composition of DPPC:CHOL:DSPE-PEG-NH2:NBD-PE (56:40:3:1, mol%) was used. After ethanol dissolution the lipids were hydrated with 150 mM NaCl solution. The final lipid content of the liposomes was 50 mM. The liposomes were extruded through a 400 nm polycarbonate membrane and measured by Dynamic Light Scattering (DLS).
Agglomeration of liposomes with fluorescent tagged lipids
Liposomes with a lipid content of 50 mM were agglomerated using DTSSP as the linker. The amount of DTSSP used for the agglomeration was a 20 fold molar excess over the PEG-NH2 groups on the outer leaflet of the liposomes. The crosslinking reaction was carried out at a pH of 7.4 with constant stirring for 1.5 hours. The size distribution of the agglomerates was determined using the Fraunhofer diffraction technique.
Pharmacokinetic studies of ciprofloxacin-loaded liposomes and agglomerates
The rabbits were anesthetized by a subcutaneous injection of ketamine/xylazine (40–50/5–10 mg/kg). The rabbits were maintained on 2% isoflurane and O2 (1 lit/minute). The heart rate, temperature and O2 were monitored periodically, and remained stable throughout the course of the experiment.
The marginal ear vein was accessed via an i.v. catheter (24 G). The catheter was taped to the ear. Using a laryngoscope, an endotracheal tube (size 3) was inserted into the trachea, with a urinary catheter acting as a guide. Insertion into trachea was confirmed with a stethoscope, by pumping air into the lungs. The cuff of the endotracheal tube was filled with air to maintain the tube in the trachea. Additionally, the tube was tied to the mouth to retain it in place.
The rabbit’s head was maintained in an upright position and drug formulation was instilled into the lungs with a syringe. The formulation was pushed down by forcing air into the lungs with the syringe several times. 2.2 to 2.8 ml of formulation (corresponding to ciprofloxacin doses of 17 ± 1.6 mg/kg) was instilled.
Blood was drawn from the ear vein before (time 0) and after instillation of drug formulation. Blood samples were centrifuged at 13,400 rpm for 15 minutes to separate the plasma from cell components. The plasma was then diluted 1:4 times with methanol: phosphate buffer (9:1 v/v) to precipitate the proteins present in the sample. The resulting suspension was then centrifuged at 13,400 rpm in a micro-centrifuge for 15 minutes and the supernatant was assayed for ciprofloxacin content by HPLC. Standard curve for Ciprofloxacin was determined by spiking known amounts of ciprofloxacin in rabbit plasma. The lowest detection limit by HPLC was 0.05 μg/ml.
After instillation of the formulations, the rabbits were maintained on 1.5% isoflurane and O2 (1 lit/minute). The isoflurane was stopped 2 hours after the instillation and the rabbits were maintained on O2 (1 lit/minute) and recovered from the anesthesia. The rabbits were fully awake after 5–6 hours from sedation and returned to their cages.
The following formulations were instilled into the lungs:
Free ciprofloxacin solution
Ciprofloxacin was dissolved in normal saline at a concentration of 21.9 mg/ml. The pH of the solution was adjusted to approximately 3.1 to maintain ciprofloxacin in solution. This solution was then instilled into the lungs of the rabbits as described above.
Liposome-encapsulated ciprofloxacin
The instillation of the Stealth® liposomes was as described by the procedure above. The final lipid content of the liposomes that were instilled was ~110 mM.
AVT 1
AVT particles made with the crosslinker DTBP are referred to as AVT 1 in this paper. These agglomerates are not susceptible to cysteine cleavage. They were instilled according to the procedure described above. The final lipid content of the AVT 1 that was instilled was ~50 mM.
AVT 2
AVT particles made with the aliphatic disulfide linker DTSSP are referred to as AVT 2 in this text. Two sets of studies were done with these agglomerates. In the first study the agglomerates were instilled as described above, and their drug release monitored. In the second study, 1 ml cysteine (60 mg/ml) was introduced in to the lungs 90 minutes after instillation of AVT 2. This was done in order to cleave the agglomerates and alter the release of ciprofloxacin into the blood. The rabbits recovered from anesthesia and were returned to their cages after 6 hours.
After 24 hours, the rabbits were anesthetized with ketamine/xylazine (40–50/5–10 mg/kg) and intubated again and cysteine solution was instilled. Blood was drawn at all the time points as described earlier.
Lipid assay of lungs after instillation of particles with fluorescent tagged lipid
Male Sprague Dawley rats were anesthetized with an injection of ketamine (80 mg/kg body wt.) and xylazine (10 mg/kg body wt.) given intraperitoneally. The rats were endotracheally intubated with a 16 G catheter. The endotracheal tube was then connected to a ventilator and under forced ventilations; successful intubation was confirmed if the chest of the rat expanded with the same frequency as the ventilator. 200 μL of the liposomal and agglomerate formulations (50 mM lipid content) with the fluorescent tagged lipids were instilled into the lungs of the rats. The total lipid dosed was 4078.78 ± 506.07 μg/gm of lung wt. In a separate study, 100 μL of cysteine (60 mg/ml) was instilled after 4 minutes from the instillation of the cleavable agglomerates. At 2, 24, and 48 hours post-instillation of the liposomes and agglomerates, the rats were euthanized with an overdose of pentobarbital (200 mg/kg) administered i.p. and the lungs were collected for the lipid assay.
The collected lungs were refrigerated and processed within a few hours of extraction. For the lipid extraction a modification of the method of CitationMatot et al (2003) was used. Briefly, the lungs were weighed, chopped and then homogenized in cold chloroform/methanol (2:1 vol.) mixture. The homogenized tissue was filtered through Whatman filter paper no. 1. The collected filtrate was then assayed for fluorescence in a SpectraMax GeminiXS spectrofluorometer (Molecular Devices Corp, Sunnyvale, CA, USA) with filter settings of 463 nm excitation and 530 nm emission wavelengths. (calibration standards were made by spiking known amounts of the tagged liposomes into the filtrate collected from the lungs of untreated rats.) The fluorescence intensity associated with the known lipid from the liposomes was used to determine the amount of lipid that remained in the lungs after instillation of the liposomes and the agglomerates.
Results and discussion
Characterization of liposomes by DLS
The size of the Stealth® liposomes encapsulating ciprofloxacin was in the range of 160 to 400 nm with a mean of 330 nm. The size of liposomes encapsulating ciprofloxacin that were agglomerated later with DTBP to form AVT 1, was in the range of 150–300 nm with mean of 260 nm. The size of the liposomes that were agglomerated later with DTSSP to form AVT 2, were in the range of 140–350 nm with a mean at 210 nm. The liposomes incorporating fluorescent tagged lipid were in the range of 177–460 nm with a mean at 290 nm. The effective encapsulation and release rate of drug from these preparations was not expected to be significantly different.
Characterization of agglomerates
The particle size distribution, by Fraunhofer Diffraction, of AVT 1 and AVT 2 that were used for the pharmacokinetic studies, and that of the agglomerates used for the lipid assay studies, are shown in . The agglomerate populations for AVT 1 and AVT 2 are broadly distributed from 1–140 μm with somewhat different modes.
Figure 2 Particle size distribution (measured by Fraunhofer Diffraction) of the agglomerates used in the study.
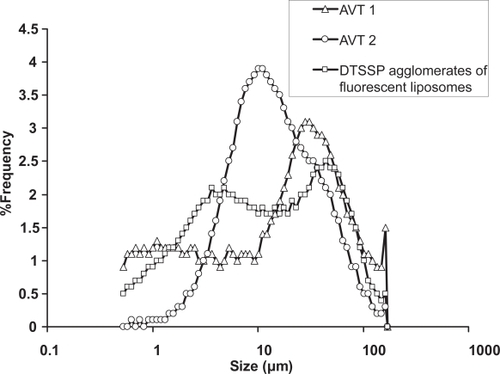
The agglomerates of DTSSP that were used for the lipid assay studies show a bimodal distribution from 1–125 μm, with one mode at 5 μm and the second one at 44 μm. Factors such as pH, buffer, the linker: lipid ratio, stirring rate, etc could affect the agglomeration process ultimately affecting the agglomerate size and the nanostructure of the agglomerate (CitationBhavane et al 2003; CitationKarathanasis et al 2005). The differences in size and the nanostructure could in turn affect the release properties of the microparticle formed. A compact structure of the agglomerate would represent a high concentration of drug-bearing nanoparticles, which may not promote diffusive transport. On the other hand, a more porous, open structure would represent a lower concentration of drug-bearing particles, but a higher diffusive transport rate. Also, due to the dynamic nature of the lungs, large and loose agglomerates maybe more susceptible to rupture and consequent increase in drug release in comparison to smaller compact agglomerates. This would mean more drug being released into the systemic circulation, and a possible improvement in the bioavailability, as the pharmacokinetic studies seem to suggest so.
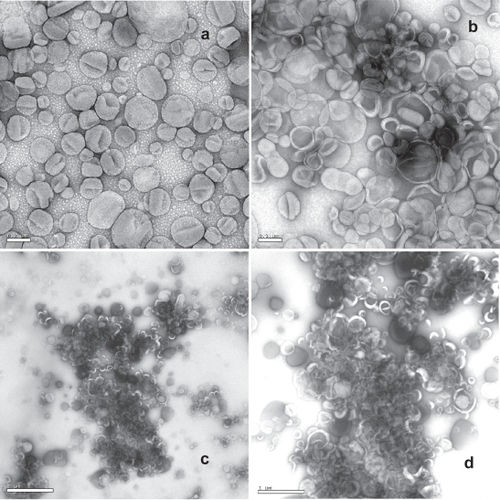
shows the negative stain electron microscope images of liposomes and AVT particles encapsulating ciprofloxacin. is the EM image of the parent liposomes with diameters around 200 nm. Since the length of the PEG anchor (~7 nm) is very small compared to the size of the liposome, linked liposomes are in close proximity as shown in –d. The agglomerates clearly consisted of clusters of cross-linked liposomes incorporating high amounts of the negative stain suggesting high accessibility of the aqueous phase to the interior of the agglomerate structure. A heterogeneous structure can be seen including highly porous as well as compact domains within the agglomerate. As the compact structures were nucleated rapidly, diffusion-limitations dominated during the agglomeration reaction, resulting in the formation of porous structures of the compact “nuclei”. Similar structures are expected for all the agglomerates formed, since the reaction conditions for each were very similar. One therefore expects that drug release properties of each of the agglomerates would also be very similar to each other.
Figure 3 Negative stain electron microscope images (1% uranyl acetate) taken on a JEOL JEM 1230. (a) liposomes at 10 K magnification, scale bar 0.2 μm; (b) agglomerate at 10 K magnification, scale bar 0.2 μm; (c) agglomerate at 2 K magnification, scale bar 2 μm; (d) agglomerate at 4 K magnification, scale bar 1 μm.
Pharmacokinetic studies with ciprofloxacin-loaded liposomes and agglomerates
shows the blood levels of ciprofloxacin after intratracheal instillation of the free ciprofloxacin, liposome-encapsulated ciprofloxacin, AVT 1, AVT 2, and AVT 2 + cysteine treatment. Each data point represents the mean and standard deviation of the group (3 rabbits per group). The difference of the means between groups was verified to be significant by variance analysis (Student’s t-test). A p-value less than 0.05 was used to confirm significant differences at the 95% confidence level.
Figure 4 Pharmacokinetic data for the different formulations tested in rabbits. Arrows indicate time points when cysteine was instilled into the lungs. Inset–detail of the first 500 minutes for the liposomes, AVT 1, AVT 2, and AVT 2 + cysteine (n = 1 for the free ciprofloxacin study. And n = 3 for the other studies). Study with liposomes and free ciprofloxacin was done over 2 days. The AVT studies were done over 1 day. AVT 2 + Cysteine treatment resulted in blood levels of ciprofloxacin different from AVT 2 treatment alone (p < 0.05).
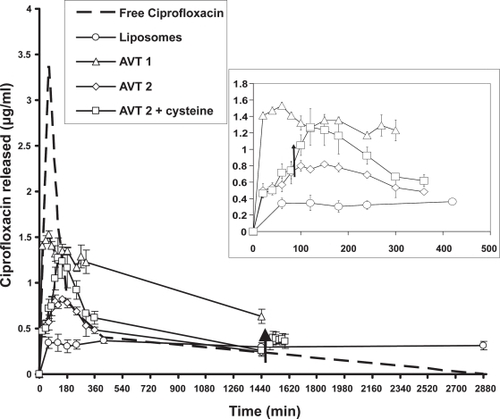
The doses of the drug formulations instilled in the rabbits are tabulated in . It should be noted that ciprofloxacin has a very low solubility at neutral pH. Ciprofloxacin at a concentration of 21.9 mg/ml was instilled in the lungs in this study to compare with the concentrations used in experiments involving liposomes and the agglomerates. To keep ciprofloxacin in solution at this concentration, the pH of the formulation was maintained at ~3.1. The rabbit treated with the free ciprofloxacin formulation showed signs of distress (heavy panting) after instillation. When the lungs were observed post mortem, there were signs of extensive injury highlighted by the darkening of the lung tissue. Hence, no more rabbits were used to evaluate free ciprofloxacin. The lung injury may be attributed to the high concentration of ciprofloxacin that is in immediate contact with the tissue and the low pH of the formulation. To the authors’ knowledge there are no studies reporting inflammation of the lungs or toxicity effects due to administration of ciprofloxacin to the lungs. No such signs of damage were observed in the lungs of the rabbits that were used to evaluate the liposomes and AVT particles. It can well be argued that the damage caused to the lungs by the free ciprofloxacin solution may alter the clearance of the drug and is not a valid comparison to the liposomal and AVT treatment. CitationWong et al (2003) showed the advantage of liposomal ciprofloxacin treatment over free ciprofloxacin in a diseased animal model. For consistency with that study, free ciprofloxacin was evaluated in our work, to show the extended and modulated capabilities of the AVT. Clearly the liposomes and AVT seem to protect the lung from considerable damage at these high levels.
Table 1 Formulation of ciprofloxacin instilled in the lungs of rabbits
A burst in release was observed in the case of the free ciprofloxacin during the 1st hour, which then rapidly dropped to low values and eventually went to zero at the end of 48 hours. Cmax in blood was measured to be 3.38 μg/ml. This rapid release can be attributed to the fact that ciprofloxacin is in direct contact with the tissue and is able to quickly diffuse out in the blood. An assay of the lungs of the rabbit at the end of 48 hours indicated no ciprofloxacin remained in the lungs.
In the case of liposomes, AVT 1, and AVT 2, though an initial burst in release of drug was observed, it was much lower than that for free ciprofloxacin. However, the AVT and liposomes show that they are capable of extended drug release in the blood. Cmax for the liposomal formulation was 0.34 ± 0.06 μg/ml and this level of drug in the blood was maintained through the 48 hour period. Cmax for AVT 1 was observed to be 1.53 ± 0.04 μg/ml with the level gradually falling to 0.63 ± 0.08 μg/ml over 24 hours. Thus the entire curve for AVT 1 was higher than that for liposomes, indicating a much higher bioavailability. AVT 2 exhibited a Cmax between that for AVT 1 and liposomes (0.8 ± 0.07 μg/ml) followed by a gradual drop to 0.24 ± 0.04 μg/ml in 24 hours. The lower initial release-burst for the liposomes and AVT particles is consistent with the fact that the ciprofloxacin is encapsulated within the particles and is not in direct contact with the lung environment. As the drug leaks out of the particles, it is transported rapidly to the bloodstream, but at no point is the free drug concentration in the lung high enough to cause a large spike in the blood levels as observed in the free ciprofloxacin case.
After instillation of cysteine at 90 minutes into the lungs of the rabbits treated with AVT 2, an elevation in release rate was observed. The Cmax attained due to cysteine instillation was 1.27 ± 0.22 μg/ml. The blood levels of drug in animals treated with AVT 2 and then with cysteine are significantly different (p < 0.05) from those for the same formulation with no cysteine treatment. The bioavailability of the AVT 2 particles is clearly improved after cysteine instillation as indicated by the increase in the AUC.
A 40% increase in total release (as measured by the area under the curve) was caused by the cysteine instillation. After 24 hours a repeat cysteine instillation resulted in further triggered release. However, smaller drug amounts were released consistent with overall lower residual drug content in the particles at this extended time post administration. The triggered release due to cysteine addition may be attributed to the cleaving of the disulfide linkages on the DTSSP that links two liposomes. This breakage would result in the rupture of the structure of the microparticle resulting in accelerated drug release. The areas under the concentration-time curves (AUC0 – 24) for AVT 1, AVT 2, AVT 2 + cysteine, and liposomes for 24 hours were computed and are listed in . The AUC’s increase with the size of the particles, suggesting that the larger particles have an improved systemic bioavailability in comparison to the smaller particles.
Table 2 AUC0 – 24 for pharmacokinetic data
Lipid assay of lungs after instillation of particles with fluorescent tagged lipid
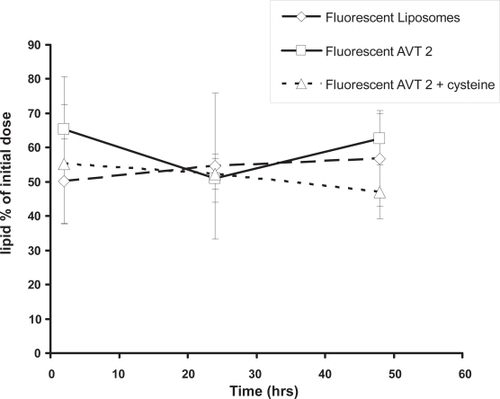
The measurement of lipid retained in the lungs indicated that there is no significant difference between liposomes and agglomerates (). Further, there is no systematic change in lipid levels during the 48 hour observation period; 50%–60% of the initial amount of lipids dosed is observed in the lungs at all times. This result is similar to those of CitationWebb et al (1998) who showed that after instillation of liposomes the amount of lipid observed in the lungs during 24 hours post administration was between 76%–106% of initial lipid dose suggesting that the lipids are not transported into the circulation. We therefore believe that the total lipid (as particles) is remaining in the lungs for the entire time of the experiment (48 hours), and that drug release from the particles into the lungs then results in the transport of free drug into the systemic circulation.
Figure 5 Percentage of original dosed lipid remaining in the lungs on the basis of fluorescence assay of the tagged lipid. n = 3 animals.
Conclusions
The aliphatic linker DTSSP creates a disulfide link in AVT particles that is cleavable by cysteine in vivo, triggering a ciprofloxacin release. Both the AVT particles and unagglomerated liposomes appear to remain in the lung for at least 48 hours after administration. The mechanism by which ciprofloxacin is delivered to the blood therefore appears to be (1) a release of ciprofloxacin from the particles into the lung fluid, followed by (2) transport of the free ciprofloxacin into the bloodstream. Specifically, the transport of encapsulated ciprofloxacin into the blood is not suggested by our results. In contrast, previous work (CitationWong et al 2003) showed that liposomal ciprofloxacin was more effective than free ciprofloxacin against intracellular pathogens such as F. tularensis, suggesting that liposomal drug was possibly transported intact into macrophages. However, the liposomal formulations used by Wong were substantially different from those used in this work, and it is possible that changes in the bilayer composition affect the route of clearance from the lung.
While free ciprofloxacin administered to the lung resulted in significant inflammation, the liposomal and AVT formulations did not. This suggests that the encapsulation effectively prevented the free drug from contacting the lung tissue in large amounts. Progressive release of the drug, however, does not cause significant inflammation either, since the overall free drug concentration at any time is low.
AVT particles also exhibited higher bioavailability than the liposomes, as indicated by the higher AUC for the AVT particles. This is somewhat counterintuitive, since one expects the higher surface area of the parent liposomes to result in higher release rates. However, we have previously observed this phenomenon in vitro, and it is believed that the larger AVT particles, being subject to larger shear forces in the lung, are being disrupted easier than the small liposomes. Thus, AVT particles are expected to have two mechanisms for drug release (1) diffusion of the drug out of the liposomes themselves, and (2) bulk leakage due to bilayer disruption. This mechanism is consistent with the repeated observation that AVT particles have higher systemic bioavailability than liposomes.
The two AVT formulations tested also had somewhat different bioavailabilities. While the reasons for this are yet unclear, they are probably related to the different size distributions of the two AVT particle formulations. The non-cleavable formulation AVT1 had larger particles than the cleavable AVT2. Thus, AVT1 had a higher release and bioavailability, while AVT2 had lower release and bioavailability.
Cleavage of the links in AVT2 by the administration of exogenous cysteine causes an immediate increase in ciprofloxacin release. Further, the release rate appears to remain at this elevated level, reflected in an increased blood concentration of ciprofloxacin at subsequent times. The cumulative release, measured by the AUC, is also higher for the cleaved formulation, consistent with this overall increased release rate.
The ability to accelerate release in vivo by the instillation of exogenous cysteine, cleaving the aliphatic disulfides linking the AVT particles, is the most significant finding of this work, facilitating post-administration modulation of the drug release rate. Thus, one envisions a treatment regimen wherein a patient could inhale a dose sufficient for an extended period of time at a basal release rate, and periodically accelerate this release by the inhalation of a cleaving agent, thus achieving periodic elevations of the blood level of drug, without increasing the overall drug or excipient load.
Abbreviations
| DTSSP | = | Dithiobis [succinimidylpropionate] |
| DTBP | = | dimethyl 3,3′-dithiobispropionimidate |
| DSPE-PEG-NH2 | = | DistearoylPhosphoethanolamine [Amino (Polyethylene Glycol) 2000] |
| DPPC | = | dipalmitoylphosphatidylcholine |
| MPEG-DSPE | = | methoxy poly(ethylene glycol) distearoylphosphatidylethanolamine |
| PEG | = | polyethylene glycol |
| CHOL | = | cholesterol |
| DTT | = | dithiothreitol |
| NBD-PE | = | N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn- glycero-3-phosphoethanolamine |
| DLS | = | dynamic light scattering. |
Acknowledgements
The authors thank the veterinary staff at the University of Texas Health Science Center at Houston for their help in assisting with the animal procedures. The authors also acknowledge the help of Dr. Kim Dunn and Dr. Elmer Bernstam. Financial assistance was provided by the Whitaker Foundation.
References
- BhavaneRKarathanasisEAnnapragadaAV2003Agglomerated vesicle technology: a new class of particles for controlled and modulated pulmonary drug deliveryJournal of Controlled Release9315 2814602418
- FenlonCHCynamonMH1986Comparative in vitro activities of ciprofloxacin and other 4-quinolones against Mycobacterium tuberculosis and Mycobacterium intracellulare. Antimicrob Agents Chemother29386882940969
- GayJDDeYoungDRRobertsGD1984In vitro activities of norfloxacin and ciprofloxacin against Mycobacterium tuberculosis, M. avium complex, M. chelonei, M. fortuitum, and M. kansasiiAntimicrob Agents Chemother269466236748
- KarathanasisEAyyagariABhavaneR2005Preparation of in vivo cleavable agglomerated liposomes suitable for modulated pulmonary drug deliveryJournal of Controlled Release10311597515710508
- KarathanasisEBhavaneRAnnapragadaAV2006Triggered Release of Inhaled Insulin from the Agglomerated Vesicles: Pharmacodynamic Studies in RatsJournal of Controlled Release11321172716765471
- LasicDDCehBStuartMCA1995Transmembrane gradient driven phase transitions within vesicles: lessons for drug deliveryBiochimica Biophysica Acta123914556
- MatotIManevichYAl-MehdiA2003Fluorescence imaging of lipid peroxidation in isolated rat lungs during nonhypoxic lung ischemiaFree Radical Biology & Medicine3467859012633755
- SandersCCSandersWEJrGoeringRV1987Overview of preclinical studies with ciprofloxacinAm J Med822113646829
- WebbMSBomanNLWisemanDJ1998Antibacterial efficacy against an in vivo Salmonella typhimurium infection model and pharmacokinetics of a liposomal Ciprofloxacin formulationAntimicrobial Agents and Chemotherapy42145529449259
- WongJPYangHBlasettiKL2003Liposome delivery of ciprofloxacin against intracellular Francisella tularensis infectionJ Controlled Release9226573
- ZalipskyS1993Synthesis of an end-group functionalized polyethylene glycol-lipid conjugate for preparation of polymer-grafted liposomesBioconjugate Chem442969