
Abstract
Background
The RAS family of oncogenes (KRAS, HRAS, NRAS) are the most frequent mutations in cancers and regulate key signaling pathways that drive tumor progression. As a result, drug delivery targeting RAS-driven tumors has been a long-standing challenge in cancer therapy. Mutant RAS activates cancer cells to actively take up nutrients, including glucose, lipids, and albumin, via macropinocytosis to fulfill their energetic requirements to survive and proliferate.
Purpose
We exploit macropinocytosis pathway to deliver nanoparticles (NPs) in cancer cells harboring activating KRAS mutations.
Methods
NPs were synthesized by the desolvation method. The physicochemical properties and stability of NPs were characterized by dynamic light scattering and transmission electron microscopy. Uptake of fluorescently labelled NPs in wild-type and mutant KRAS cells were quantitively determined by flow cytometry and qualitatively by fluorescent microscopy. NP uptake by KRAS-driven macropinocytosis was confirmed by pharmacological inhibition and genetic knockdown.
Results
We have synthesized stable albumin NPs that demonstrate significantly greater uptake in cancer cells with activating mutations of KRAS than monomeric albumin (ie, dissociated form of clinically used nab-paclitaxel). From pharmacological inhibition and semi-quantitative fluorescent microscopy studies, these NPs exhibit significantly increased uptake in mutant KRAS cancer cells than wild-type KRAS cells by macropinocytosis.
Conclusions
The uptake of albumin nanoparticles is driven by KRAS. This NP-based strategy targeting RAS-driven macropinocytosis is a facile approach toward improved delivery into KRAS-driven cancers.
Background
Mutations of the RAS oncogenes (HRAS, NRAS, and KRAS) are the most frequent mutations in human cancers and are present in 25% of all cancers. Among the three isoforms of RAS genes, KRAS is the most frequent mutated (85% in all RAS-driven cancers). In particular, hyperactivated mutations of RAS oncogenes initiate and drive tumor progression in a significant subset of lung, colorectal, and pancreatic cancers.Citation1 Patients with oncogenic RAS mutations have poor prognosis in colorectal and pancreatic cancers.Citation2,Citation3 As a result, drug delivery targeting RAS-driven tumors has been a long-standing goal for cancer therapy.Citation1,Citation4 However, targeting cancers with RAS mutations has been a significant challenge due to the poor therapeutic index of existing RAS inhibitors.Citation1 Consequently, approaches that enhance delivery and accumulation of RAS-targeting therapeutics would greatly advance and significantly improve patient outcomes.
RAS hyperactivation drives cancer cell survival and proliferation by altering metabolic requirements of the cells to upregulate intracellular uptake; as a result, mutant RAS drives the uptake of numerous solutes. RAS proteins are GTPases that act as “molecular switches”, effectively cycling between binding to guanosine triphosphatase (GTP) and guanosine diphosphatase (GDP).Citation5 During homeostasis, RAS protein toggle between binding to GTP in its active state and GDP in its non-stimulated, inactive state. At rest, RAS protein is bound to GDP in its inactive state. Upon stimulation by growth factor cues, GDP is released and RAS binds to GTP, which subsequently activates downstream RAF/MEK/ERK signaling axis, resulting in cell proliferation. RAS activation also spurs on PI3K and RalGDS effectors, which also stimulate cell proliferation, migration, and survival.Citation6 Then, GTPase activation protein stimulates the breakdown of GTP via hydrolysis, producing GDP to bind and inactivate RAS. During tumorigenesis, specific mutations of RAS cause constitutive RAS-GTP binding and subsequent constitutive activation of downstream effectors resulting in uncontrollable cell proliferation and survival.Citation6
Here, oncogenic RAS reprograms downstream signaling and alters cellular metabolism to fulfill the nutrient requirements of these actively proliferating cancer cells. RAS-transformed cells activate RAF/MEK/ERK signaling to increase glycolysis,Citation7,Citation8 non-oxidative pentose phosphate pathway,Citation7 and hexosamine biosynthesis pathways,Citation9 which increase biomass synthesis needed for cell survival and proliferation. In addition, cells have evolved to use available sources of lipids, proteins, and nutrients for cell survival and proliferation. RAS-driven cancers can scavenge nutrients intracellularly and extracellularly for their survival. Oncogenic RAS proteins stimulate macropinocytosis in quiescent fibroblastsCitation10 and cancer cellsCitation11–Citation13 to “drink” in surrounding bulk fluid and scavenge extracellular lipids and proteins. HRAS overexpressing embryonic fibroblasts and RAS-transformed cells demonstrate increased membrane ruffling characteristic of macropinocytosis and higher intracellular content of lysophospholipids.Citation10,Citation14 Macropinocytosis is a fluid-phase endocytic process whereby cells form membrane ruffles upon extracellular or intracellular cues, resulting in the formation of large diameter vacuoles (0.2–5 μm), or macropinosomes, that are able to transport solutes intracellularly.Citation15,Citation16 RAS-transformed cells use the macropinocytosis program to fulfill their metabolic dependency to maintain their growth and survival. Lipids, glutamine, and in particular albumin have been actively scavenged by RAS-transformed fibroblasts, breast, and pancreas cancers harboring activating RAS mutations.Citation11–Citation13,Citation17,Citation18 Collectively, these findings strongly indicate that albumin is actively macropinoctyosed by RAS-transformed cells in vivo for metabolic needs. Toward this end, is it feasible to exploit this vulnerability of nutrient transport to deliver drug carriers?
Attempts have been reported to deliver therapeutic payloads encapsulated in exosomes and lipoprotein nanostructures to cancer cells via macropinocytosis.Citation19–Citation21 We hypothesize that albumin-based particles can exploit the macropinocytosis pathway of RAS-driven cancer cells for intracellular delivery. Albumin has been used as a carrier to deliver different drugs for various diseases including inflammation and cancer.Citation22 In particular nab-paclitaxel, or albumin-complexed paclitaxel, has been shown in combination therapy to improve overall survival compared to monotherapy and is standard of care for the treatment of advanced pancreatic cancer.Citation23 While albumin has been used, it has not been developed to actively and explicitly used in mechanistic, macropinocytosis-driven delivery into mutant KRAS-specific cancers.
Here, cross-linked albumin nanoparticles (NPs) demonstrate enhanced uptake in oncogenic RAS cells compared to control cells with wild-type RAS by non-ligand mediated macropinocytosis. The physicochemical properties of the NPs are tunable and they are colloidally and physiologically stable. Interestingly, these particles exhibit greater uptake than equivalent amounts of monomeric albumin (ie, present in in vivo dissociated nab-paclitaxel). Through microscopy-based quantification, these NPs colocalize in macropinosomes. Through pharmacological inhibition and genetic knockdown experiments, we demonstrate that these NPs can be endocytosed via RAS-driven macropinocytosis. These collective findings demonstrate that the macropinocytosis pathway of oncogenic RAS cancer cells can be exploited for NP delivery. By understanding this mechanism between the specific cancer pathway and its impact on delivery, it will be feasible to develop drug carriers for pathway-specific, targeted delivery. This work has the impact to greatly improve upon drug delivery and targeting to RAS-driven cancers.
Materials and methods
Synthesis of serum albumin NPs
NPs were synthesized by modified desolvation methods.Citation24 Briefly, bovine serum albumin (BSA, Fraction V, Fisher Scientific) was dissolved in 10 mM sodium chloride solution to make a 1.5% (w/v) BSA solution. The pH of the solution was adjusted to 9.0 with sodium hydroxide. The desolvation agent was a mixture of methanol and ethanol at the ratio of 7:3 (v/v). Then, 4 mL of the desolvation agent was added into 1 mL BSA solution using a syringe pump (KD Scientific) at 1 mL/min under constant stirring. Subsequently, 8% glutaraldehyde solution (Sigma-Aldrich) was added to the system to induce particle cross-linking. Cross-linking process was allowed under stirring at room temperature for 12 hrs. The synthesized NPs were washed with water for three times, using centrifugal filter membrane units (molecular weight cutoff 100 kDa, Amicon). Fluorescent fluorescein isothiocyanate (FITC, ThermoFisher Scientific) and Cyanine 7 (Cy7, Lumiprobe) was conjugated to monomeric BSA according to the manufacturer’s protocol, respectively. Fluorescently labeled nanoparticles (FITC-NP and Cy7-NP) were synthesized by the same procedures as described above using FITC-BSA or Cy7-BSA instead of BSA.
Characterization of NPs
The hydrodynamic diameter and zeta potential of the synthesized NPs were characterized using Zetasizer Nano ZS (Malvern) with 173° backscatter angle. The morphology of the NPs was observed by a transmission electron microscope (TEM). NPs solution was spread on a carbon-coated grid and negatively stained with 2% uranyl acetate. The grid was air-dried and then observed by TEM (FEI Tecnai).
Cell lines and cell culture
Human breast cancer cell lines, MDA-MB-231 harboring oncogenic KRAS mutation and MDA-MB-468 with wild-type KRAS, were purchased from American Type Culture Collection. DMEM/high glucose medium (Corning) supplemented with 10% fetal bovine serum (Gibco) and 100 U/mL penicillin-streptomycin (Gibco) was used to maintain both cell lines. Cells were kept in a humidified atmosphere with 5% carbon dioxide at 37°C.
Measurement of intracellular uptake of NPs by flow cytometry
MDA-MB-231 and MDA-MB-468 cells were seeded in 24-well plates at a density of 4×105cells/well, respectively. After attachment, cells were starved in serum-free medium overnight. To compare the difference in uptake of monomeric albumin and NPs, cells were, respectively, incubated with FITC-BSA and FITC-NP for 2 hrs at different concentrations of 0.5, 1.0, and 1.5 mg/mL (equivalent amount of albumin). To evaluate the inhibitory effect of macropinocytic inhibitor 5-(N-ethyl-N-isopropyl) amiloride (EIPA, Sigma–Aldrich) on the uptake of NPs, cells were pre-treated with 25, 50, and 75 µM EIPA for 30 mins, respectively. Then, cells were incubated with 500 µg/mL FITC-NP for another 30 mins. After each treatment, cells were placed on ice and washed with ice-cold PBS for three times. Cells were collected in PBS buffer and stained with propidium iodide (PI, Sigma–Aldrich). Samples were then analyzed by a flow cytometer (Accuri, BD Biosciences). PI-positive cells were excluded as dead cells.
Measurement of macropinocytic index
Macropinocytotic index was measured by an image-based method with slight modification.Citation25,Citation26 Cells were plated in a 24-well plate with a circular cover glass in each well. Cells were incubated with serum-free medium overnight after reaching 60–70% confluency. The cells were incubated with 1 mg/mL tetramethylrhodamine (TMR)-dextran in serum-free medium for 30 mins. After treatment, cells were washed with ice-cold PBS for five times and fixed with 3.7% formaldehyde solution for 30 mins at room temperature. DAPI solution was added to stain the nucleus of the cells. The cover glass was then placed cell side down onto a glass slide with a drop of mounting medium. Cell images were randomly captured using an Olympus IX-83 inverted fluorescence microscope with a 100× phase objective. A z-stack of frames throughout the entire height of cell monolayers was aquored. The z-stack images were then collapsed to a single image using extended focus imaging projection (CellSens 1.16). To calculate the macropinocytic index, the total cell area was first selected from phase contrast images using the polygon selection tool of ImageJ. Then, the region of interest was applied onto the corresponding TMR-dextran image with thresholding for macropinosomes. The total area of macropinosomes was computed. The macropinocytic index was calculated as the following: macropinocytotic index=total area of macropinosomes/cell number.
Colocalization of NPs with macropinosomes
To quantify the colocalization of NP with macropinosomes, cells were plated in the same way as described in the measurement of the macropinocytic index. Then, cells were incubated with 1 mg/mL TMR-dextran and 1 mg/mL Cy7 labeled NPs simultaneously. A group of cells were treated with 25 µM EIPA to evaluate the effect of macropinocytic inhibition on colocalization. After incubation, cells were fixed and sealed onto glass slides. Cell images were captured using an Olympus IX-83 inverted fluorescence microscope with a 100× phase objective. A z-stack of frames throughout the entire height of cell monolayers was acquired. The z-stack images were then collapsed to a single image using extended focus imaging projection. Background was subtracted with a constant of 1500. For each channel, the contrast was adjusted to the same scale. The Pearson correlation coefficient (PCC) between pixels of TMR-dextran and pixels of Cy7 NPs was analyzed by Cellsense 1.16 (Olympus).
Knockdown of KRAS protein expression and NP uptake
MDA-MB-231 cells were seeded in six-well plates at a density of 1×106cells/well. After 24 hrs, cells were transfected with siRNAs against KRAS gene (SMARTpool: Accell KRAS siRNA, Dharmacon) at a final concentration of 0.5, 1.0, and 1.5 µM in Accell siRNA delivery media, respectively. A non-targeting siRNA was used as a negative control. One hundred and twenty hours after transfection, cells were harvested for Western blot analysis. Cells were lysed using RIPA buffer (Thermo Scientific) with protease inhibitor (Roche). The protein concentrations were determined by BCA protein assay reagent kit (Thermo Scientific). Equivalent amounts of lysates (20 µg total protein per lane) were loaded and separated by 10% SDS-PAGE (Invitrogen Bolt Bis-Tris Plus gel). Then, proteins were transferred onto a low fluorescence PVDF membrane (Invitrogen). In order to probe KRAS and β-actin separately, the membrane was cut into two pieces according to the protein ladder and blocked with 5% non-fat milk. Then, the membranes were incubated with anti-KRAS antibody (Abcam 55,391) and anti-β-actin antibody (Sigma AC-40) at 4°C overnight, respectively, followed by washing and incubating with secondary antibody (IRDye 800 CW, LI-COR) at room temperature for 2 hrs. Finally, the protein bands were visualized using the Odyssey Clx imaging system (LI-COR). Densitometry measurements were calculated using the gel analysis tool in ImageJ.
To evaluate the intracellular uptake of NPs in cells with decreased KRAS expression, MDA-MB-231 cells were seeded in 24-well plates and transfected with 1.0 and 1.5 µM Accell siRNAs, respectively. One hundred and twenty hours after transfection, cells were incubated with 500 µg/mL FITC-NP for 30 mins and subsequently analyzed by flow cytometry using the same method as described above.
Statistical analysis
All experiments were performed in triplicate at minimum. Experiments to measure macropinocytic index and colocalization of NPs with macropinosomes were repeated 10 times. The results were expressed as means ± standard deviation. Statistical significance was analyzed using Students’ t-test for mean differences among the samples.
Results
Physicochemical properties of NPs
Cross-linked albumin NPs were synthesized by desolvation method.Citation24,Citation27 The hydrodynamic size and charge of the NPs were characterized by dynamic light scattering and zeta potential measurements, respectively. The sizes of the NPs were controlled by changing the ratio of methanol and ethanol in the desolvation agent. As shown in , increasing the percentage of ethanol from 0% to 100% increased the mean diameter of the NPs from 36.13±0.27 to 252±0.172 nm. The polydispersity indexes of the NPs were all below 0.25, which suggest that the synthesis resulted in relatively monodisperse populations. The zeta potentials of the NPs ranged from −37.60±0.53 to −46.67±0.32 mV. There is no obvious trend correlating desolvation agent with charge of NPs (). The mean size of nanoparticles synthesized with desolvation agent of methanol: ethanol at the ratio of 7:3 (v/v) was 69.78±0.43 nm with PDI of 0.13±0.01. The zeta potential of the NPs was −42.73±1.20 mV. If the methanol:ethanol ratio was changed, either the size of NPs increased, or the size distribution broadened. Based on published literature regarding the size effects of NPs, small NPs not only extravasate and travel deep into tumor, but also have a higher probability to be internalized by cells.Citation28,Citation29 Thus, cross-linked NPs synthesized with the 7:3 methanol:ethanol ratio were chosen for subsequent experiments.
Figure 1 (A) Size and polydispersity index of nanoparticles synthesized at different EtOH (%). (B) Zeta potential of nanoparticles synthesized at different EtOH (%). (n=3). TEM images of (C) albumin nanoparticles and (D) FITC labeled nanoparticles.
Abbreviations: TEM, transmission electron microscope; FITC, fluorescent fluorescein isothiocyanate; EtOH, ethanol; PDI, polydispersity index.
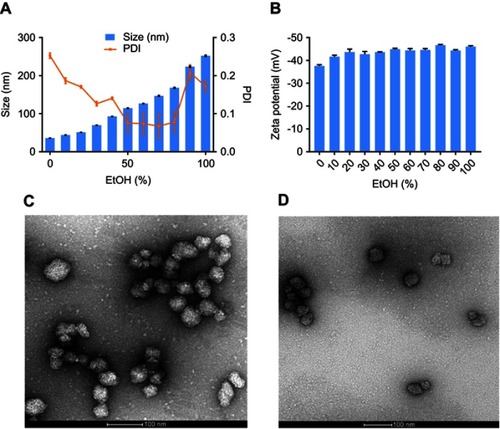
For subsequent studies to investigate cell uptake of particles, cross-linked albumin nanoparticles (FITC-NPs) were prepared using fluorescently labeled monomeric albumin to ensure cross-linked NPs had equivalent amount of FITC/albumin ratio as FITC-BSA. The mean size and zeta potential of the FITC-NPs were 71.41±0.64 nm (PDI 0.10±0.004) and −42.5±0.36 mV, respectively. These measurements indicate that the conjugation of FITC to albumin prior to nanoprecipitation of cross-linked NPs did not impact their physicochemical properties. Both non-labeled and FITC-labeled NPs were observed by transmission electron microscopy (, respectively).
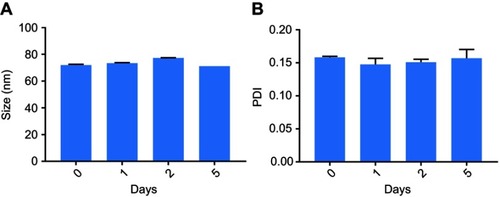
The NPs had a spherical morphology and were evenly distributed. To confirm their physiological stability in vitro, particles were incubated in complete media with 10% FBS at 37°C. The size and PDI of NPs (, respectively) had a negligible change up to 5 days, which confirms their serum stability.
Figure 2 Stability of NPs incubated with 10% FBS in DMEM, (A) size and (B) PDI measured at predetermined time-points. (n=3).
Abbreviations: NPs, nanoparticles; FBS, fetal bovine serum; DMEM, Dulbecco's Modified Eagle's Medium; PDI, polydispersity index.
NPs demonstrate higher uptake than monomeric albumin
Intracellular uptake of FITC-BSA and FITC-NPs was evaluated in MDA-MB-231 cells with oncogenic KRAS mutation G13D () and control MDA-MB-468 cells with wild-type KRAS alleles (). Equivalent amounts of FITC-BSA and FITC-NPs were incubated, and uptake of these fluorescent particles was quantified by flow cytometry. As shown in , the mean fluorescence intensity (MFI) of FITC-BSA and FITC-NPs in both cell lines increased in a dose-dependent manner. However, at each dose, the MFI of cells treated by FITC-NPs was significantly higher than those of cells treated by FITC-BSA. In mutant KRAS MDA-MB-231 cells incubated with 0.5, 1.0, and 1.5 mg/mL monomeric albumin or NPs, the uptake of albumin NPs was 1.37, 1.31, and 1.29-fold higher than that of monomers, respectively. In MDA-MB-468 cells incubated with 0.5, 1.0, and 1.5 mg/mL monomeric albumin or NPs, the uptake of albumin NPs was 1.15, 1.25, and 1.76-fold higher than that of monomeric albumin, respectively. The results indicated that cross-linked NPs demonstrate greater uptake than monomeric albumin.
Figure 3 In vitro uptake of FITC-BSA and FITC-NPs in (A) MDA-MB-231 and (B) MDA-MB-468 cells. (n=3, *p<0.05, **p<0.01).
Abbreviations: FITC, fluorescein isothiocyanate; BSA, bovine serum albumin; NP, nanoparticles.
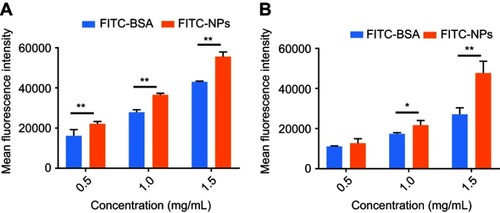
Cross-linked albumin may stabilize and improve upon existing nab-paclitaxel; upon systemic administration, nab-paclitaxel dissociates from particulate form into monomeric albumin and thereby limits the amount of drug delivery without incurring systemic toxicities.Citation17,Citation18,Citation30 From the MFI values in , oncogenic KRAS MDA-MB-231 cells exhibited greater uptake of both monomeric albumin and cross-linked albumin NPs than control MDA-MB-468 cells (p<0.01). The uptake of FITC-NPs by MDA-MB-231 cells was 1.74, 1.69, and 1.16-fold as the uptake in MDA-MB-468 cells at 0.5, 1.0, and 1.5 mg/mL, respectively. Subsequent experiments were performed to support that activating mutations of RAS stimulate greater particle uptake than in cells with wild-type KRAS alleles.
Decreased KRAS protein expression resulted in reduced intracellular uptake of NPs
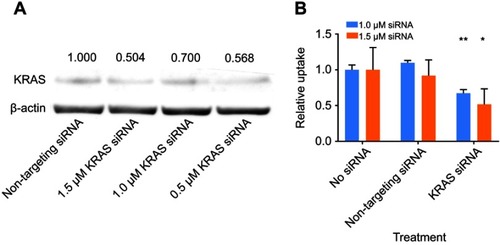
It was next tested if activating KRAS stimulates NP uptake in cancer cells. MDA-MB-231 cells were transfected with siRNA targeting KRAS, and knockdown resulted in decreased KRAS protein expression, as indicated by immunoblotting (). As shown in , siRNA knockdown of KRAS in cells at different concentrations decreased KRAS expression compared to non-targeting siRNA treated cells. Using densitometry to semi-quantify protein expression, KRAS protein expression decreased to 50.4% with 1.5 µM siRNA treatment. Subsequently, the intracellular uptake of FITC-NPs was determined in MDA-MB-231 cells with siRNA-mediated knockdown of KRAS. As shown in , there was no difference between cells without any treatment and cells treated with non-targeting siRNA. The addition of control siRNA did not affect the uptake of FITC-NPs. However, the uptake of FITC-NPs was significantly decreased in cells treated with KRAS- targeting siRNAs, which indicates that direct inhibition of KRAS can negatively impact uptake of FITC-NPs. This finding supports previous studies that demonstrated hyperactivating RAS in cells stimulates their uptake of macromolecules.Citation10,Citation11,Citation13
Figure 4 (A) Protein expression of KRAS and β-actin in MDA-MB-231 cells harvested 120-hr post-transfection by siRNA. Normalized values of the densitometric measurements are listed in the figure. (B) Relative uptake of FITC-NPs by MDA-MB-231 cells 120-hr post-transfection by siRNA. (n=3, *p<0.05, **p<0.01).
Elevated macropinocytosis of NP uptake in oncogenic KRAS cells
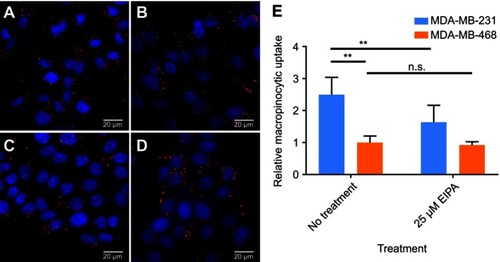
It was then confirmed that oncogenic KRAS MDA-MB-231 exhibited increased macropinocytosis compared to cells with wild-type RAS. The macropinocytic activity of MDA-MB-231 cells and MDA-MB-468 cells was visualized by imaging the uptake of TMR-dextran, a fluorescent tracer for macropinocytosis. Here, TMR-dextran was internalized into cells via macropinosomes, shown as red puncta in (Supplementary material). The amount of macropinosomes, which correlates with the extent of macropinocytosis, was quantified by calculating their macropinocytic index, as developed by Commisso et al.Citation25 As shown in , the relative uptake of TMR-dextran in MDA-MB-231 cells was 2.5-fold higher than in MDA-MB-468 cells (p<0.01). Greater uptake of TMR-dextran in oncogenic KRAS cells was due to macropinocytosis, as confirmed with pharmacological inhibition of EIPA, a canonical inhibitor of macropinocytosis (). After treatment with 25 µM EIPA, the relative macropinocytic index of MDA-MB-231 cells significantly decreased (p<0.01). However, there was no statistical difference in TMR-dextran uptake of MDA-MB-468 cells with or without EIPA (). These results confirm that mutant KRAS cancer cells exhibit greater macropinocytosis than cells with wild-type KRAS alleles.
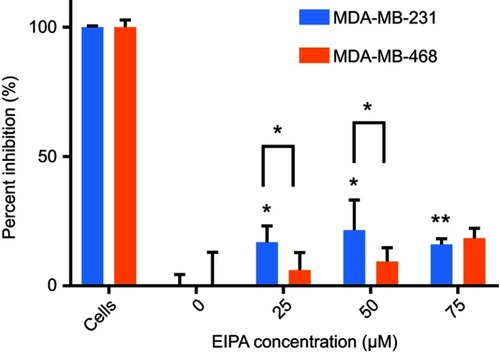
After confirming oncogenic KRAS MDA-MB-231 macropinocytosis tracer TMR-dextran, it was next confirmed that the uptake of our cross-linked albumin NPs is inhibited by a similar mechanism. As shown in , when MDA-MB-231 cells were treated with 25, 50, and 75 µM EIPA, the uptake of FITC-NPs was significantly inhibited by 16.79%, 21.50%, and 16.03%, respectively. For MDA-MB-468 cells (), the inhibition percentages were 6.09%, 9.39%, and 18.40%, respectively. A larger percentage of FITC-NPs was inhibited by EIPA in MDA-MB-231 cells harboring oncogenic KRAS mutation compared to MDA-MB-468 cells with wild-type KRAS. The decrease in uptake due to the EIPA inhibition indicates that cross-linked albumin NPs can be endocytosed by macropinocytosis, and this decrease is more pronounced in mutant KRAS cells; this finding is comparable to other reports demonstrating increased albumin uptake by oncogenic KRAS cancer cells and tumors via macropinocytosis.Citation7,Citation11,Citation12
Figure 5 EIPA inhibited uptake of FITC-NPs in MDA-MB-231 cells and MDA-MB-468 cells. (n=3, *p<0.05, **p<0.01).
Colocalization of NPs with macropinosomes in oncogenic KRAS cells
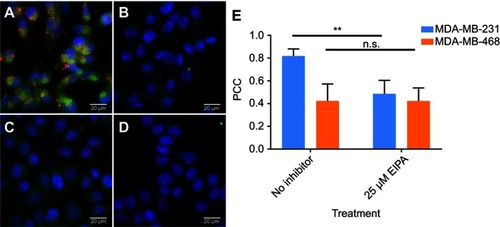
To further confirm that albumin NPs were present in macropinosomes, which are indicative of macropinocytic uptake, Cy7 labeled NPs were synthesized for the colocalization analysis. The hydrodynamic size and zeta potential of Cy7 NPs were 62.22±0.55 nm and −45.70±0.42 mV. Colocalization of Cy7 NPs with TMR-dextran is shown in . As shown in , Cy7 NPs (green) and TMR-dextran (red) were both taken up by mutant KRAS MDA-MB-231 cells. After treatment with EIPA (), the amount of red and green puncta both decreased, indicating the uptake of TMR-dextran and Cy7 NPs was decreased. In , the uptake level of Cy7 NPs and TMR-dextran was similar in MDA-MB-468 cells, regardless of the EIPA inhibition. To determine the localization of albumin NPs in macropinosomes, we quantitatively correlated the colocalization of NPs with TMR-dextran marker via image analysis and calculation of the PCC. The PCC values between the two channels were summarized in . The PCC value for MDA-MB-231 cells was 0.82, which indicates good correlation of pixel intensity distribution between red and green channels. After EIPA inhibition, the PCC value for MDA-MB-231 significantly decreased to 0.49 (p<0.05), which means there was a significant decrease of NP uptake by macropinocytosis. The relative low PCC values in MDA-MB-468 cells indicated that the Cy7 NPs did not colocalize well with TMR-dextran as those in MDA-MB-231 cells. In other words, the fraction of NPs taken up by macropinocytosis was lower than that in oncogenic KRAS MDA-MB-231 cells.
Figure 6 Extended focus images of intracellular uptake of TMR-dextran (red) and Cy7-NPs (green): (A) MDA-MB-231 cells; (B) MDA-MB-231 cells treated with 25 µM EIPA; (C) MDA-MB-468 cells; (D) MDA-MB-468 cells treated with 25 µM EIPA. (E) Pearson correlation coefficient of Cy7-NPs vs TMR-dextran in MDA-MB-231 and MDA-MB-468 cells. (n=10, **p<0.01).
Abbreviation: n.s, no significant difference.
Discussion
Oncogenic RAS, the most abundant overall mutation in cancers, programs cell signaling pathways and tumor progression; however, drugging this difficult-to-target oncoprotein has been the promise and a long-standing goal in cancer therapy. Hyperactive RAS triggers intake of extracellular nutrients needed for biogenesis, cancer cell survival, and proliferation to worsen tumor progression. Studies demonstrated that mice with pancreatic tumors possessing KRAS mutations demonstrated greater uptake of radiolabeled mouse serum albumin than healthy pancreas and wild-type KRAS pancreatic cancers.Citation11,Citation12 This increased uptake of albumin has also been previously observed with RAS-transformed fibroblastsCitation10,Citation11 and glioblastoma.Citation31 Further, it was found that scavenged and catabolized extracellular albumin was the only source of amino acids present in oncogenic KRAS tumors and was needed for cancer cell proliferation and DNA synthesis.Citation12 Davidson et al, confirmed that albumin was macropinocytosed by oncogenic KRAS tumors and then degraded in lysosomes in vivo.Citation12 In addition to albumin and lipid uptake by oncogenic KRAS cells via macropinocytosis,Citation11-13,Citation17 it has been recently demonstrated that larger-sized solutes, including antibodiesCitation21 and lipoprotein NPs,Citation20 also exhibit increased uptake in hyperactive RAS-stimulated cells via macropinocytosis. Constitutively active RAS stimulates intracellular uptake of lipids and proteins through the endocytic route of macropinocytosis, which facilitates transport of large solutes in vesicles up to 5 μm in diameter.
Leveraging the metabolic needs of cancers for solute uptake via macropinocytosis, we wanted to develop NPs that specifically target this mechanism present in RAS-transformed or oncogenic cancer cells toward drug delivery. Building on prior studies demonstrating albumin uptake and catabolism in mutant KRAS cancers, we synthesized stable albumin NPs and tested their ability to enter oncogenic KRAS cancer cells for intracellular delivery. We confirmed that albumin NPs exhibit significantly enhanced uptake in oncogenic KRAS cancer cells compared with control cells with wild-type KRAS. Through extensive pharmacological inhibition, genetic knockdown, and microscopy studies, we demonstrated hyperactivated KRAS is responsible for stimulating macropinocytosis to engulf albumin NPs. By targeting oncogenic RAS-driven macropinocytosis for delivery, there is no need for conjugated ligands on the drug delivery system to facilitate cell binding and internalization. This simplicity of the nanoprecipitation synthesis and lack of conjugation chemistries avoids the challenges of chemistry and scalability and highlights the potential of this carrier for translation.Citation32,Citation33
Interestingly, albumin NPs demonstrated significantly improved uptake compared to the equivalent amount of albumin monomers in cancer cell lines and in particular oncogenic KRAS MDA-MB-231 cells. The current gold standard in albumin-drug carriers, nab-paclitaxel, is a 130 nm paclitaxel-loaded albumin NP in formulation;Citation34 however, upon systemic administration, the particle rapidly dissociates into albumin monomers equivalent to endogenous albumin, and the drug prematurely releases, resulting in promiscuous accumulation in non-tumor tissues and organs and off-target toxicities.Citation17,Citation18,Citation30 Even though macropinocytic activity and uptake of monomeric albumin-drug conjugate were observed in noncancerous and cancer cells with wild-type RAS, both were significantly lower than oncogenic KRAS cells. An albumin-drug conjugate was shown to demonstrated improved therapeutic efficacy in mutant KRAS xenografts compared to wild-type KRAS xenografts via macropinocytosis.Citation35 Coupled with the information that the cross-linked albumin NPs are stable in serum for several days, these findings suggest that cross-linked albumin NPs could deliver a higher amount of albumin and potentially, drug, than non-covalent, monomeric albumin-associated nab-paclitaxel. While these albumin NPs improved uptake in mutant RAS cancer cells compared to controls, future studies against noncancerous cells are needed to confirm cell specificity. Additional studies will be needed to confirm drug encapsulation and stability of the cross-linked particles and compare its efficacy to nab-paclitaxel in cell culture and tumors.
Here, breast cancer cell line MDA-MB-231 is known to harbor a single point mutation at KRAS G13D, while MDA-MB-468 possess wild-type KRAS. The expression levels of total RAS are similar between the two cell lines.Citation36 As a result, from our studies, increased uptake of NPs in MDA-MB-231 is likely due to the mutant KRAS. Although the KRAS mutation is not present in the majority of breast cancers (<5%), considerable evidence has been reported that aberrant RAS activation and signaling can promote breast cancer development.Citation36 Finally, these NPs were formulated to actively target the macropinocytic pathway for intracellular uptake in oncogenic RAS cancers; to fulfill the promise of these NPs for RAS-targeted therapy, it will be necessary to extend delivery to additional cancers with more canonical RAS mutations (eg, KRAS G12V, G12C; NRAS G12D) than the cells used in this current study and to use RAS-targeted therapeutics such as covalent inhibitors,Citation37,Citation38 stapled peptides,Citation39 and other downstream Raf/MEK/ERK inhibitors.Citation1
Conclusion
In this work, the synthesized cross-linked albumin NPs exhibit significant uptake in oncogenic KRAS cancer cells compared with control wild-type KRAS cells. Oncogenic KRAS mutation is responsible for driving macropinocytosis to engulf albumin NPs. This initial step to use stable albumin-based particles to exploit the metabolic vulnerability of oncogenic RAS for intracellular uptake opens new avenues for macropinocytic-driven drug delivery targeting the formidable barrier of oncogenic RAS-driven cancers.
Disclosure
The authors report no conflicts of interest in this work.
Acknowledgment
This work was supported by startup funds generously provided by The University of Texas at Austin College of Pharmacy.
Supplementary material
Figure S1 Macropinocytosis of TMR-dextran (red) in cells (nuclei in blue) observed by fluorescence microscopy. (A) MDA-MB-231 cells without EIPA treatment; (B) MDA-MB-231 cells treated with 25 µM EIPA; (C) MDA-MB-468 cells without EIPA treatment; (D) MDA-MB-468 cells treated with 25 µM EIPA. (E) Relative macropinocytotic index of cells. (n=10, **p<0.05, n.s. is no significant difference)
Abbreviations: TMR, tetramethylrhodamine; EIPA, 5-(N-ethyl-N-isopropyl)amiloride.
References
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13(11):828–851. doi:10.1038/nrd438925323927
- Tao LY, Zhang LF, Xiu DR, Yuan CH, Ma ZL, Jiang B. Prognostic significance of K-ras mutations in pancreatic cancer: a meta-analysis. World J Surg Oncol. 2016;14:146. doi:10.1186/s12957-016-0888-327183870
- Rui Y, Wang C, Zhou Z, Zhong X, Yu Y. K-Ras mutation and prognosis of colorectal cancer: a meta-analysis. Hepatogastroenterology. 2015;62(137):19–24.25911860
- McCormick F. K-Ras protein as a drug target. J Mol Med (Berl). 2016;94(3):253–258. doi:10.1007/s00109-016-1382-726960760
- Milburn MV, Tong L, deVos AM, et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science. 1990;247(4945):939–945. doi:10.1126/science.24069062406906
- Simanshu DK, Nissley DV, McCormick F, Proteins RAS. Their regulators in human disease. Cell. 2017;170(1):17–33. doi:10.1016/j.cell.2017.06.00928666118
- Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149(3):656–670. doi:10.1016/j.cell.2012.01.05822541435
- Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi:10.1038/nature1204023535601
- Dall’Olio F, Malagolini N, Trinchera M, Chiricolo M. Mechanisms of cancer-associated glycosylation changes. Front Biosci (Elite Ed). 2012;17:670–699.
- Bar-Sagi D, Feramisco JR. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 1986;233(4768):1061–1068. doi:10.1126/science.30906873090687
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497(7451):633–637. doi:10.1038/nature1213823665962
- Davidson SM, Jonas O, Keibler MA, et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat Med. 2017;23(2):235–241. doi:10.1038/nm.425628024083
- Kamphorst JJ, Nofal M, Commisso C, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75(3):544–553. doi:10.1158/0008-5472.CAN-14-221125644265
- Kamphorst JJ, Cross JR, Fan J, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A. 2013;110(22):8882–8887. doi:10.1073/pnas.130723711023671091
- Hewlett LJ, Prescott AR, Watts C. The coated pit and macropinocytic pathways serve distinct endosome populations. J Cell Biol. 1994;124(5):689–703. doi:10.1083/jcb.124.5.6898120092
- Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi:10.1146/annurev.biochem.78.081307.11054019317650
- Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell. 2015;162(2):259–270. doi:10.1016/j.cell.2015.06.01726144316
- Tanei T, Leonard F, Liu X, et al. Redirecting transport of nanoparticle albumin-bound paclitaxel to macrophages enhances therapeutic efficacy against liver metastases. Cancer Res. 2016;76(2):429–439. doi:10.1158/0008-5472.CAN-15-157626744528
- Kamerkar S, LeBleu VS, Sugimoto H, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546(7659):498–503. doi:10.1038/nature2234128607485
- Huang JL, Jiang G, Song QX, et al. Lipoprotein-biomimetic nanostructure enables efficient targeting delivery of siRNA to Ras-activated glioblastoma cells via macropinocytosis. Nat Commun. 2017;8:15144. doi:10.1038/ncomms1514428489075
- Ha KD, Bidlingmaier SM, Zhang Y, Su Y, Liu B. High-content analysis of antibody phage-display library selection outputs identifies tumor selective macropinocytosis-dependent rapidly internalizing antibodies. Mol Cell Proteomics. 2014;13(12):3320–3331. doi:10.1074/mcp.M114.03976825149096
- Larsen MT, Kuhlmann M, Hvam ML, Howard KA. Albumin-based drug delivery: harnessing nature to cure disease. Mol Cell Ther. 2016;4:3. doi:10.1186/s40591-016-0048-826925240
- Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703. doi:10.1056/NEJMoa130436924131140
- von Storp B, Engel A, Boeker A, Ploeger M, Langer K. Albumin nanoparticles with predictable size by desolvation procedure. J Microencapsul. 2012;29(2):138–146. doi:10.3109/02652048.2011.63521822329480
- Commisso C, Flinn RJ, Bar-Sagi D. Determining the macropinocytic index of cells through a quantitative image-based assay. Nat Protoc. 2014;9(1):182–192. doi:10.1038/nprot.2014.00424385148
- Wang JTH, Teasdale RD, Liebl D. Macropinosome quantitation assay. MethodsX. 2014;1:36–41. doi:10.1016/j.mex.2014.05.00226150932
- Weber C, Coester C, Kreuter J, Langer K. Desolvation process and surface characterisation of protein nanoparticles. Int J Pharm. 2000;194(1):91–102.10601688
- Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng. 2012;14:1–16. doi:10.1146/annurev-bioeng-071811-15012422524388
- Shang L, Nienhaus K, Nienhaus GU. Engineered nanoparticles interacting with cells: size matters. J Nanobiotechnology. 2014;12:5. doi:10.1186/1477-3155-12-524491160
- Bhattacharyya J, Bellucci JJ, Weitzhandler I, et al. A paclitaxel-loaded recombinant polypeptide nanoparticle outperforms abraxane in multiple murine cancer models. Nat Commun. 2015;6:7939. doi:10.1038/ncomms893926239362
- Overmeyer JH, Kaul A, Johnson EE, Maltese WA. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol Cancer Res. 2008;6(6):965–977. doi:10.1158/1541-7786.MCR-07-203618567800
- Elias DR, Poloukhtine A, Popik V, Tsourkas A. Effect of ligand density, receptor density, and nanoparticle size on cell targeting. Nanomedicine. 2013;9(2):194–201. doi:10.1016/j.nano.2012.05.01522687896
- Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST. Challenges and strategies in anti-cancer nanomedicine development: an industry perspective. Adv Drug Deliv Rev. 2017;108:25–38. doi:10.1016/j.addr.2016.04.02527137110
- Desai N, Trieu V, Yao Z, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res. 2006;12(4):1317–1324. doi:10.1158/1078-0432.CCR-05-163416489089
- Liu H, Sun M, Liu Z, et al. KRAS-enhanced macropinocytosis and reduced FcRn-mediated recycling sensitize pancreatic cancer to albumin-conjugated drugs. J Controlled Release. 2019;296:40–53. doi:10.1016/j.jconrel.2019.01.014
- Eckert LB, Repasky GA, Ülkü AS, et al. Involvement of Ras activation in human breast cancer cell signaling, invasion, and anoikis. Cancer Res. 2004;64(13):4585–4592. doi:10.1158/0008-5472.CAN-04-039615231670
- Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov. 2016;15(11):771–785. doi:10.1038/nrd.2016.13927469033
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–551. doi:10.1038/nature1279624256730
- Leshchiner ES, Parkhitko A, Bird GH, et al. Direct inhibition of oncogenic KRAS by hydrocarbon-stapled SOS1 helices. Proc Natl Acad Sci U S A. 2015;112(6):1761–1766. doi:10.1073/pnas.141318511225624485